
Discovery of Novel Delta Opioid Receptor (DOR) Inverse Agonist and Irreversible (Non-Competitive) Antagonists

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Rational Design of DOR Ligands
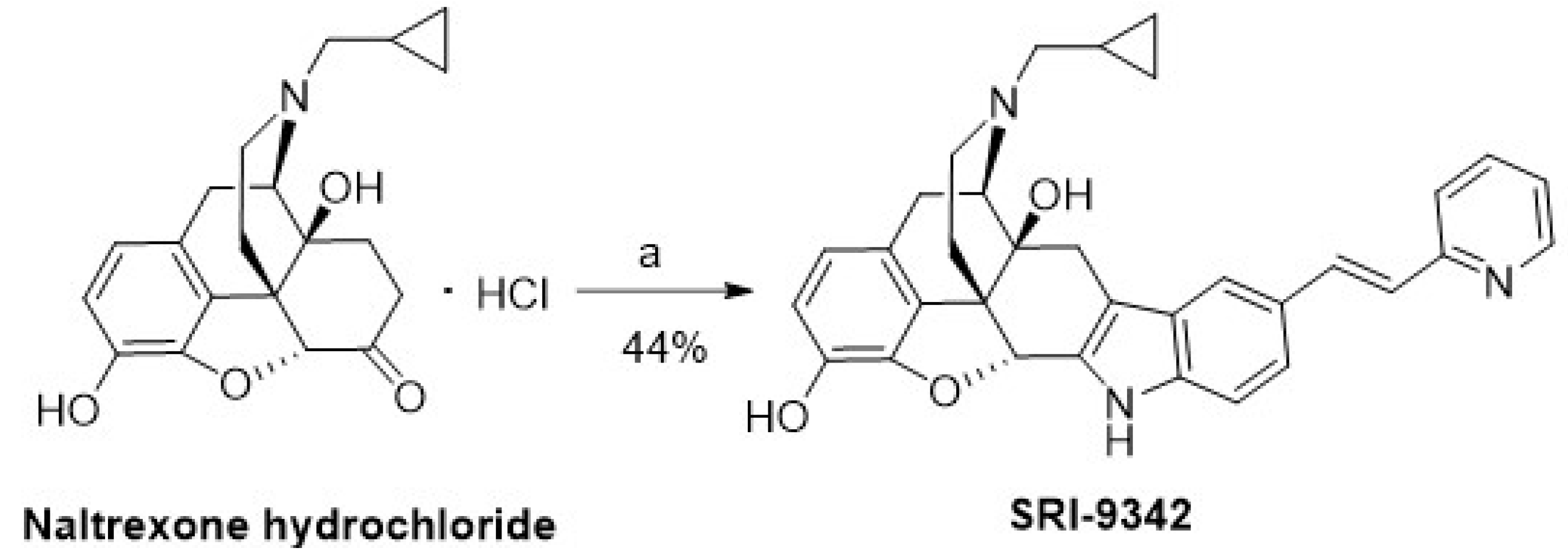
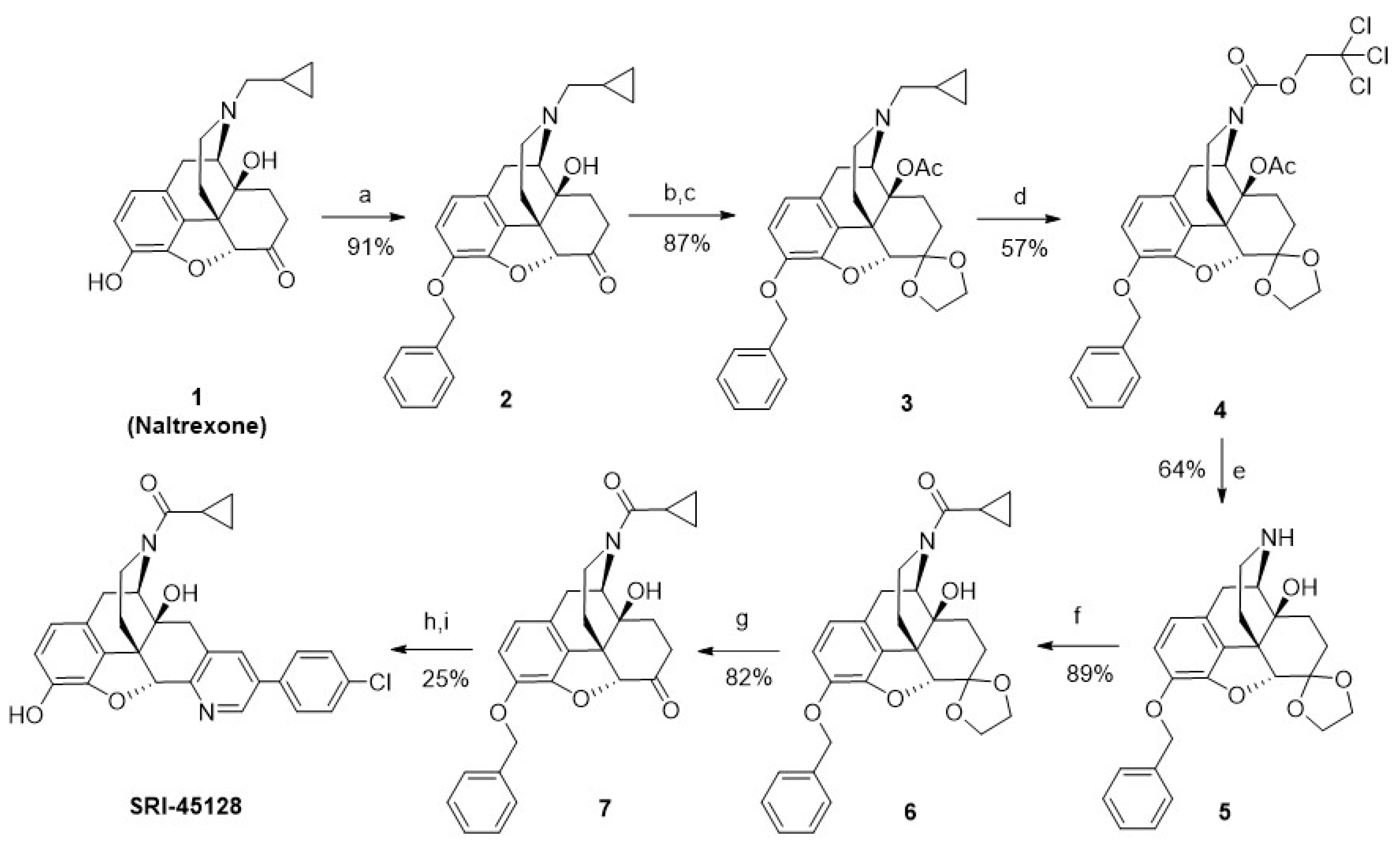
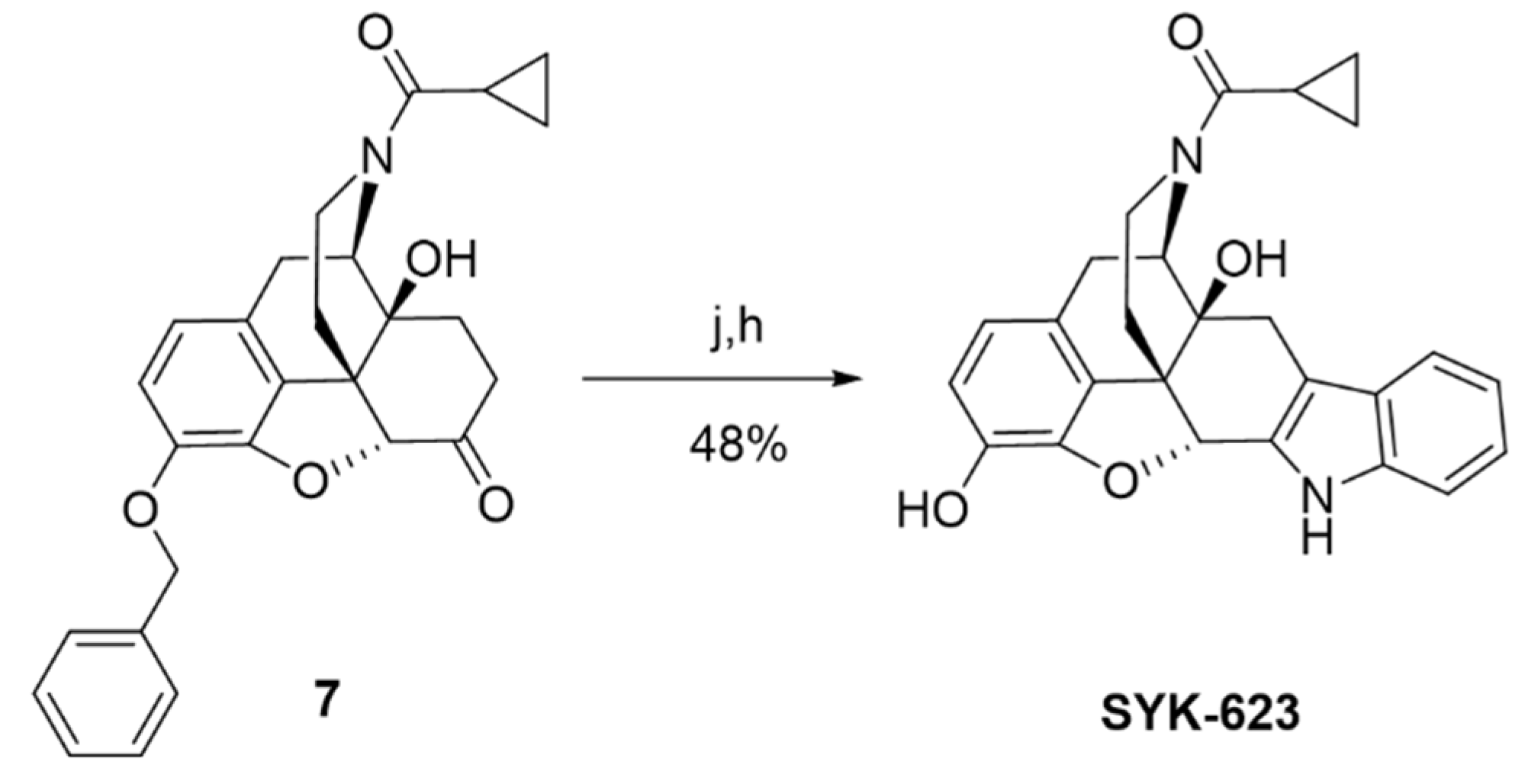
2.2. Synthesis of Novel DOR Irreversible Antagonist and Inverse Agonists
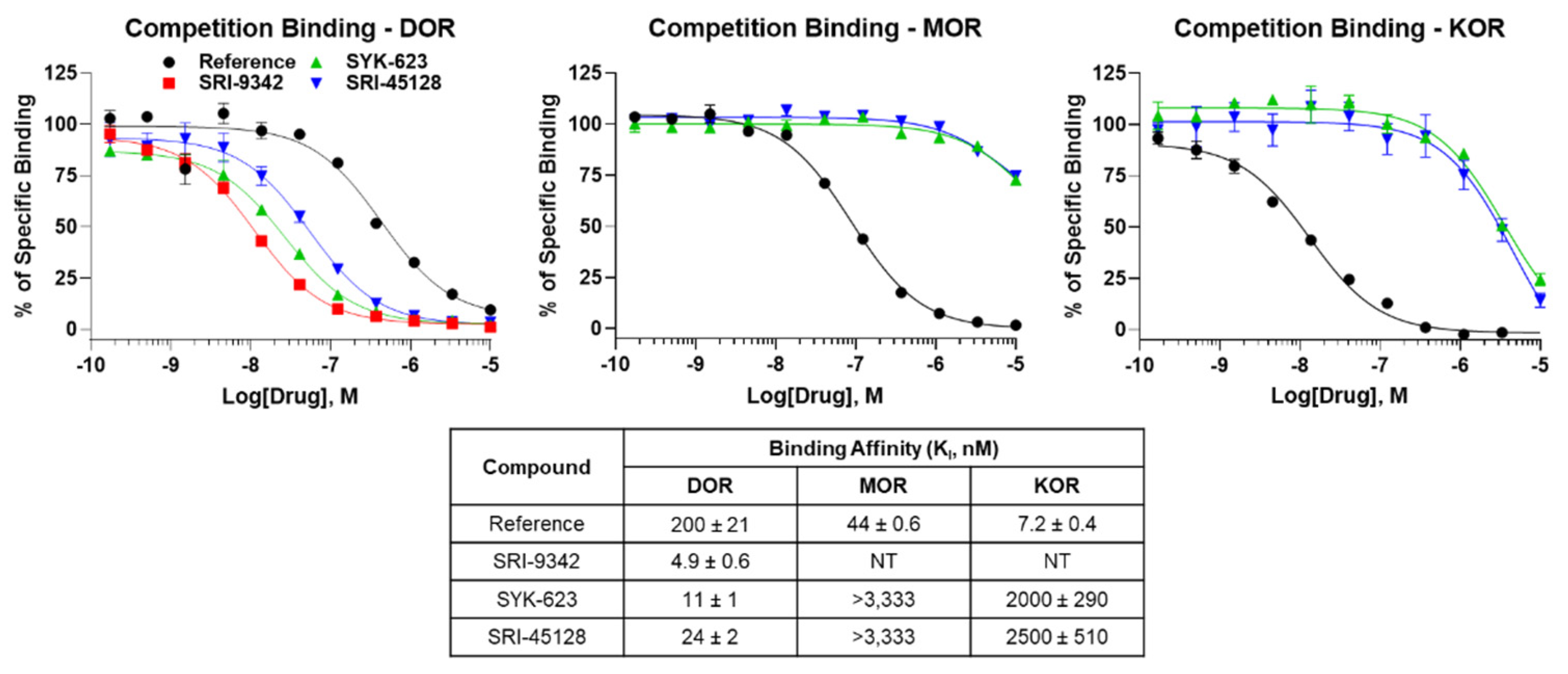
2.3. All Compounds Display High DOR Binding Affinity
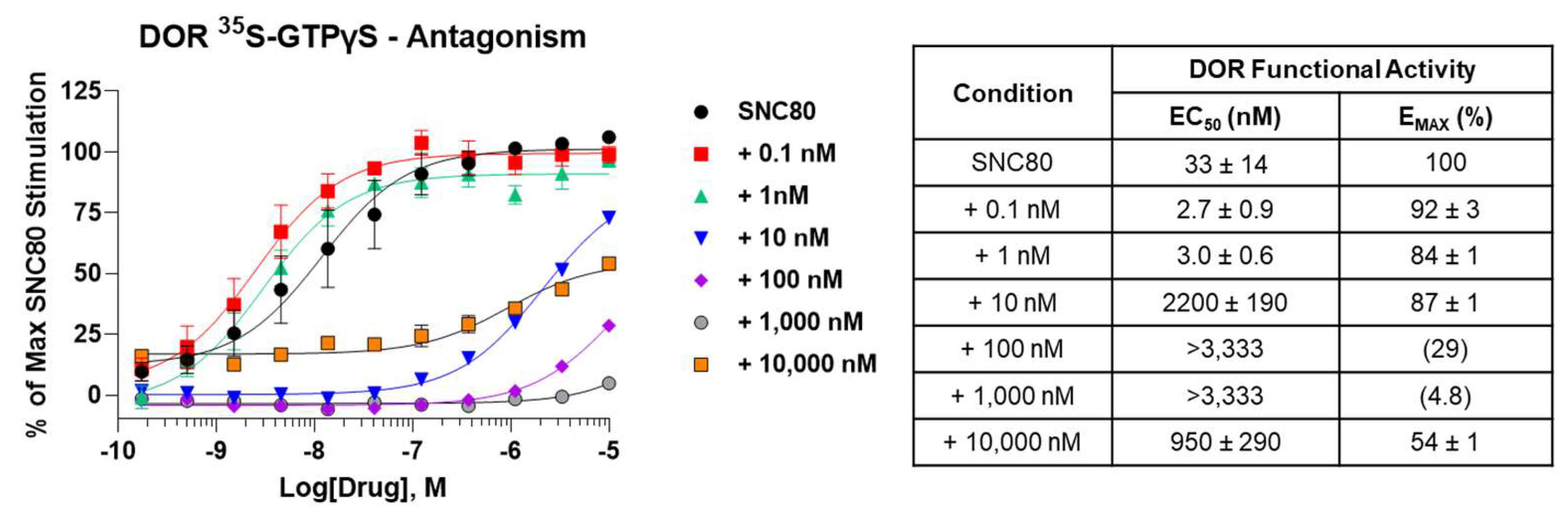
2.4. SRI-9342 Displays Irreversible Antagonism at the DOR at Low Concentrations
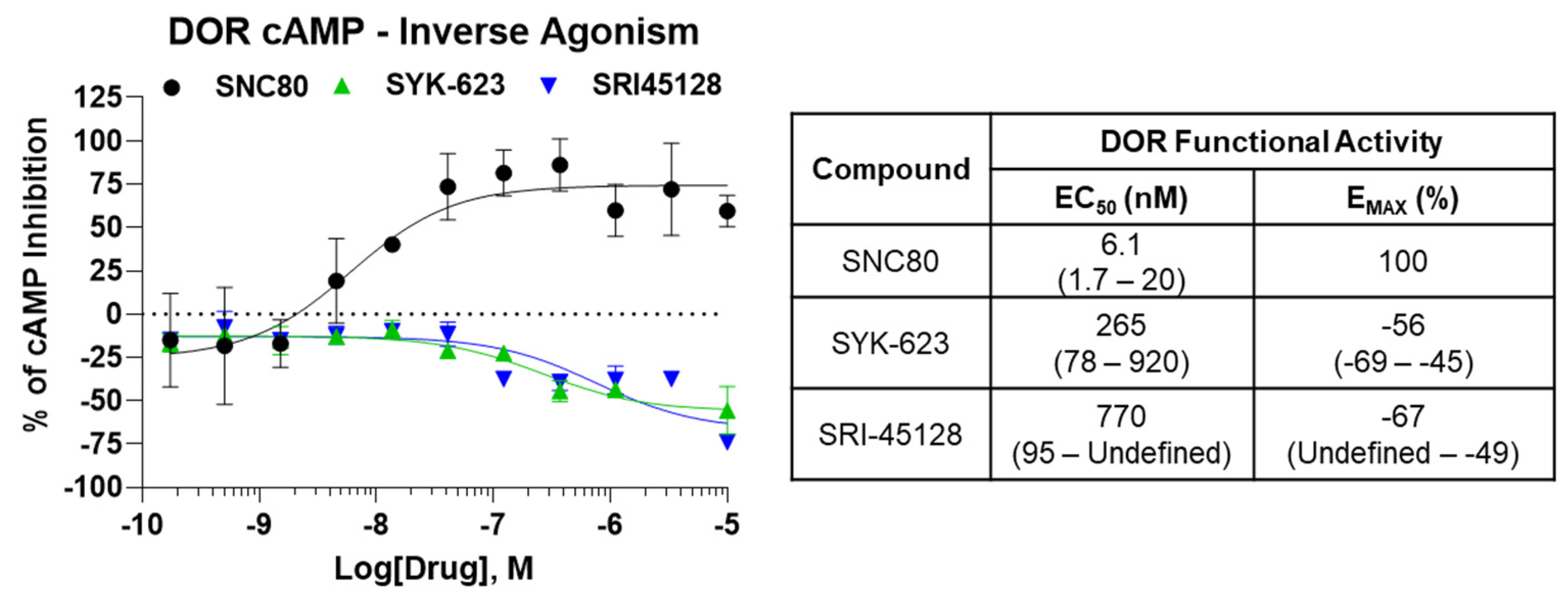
2.5. SRI-45128 Displays DOR Inverse Agonism
3. Discussion
4. Materials and Methods
4.1. Chemical Synthesis and Characterization
4.1.1. (4bS,8R,8aS,14bR)-7-(Cyclopropylmethyl)-11-((E)-2-(pyridin-2-yl)vinyl)-5,6,7,8,14,14b-hexahydro-4,8-methanobenzofuro [2,3-a]pyrido[4,3-b]carbazole-1,8a(9H)-diol (SRI-9342)
4.1.2. ((4bS,8R,8aS,13bR)-11-(4-Chlorophenyl)-1,8a-dihydroxy-5,6,8,8a,9,13b-hexahydro-7H-4,8-methanobenzofuro[3,2-h]pyrido[3,4-g]quinolin-7-yl)(cyclopropyl)methanone (SRI-45128)
4.1.3. 2,2,2-Trichloroethyl (4′R,7a′R,12b′S)-9′-(benzyloxy)-1′,2′,4′,6′-tetrahydro-3′H,7a′H-spiro[[1,3]dioxolane-2,7′-[4,12]methanobenzofuro[3,2-e]isoquinoline]-3′-carboxylate (2)
4.1.4. (4′R,4a′S,7a′R,12b′S)-9′-(Benzyloxy)-3′-(cyclopropylmethyl)-1′,2′,3′,4′,5′,6′-hexahydro-4a′H,7a′H-spiro[[1,3]dioxolane-2,7′-[4,12]methanobenzofuro[3,2-e]isoquinolin]-4a′-yl acetate (3)
4.1.5. 2,2,2-Trichloroethyl (4′R,4a′S,7a′R,12b′S)-4a′-acetoxy-9′-(benzyloxy)-1′,2′,4′,4a′,5′,6′-hexahydro-3′H,7a′H-spiro[[1,3]dioxolane-2,7′-[4,12]methanobenzofuro[3,2-e]isoquinoline]-3′-carboxylate (4)
