
Understanding the Mechanism of Action of NAI-112, a Lanthipeptide with Potent Antinociceptive Activity

 , ,
, , 
Abstract
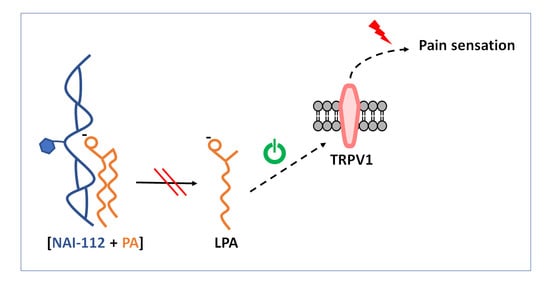
:
1. Introduction
2. Results
2.1. Lipidome Analysis in the Spinal Cord
2.2. Effects on Enzymes in the TPRV1 Pathway
2.3. Binding Experiments
2.4. Isolation of NAI-112-Resistant Mutants
2.5. Genome Analysis of Resistant Strains
3. Discussion
4. Materials and Methods
4.1. NAI-112 Source
4.2. Mice Study
4.3. Untargeted Lipidomics Sample Preparation
4.4. Mass Spectrometer Data Acquisition
4.5. Multivariate Data Analysis and Lipid Identification
4.6. Pain-Related Receptor Antagonist Effect
4.7. Enzymatic Assays
4.8. Binding Experiments
4.9. Selection of Resistant Strains by Direct Plating and Serial Passages
4.10. Antibacterial Assays
4.11. Genome Sequencing and Bioinformatic Analysis
4.12. Nucleotide Sequence Accession Numbers
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; van der Donk, W.A. New Insights into the Biosynthetic Logic of Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products. Cell Chem. Biol. 2016, 23, 31–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, M.; Bruner, S.D.; Ding, Y. Heterologous Production of Microbial Ribosomally Synthesized and Post-Translationally Modified Peptides. Front. Microbiol. 2018, 9, 1801. [Google Scholar] [CrossRef]
- Wang, H.; van der Donk, W.A. Biosynthesis of the Class III Lantipeptide Catenulipeptin. ACS Chem. Biol. 2012, 7, 1529–1535. [Google Scholar] [CrossRef]
- Zdouc, M.M.; Alanjary, M.M.; Zarazúa, G.S.; Maffioli, S.I.; Crüsemann, M.; Medema, M.H.; Donadio, S.; Sosio, M. A Biaryl-Linked Tripeptide from Planomonospora Reveals a Widespread Class of Minimal RiPP Gene Clusters. Cell Chem. Biol. 2021, 28, 733–739.e4. [Google Scholar] [CrossRef]
- Iorio, M.; Sasso, O.; Maffioli, S.I.; Bertorelli, R.; Monciardini, P.; Sosio, M.; Bonezzi, F.; Summa, M.; Brunati, C.; Bordoni, R.; et al. A Glycosylated, Labionin-Containing Lanthipeptide with Marked Antinociceptive Activity. ACS Chem. Biol. 2014, 9, 398–404. [Google Scholar] [CrossRef] [Green Version]
- Meindl, K.; Schmiederer, T.; Schneider, K.; Reicke, A.; Butz, D.; Keller, S.; Gühring, H.; Vértesy, L.; Wink, J.; Hoffmann, H.; et al. Labyrinthopeptins: A New Class of Carbacyclic Lantibiotics. Angew. Chem. Int. Ed. Engl. 2010, 49, 1151–1154. [Google Scholar] [CrossRef]
- Yekkirala, A.S.; Roberson, D.P.; Bean, B.P.; Woolf, C.J. Breaking Barriers to Novel Analgesic Drug Development. Nat. Rev. Drug Discov. 2017, 16, 810. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, V. New Approaches and Challenges to Targeting the Endocannabinoid System. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Rashid, M.H.; Fujita, R.; Contos, J.J.A.; Chun, J.; Ueda, H. Initiation of Neuropathic Pain Requires Lysophosphatidic Acid Receptor Signaling. Nat. Med. 2004, 10, 712–718. [Google Scholar] [CrossRef]
- Ueda, H.; Matsunaga, H.; Olaposi, O.I.; Nagai, J. Lysophosphatidic Acid: Chemical Signature of Neuropathic Pain. Biochim. Biophys. Acta 2013, 1831, 61–73. [Google Scholar] [CrossRef]