4.1.6. (4′R,4a′S,7a′R,12b′S)-9′-(Benzyloxy)-1′,2′,3′,4′,5′,6′-hexahydro-4a′H,7a′H-spiro[[1,3]dioxolane-2,7′-[4,12]methanobenzofuro[3,2-e]isoquinolin]-4a′-ol (5)
4.1.7. ((4′R,4a′S,7a′R,12b′S)-9′-(Benzyloxy)-4a′-hydroxy-1′,2′,4′,4a′,5′,6′-hexahydro-3′H,7a′H-spiro[[1,3]dioxolane-2,7′-[4,12]methanobenzofuro[3,2-e]isoquinolin]-3′-yl)(cyclopropyl)methanone (6)
4.1.8. (4R,4aS,7aR,12bS)-9-(Benzyloxy)-3-(cyclopropanecarbonyl)-4a-hydroxy-2,3,4,4a,5,6-hexahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7(7aH)-one (7)
4.1.9. ((4bS,8R,8aS,13bR)-11-(4-Chlorophenyl)-1,8a-dihydroxy-5,6,8,8a,9,13b-hexahydro-7H-4,8-methanobenzofuro[3,2-h]pyrido[3,4-g]quinolin-7-yl)(cyclopropyl)methanone (SRI-45128)
4.1.10. Cyclopropyl((4bS,8R,8aS,14bR)-1,8a-dihydroxy-5,6,8a,9,14,14b-hexahydro-4,8-methanobenzofuro[2,3−a]pyrido [4,3−b]carbazol-7(8H)-yl)methanone (SYK-623)
4.2. Cell Culture
4.3. Competition Radioligand Binding
4.4. 35S-GTPγS Coupling
4.5. cAMP Accumulation Assay
4.6. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, R.J. Endogenous opiates and behavior: 2013. Peptides 2014, 62, 67–136. [Google Scholar] [CrossRef] [PubMed]
- Neubig, R.R.; Spedding, M.; Kenakin, T.; Christopoulos, A. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on terms and symbols in quantitative pharmacology. Pharmacol. Rev. 2003, 55, 597–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craft, R.M.; Tseng, A.H.; McNiel, D.M.; Furness, M.S.; Rice, K.C. Receptor-selective antagonism of opioid antinociception in female versus male rats. Behav. Pharmacol. 2001, 12, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Haris, S.P.; Zhang, Y.; Le Bourdonnec, B.; McCurdy, C.R.; Portoghese, P.S. o-Naphthalenedicarboxaldehyde derivative of 7’-aminonaltrindole as a selective delta-opioid receptor affinity label. J. Med. Chem. 2007, 50, 3392–3396. [Google Scholar] [CrossRef] [PubMed]
- Portoghese, P.S.; Sultana, M.; Takemori, A.E. Naltrindole 5’-isothiocyanate: A nonequilibrium, highly selective delta opioid receptor antagonist. J. Med. Chem. 1990, 33, 1547–1548. [Google Scholar] [CrossRef]
- Costa, T.; Herz, A. Antagonists with negative intrinsic activity at delta opioid receptors coupled to GTP-binding proteins. Proc. Natl. Acad. Sci. USA 1989, 86, 7321–7325. [Google Scholar] [CrossRef] [Green Version]
- Carroll, F.I.; Zhang, L.; Mascarella, S.W.; Navarro, H.A.; Rothman, R.B.; Cantrell, B.E.; Zimmerman, D.M.; Thomas, J.B. Discovery of the first N-substituted 4beta-methyl-5-(3-hydroxyphenyl)morphan to possess highly potent and selective opioid delta receptor antagonist activity. J. Med. Chem. 2004, 47, 281–284. [Google Scholar] [CrossRef]
- Nemoto, T.; Iihara, Y.; Hirayama, S.; Iwai, T.; Higashi, E.; Fujii, H.; Nagase, H. Naltrindole derivatives with fluorinated ethyl substituents on the 17-nitrogen as δ opioid receptor inverse agonists. Bioorg. Med. Chem. Lett. 2015, 25, 2927–2930. [Google Scholar] [CrossRef]
- Iwamatsu, C.; Hayakawa, D.; Kono, T.; Honjo, A.; Ishizaki, S.; Hirayama, S.; Gouda, H.; Fujii, H. Effects of N-Substituents on the Functional Activities of Naltrindole Derivatives for the δ Opioid Receptor: Synthesis and Evaluation of Sulfonamide Derivatives. Molecules 2020, 25, 3792. [Google Scholar] [CrossRef]