- Santos-Nogueira, E.; López-Serrano, C.; Hernández, J.; Lago, N.; Astudillo, A.M.; Balsinde, J.; Estivill-Torrús, G.; de Fonseca, F.R.; Chun, J.; López-Vales, R. Activation of Lysophosphatidic Acid Receptor Type 1 Contributes to Pathophysiology of Spinal Cord Injury. J. Neurosci. 2015, 35, 10224–10235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, E.; Cordero-Morales, J.F.; Liu, B.; Qin, F.; Julius, D. TRPV1 Channels Are Intrinsically Heat Sensitive and Negatively Regulated by Phosphoinositide Lipids. Neuron 2013, 77, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Müller, A.; Klöckner, A.; Schneider, T. Targeting a Cell Wall Biosynthesis Hot Spot. Nat. Prod. Rep. 2017, 34, 909–932. [Google Scholar] [CrossRef] [PubMed]
- Grein, F.; Schneider, T.; Sahl, H.-G. Docking on Lipid II-A Widespread Mechanism for Potent Bactericidal Activities of Antibiotic Peptides. J. Mol. Biol. 2019, 431, 3520–3530. [Google Scholar] [CrossRef]
- Brunati, C.; Thomsen, T.T.; Gaspari, E.; Maffioli, S.; Sosio, M.; Jabes, D.; Løbner-Olesen, A.; Donadio, S. Expanding the Potential of NAI-107 for Treating Serious ESKAPE Pathogens: Synergistic Combinations against Gram-Negatives and Bactericidal Activity against Non-Dividing Cells. J. Antimicrob. Chemother. 2018, 73, 414–424. [Google Scholar] [CrossRef] [Green Version]
- Jabes, D.; Brunati, C.; Candiani, G.; Riva, S.; Romanó, G.; Maffioli, S.; Rossi, R.; Simone, M.; Gaspari, E.; Donadio, S. Pharmacological Properties of NAI-603, a Well-Tolerated Semisynthetic Derivative of Ramoplanin. Antimicrob. Agents Chemother. 2014, 58, 1922–1929. [Google Scholar] [CrossRef] [Green Version]
- Howden, B.P.; McEvoy, C.R.E.; Allen, D.L.; Chua, K.; Gao, W.; Harrison, P.F.; Bell, J.; Coombs, G.; Bennett-Wood, V.; Porter, J.L.; et al. Evolution of Multidrug Resistance during Staphylococcus Aureus Infection Involves Mutation of the Essential Two Component Regulator WalKR. PLoS Pathog. 2011, 7, e1002359. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, X.; Liu, X.; Chen, C.; Sun, B. Mechanism of Reduced Vancomycin Susceptibility Conferred by WalK Mutation in Community-Acquired Methicillin-Resistant Staphylococcus Aureus Strain MW2. Antimicrob. Agents Chemother. 2015, 59, 1352–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubrac, S.; Msadek, T. Tearing down the Wall: Peptidoglycan Metabolism and the WalK/WalR (YycG/YycF) Essential Two-Component System. Adv. Exp. Med. Biol. 2008, 631, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Graf, A.; Lewis, R.J.; Fuchs, S.; Pagels, M.; Engelmann, S.; Riedel, K.; Pané-Farré, J. The Hidden Lipoproteome of Staphylococcus Aureus. Int. J. Med. Microbiol. 2018, 308, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Peng, H.; Rao, X. Molecular Events for Promotion of Vancomycin Resistance in Vancomycin Intermediate Staphylococcus Aureus. Front. Microbiol. 2016, 7, 1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Lee, K.J. Single-Nucleotide Polymorphisms in a Vancomycin-Resistant Staphylococcus Aureus Strain Based on Whole-Genome Sequencing. Arch. Microbiol. 2020, 202, 2255–2261. [Google Scholar] [CrossRef]
- Nieto-Posadas, A.; Picazo-Juárez, G.; Llorente, I.; Jara-Oseguera, A.; Morales-Lázaro, S.; Escalante-Alcalde, D.; Islas, L.D.; Rosenbaum, T. Lysophosphatidic Acid Directly Activates TRPV1 through a C-Terminal Binding Site. Nat. Chem. Biol. 2011, 8, 78–85. [Google Scholar] [CrossRef]