- Hosohata, K.; Burkey, T.H.; Alfaro-Lopez, J.; Hruby, V.J.; Roeske, W.R.; Yamamura, H.I. (2S,3R)TMT-L-Tic-OH is a potent inverse agonist at the human delta-opioid receptor. Eur. J. Pharmacol. 1999, 380, R9–R10. [Google Scholar] [CrossRef]
- Martin, N.A.; Ruckle, M.B.; VanHoof, S.L.; Prather, P.L. Agonist, antagonist, and inverse agonist characteristics of TIPP (H-Tyr-Tic-Phe-Phe-OH), a selective delta-opioid receptor ligand. J. Pharmacol. Exp. Ther. 2002, 301, 661–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labarre, M.; Butterworth, J.; St-Onge, S.; Payza, K.; Schmidhammer, H.; Salvadori, S.; Balboni, G.; Guerrini, R.; Bryant, S.D.; Lazarus, L.H. Inverse agonism by Dmt-Tic analogues and HS 378, a naltrindole analogue. Eur. J. Pharmacol. 2000, 406, R1–R3. [Google Scholar] [CrossRef]
- Zaki, P.A.; Keith, D.E., Jr.; Thomas, J.B.; Carroll, F.I.; Evans, C.J. Agonist-, antagonist-, and inverse agonist-regulated trafficking of the delta-opioid receptor correlates with, but does not require, G protein activation. J. Pharmacol. Exp. Ther. 2001, 298, 1015–1020. [Google Scholar] [PubMed]
- Emmerson, P.J.; McKinzie, J.H.; Surface, P.L.; Suter, T.M.; Mitch, C.H.; Statnick, M.A. Na+ modulation, inverse agonism, and anorectic potency of 4-phenylpiperidine opioid antagonists. Eur. J. Pharmacol. 2004, 494, 121–130. [Google Scholar] [CrossRef]
- Thomas, J.B.; Zhang, L.; Navarro, H.A.; Carroll, F.I. Highly potent and selective phenylmorphan-based inverse agonists of the opioid delta receptor. J. Med. Chem. 2006, 49, 5597–5609. [Google Scholar] [CrossRef]
- Cheng, K.; Kim, I.J.; Lee, M.J.; Adah, S.A.; Raymond, T.J.; Bilsky, E.J.; Aceto, M.D.; May, E.L.; Harris, L.S.; Coop, A.; et al. Opioid ligands with mixed properties from substituted enantiomeric N-phenethyl-5-phenylmorphans. Synthesis of a micro-agonist delta-antagonist and delta-inverse agonists. Org. Biomol. Chem. 2007, 5, 1177–1190. [Google Scholar] [CrossRef]
- Vekariya, R.H.; Lei, W.; Ray, A.; Sainai, S.K.; Zhang, S.; Molnar, G.; Barlow, D.; Karlage, K.L.; Bilsky, E.; Houseknecht, K.; et al. Synthesis and Structure-Activity Relationships of 5’-Aryl-14-alkoxypyridomorphinans: Identification of a Mu Opioid Receptor Agonist/Delta Opioid Receptor Antagonist Ligand with Systemic Antinociceptive Activity and Diminished Opioid Side Effects. J. Med. Chem. 2020, 63, 7663–7694. [Google Scholar] [CrossRef]
- Higashi, E.; Hirayama, S.; Nikaido, J.; Shibasaki, M.; Kono, T.; Honjo, A.; Ikeda, H.; Kamei, J.; Fujii, H. Development of Novel delta Opioid Receptor Inverse Agonists without a Basic Nitrogen Atom and Their Antitussive Effects in Mice. ACS Chem. Neurosci. 2019, 10, 3939–3945. [Google Scholar] [CrossRef]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular control of delta-opioid receptor signalling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Claff, T.; Yu, J.; Blais, V.; Patel, N.; Martin, C.; Wu, L.; Han, G.W.; Holleran, B.J.; Van der Poorten, O.; White, K.L.; et al. Elucidating the active delta-opioid receptor crystal structure with peptide and small-molecule agonists. Sci. Adv. 2019, 5, eaax9115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Keenan, S.M.; Zhang, Q.; Kholodovych, V.; Welsh, W.J. 3D-QSAR comparative molecular field analysis on opioid receptor antagonists: Pooling data from different studies. J. Med. Chem. 2005, 48, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Ananthan, S.; Johnson, C.A.; Carter, R.L.; Clayton, S.D.; Rice, K.C.; Xu, H.; Davis, P.; Porreca, F.; Rothman, R.B. Synthesis, opioid receptor binding, and bioassay of naltrindole analogues substituted in the indolic benzene moiety. J. Med. Chem. 1998, 41, 2872–2881. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Yamamoto, N.; Yata, M.; Ohrui, S.; Okada, T.; Saitoh, T.; Kutsumura, N.; Nagumo, Y.; Irukayama-Tomobe, Y.; Ishikawa, Y.; et al. Design and Synthesis of Potent and Highly Selective Orexin 1 Receptor Antagonists with a Morphinan Skeleton and Their Pharmacologies. J. Med. Chem. 2017, 60, 1018–1040. [Google Scholar] [CrossRef]
- Jiang, Q.; Takemori, A.E.; Sultana, M.; Portoghese, P.S.; Bowen, W.D.; Mosberg, H.I.; Porreca, F. Differential antagonism of opioid delta antinociception by [D-Ala2,Leu5,Cys6]enkephalin and naltrindole 5’-isothiocyanate: Evidence for delta receptor subtypes. J. Pharmacol. Exp. Ther. 1991, 257, 1069–1075. [Google Scholar]
- Hirayama, S.; Iwai, T.; Higashi, E.; Nakamura, M.; Iwamatsu, C.; Itoh, K.; Nemoto, T.; Tanabe, M.; Fujii, H. Discovery of delta Opioid Receptor Full Inverse Agonists and Their Effects on Restraint Stress-Induced Cognitive Impairment in Mice. ACS Chem. Neurosci. 2019, 10, 2237–2242. [Google Scholar] [CrossRef]
- Teng, L.; Zhao, J.; Wang, F.; Ma, L.; Pei, G. A GPCR/secretase complex regulates beta- and gamma-secretase specificity for Abeta production and contributes to AD pathogenesis. Cell Res. 2010, 20, 138–153. [Google Scholar] [CrossRef]
- Burtscher, J.; Schwarzer, C. The Opioid System in Temporal Lobe Epilepsy: Functional Role and Therapeutic Potential. Front. Mol. Neurosci. 2017, 10, 245. [Google Scholar] [CrossRef]
- Jutkiewicz, E.M.; Baladi, M.G.; Folk, J.E.; Rice, K.C.; Woods, J.H. The convulsive and electroencephalographic changes produced by nonpeptidic δ-opioid agonists in rats: Comparison with pentylenetetrazol. J. Pharmacol. Exp. Ther. 2006, 317, 1337–1348. [Google Scholar] [CrossRef]
- Ling, G.S.; Simantov, R.; Clark, J.A.; Pasternak, G.W. Naloxonazine actions in vivo. Eur. J. Pharmacol. 1986, 129, 33–38. [Google Scholar] [CrossRef]
- LaVigne, J.; Keresztes, A.; Chiem, D.; Streicher, J.M. The endomorphin-1/2 and dynorphin-B peptides display biased agonism at the mu opioid receptor. Pharmacol. Rep. 2020, 72, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Stefanucci, A.; Dimmito, M.P.; Macedonio, G.; Ciarlo, L.; Pieretti, S.; Novellino, E.; Lei, W.; Barlow, D.; Houseknecht, K.L.; Streicher, J.M.; et al. Potent, Efficacious, and Stable Cyclic Opioid Peptides with Long Lasting Antinociceptive Effect after Peripheral Administration. J. Med. Chem. 2020, 63, 2673–2687. [Google Scholar] [CrossRef] [PubMed]
- LaVigne, J.E.; Hecksel, R.; Keresztes, A.; Streicher, J.M. Cannabis sativa terpenes are cannabimimetic and selectively enhance cannabinoid activity. Sci. Rep. 2021, 11, 8232. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.M.; Duron, D.I.; Womer, D.; Fell, R.; Streicher, J.M. Comprehensive molecular pharmacology screening reveals potential new receptor interactions for clinically relevant opioids. PLoS ONE 2019, 14, e0217371. [Google Scholar] [CrossRef] [Green Version]
- Stefanucci, A.; Dimmito, M.P.; Molnar, G.; Streicher, J.M.; Novellino, E.; Zengin, G.; Mollica, A. Developing Cyclic Opioid Analogues: Fluorescently Labeled Bioconjugates of Biphalin. ACS Med. Chem. Lett. 2020, 11, 720–726. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanguturi, P.; Pathak, V.; Zhang, S.; Moukha-Chafiq, O.; Augelli-Szafran, C.E.; Streicher, J.M. Discovery of Novel Delta Opioid Receptor (DOR) Inverse Agonist and Irreversible (Non-Competitive) Antagonists. Molecules 2021, 26, 6693. https://doi.org/10.3390/molecules26216693
Tanguturi P, Pathak V, Zhang S, Moukha-Chafiq O, Augelli-Szafran CE, Streicher JM. Discovery of Novel Delta Opioid Receptor (DOR) Inverse Agonist and Irreversible (Non-Competitive) Antagonists. Molecules. 2021; 26(21):6693. https://doi.org/10.3390/molecules26216693
Chicago/Turabian StyleTanguturi, Parthasaradhireddy, Vibha Pathak, Sixue Zhang, Omar Moukha-Chafiq, Corinne E. Augelli-Szafran, and John M. Streicher. 2021. "Discovery of Novel Delta Opioid Receptor (DOR) Inverse Agonist and Irreversible (Non-Competitive) Antagonists" Molecules 26, no. 21: 6693. https://doi.org/10.3390/molecules26216693
APA StyleTanguturi, P., Pathak, V., Zhang, S., Moukha-Chafiq, O., Augelli-Szafran, C. E., & Streicher, J. M. (2021). Discovery of Novel Delta Opioid Receptor (DOR) Inverse Agonist and Irreversible (Non-Competitive) Antagonists. Molecules, 26(21), 6693. https://doi.org/10.3390/molecules26216693