- Münch, D.; Müller, A.; Schneider, T.; Kohl, B.; Wenzel, M.; Bandow, J.E.; Maffioli, S.; Sosio, M.; Donadio, S.; Wimmer, R.; et al. The Lantibiotic NAI-107 Binds to Bactoprenol-Bound Cell Wall Precursors and Impairs Membrane Functions. J. Biol. Chem. 2014, 289, 12063–12076. [Google Scholar] [CrossRef] [Green Version]
- Scherer, K.M.; Spille, J.-H.; Sahl, H.-G.; Grein, F.; Kubitscheck, U. The Lantibiotic Nisin Induces Lipid II Aggregation, Causing Membrane Instability and Vesicle Budding. Biophys. J. 2015, 108, 1114–1124. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Yin, Z.; Breukink, E.; Moll, G.N.; Kuipers, O.P. An Engineered Double Lipid II Binding Motifs-Containing Lantibiotic Displays Potent and Selective Antimicrobial Activity against Enterococcus Faecium. Antimicrob. Agents Chemother. 2020, 64, e02050-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brötz, H.; Sahl, H.G. New Insights into the Mechanism of Action of Lantibiotics--Diverse Biological Effects by Binding to the Same Molecular Target. J. Antimicrob. Chemother. 2000, 46, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Märki, F.; Hänni, E.; Fredenhagen, A.; van Oostrum, J. Mode of Action of the Lanthionine-Containing Peptide Antibiotics Duramycin, Duramycin B and C, and Cinnamycin as Indirect Inhibitors of Phospholipase A2. Biochem. Pharmacol. 1991, 42, 2027–2035. [Google Scholar] [CrossRef]
- Zhao, M.; Li, Z.; Bugenhagen, S. 99mTc-Labeled Duramycin as a Novel Phosphatidylethanolamine-Binding Molecular Probe. J. Nucl. Med. 2008, 49, 1345–1352. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M. Lantibiotics as Probes for Phosphatidylethanolamine. Amino Acids 2011, 41, 1071–1079. [Google Scholar] [CrossRef] [Green Version]
- Vestergaard, M.; Berglund, N.A.; Hsu, P.-C.; Song, C.; Koldsø, H.; Schiøtt, B.; Sansom, M.S.P. Structure and Dynamics of Cinnamycin-Lipid Complexes: Mechanisms of Selectivity for Phosphatidylethanolamine Lipids. ACS Omega 2019, 4, 18889–18899. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, I.; Böttiger, T.; Bonelli, R.R.; Wiese, A.; Hagge, S.O.; Gutsmann, T.; Seydel, U.; Deegan, L.; Hill, C.; Ross, P.; et al. The Mode of Action of the Lantibiotic Lacticin 3147—A Complex Mechanism Involving Specific Interaction of Two Peptides and the Cell Wall Precursor Lipid II. Mol. Microbiol. 2006, 61, 285–296. [Google Scholar] [CrossRef]
- Medeiros-Silva, J.; Jekhmane, S.; Paioni, A.L.; Gawarecka, K.; Baldus, M.; Swiezewska, E.; Breukink, E.; Weingarth, M. High-Resolution NMR Studies of Antibiotics in Cellular Membranes. Nat. Commun. 2018, 9, 3963. [Google Scholar] [CrossRef] [Green Version]
- Prochnow, H.; Rox, K.; Birudukota, N.V.S.; Weichert, L.; Hotop, S.-K.; Klahn, P.; Mohr, K.; Franz, S.; Banda, D.H.; Blockus, S.; et al. Labyrinthopeptins Exert Broad-Spectrum Antiviral Activity through Lipid-Binding-Mediated Virolysis. J. Virol. 2020, 94, e01471-19. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.; Croixmarie, V.; Priestman, D.A.; Rosenbohm, A.; Dirrig-Grosch, S.; D’Ambra, E.; Huebecker, M.; Hussain, G.; Boursier-Neyret, C.; Echaniz-Laguna, A.; et al. Amyotrophic Lateral Sclerosis and Denervation Alter Sphingolipids and Up-Regulate Glucosylceramide Synthase. Hum. Mol. Genet. 2015, 24, 7390–7405. [Google Scholar] [CrossRef] [Green Version]
- Tautenhahn, R.; Cho, K.; Uritboonthai, W.; Zhu, Z.; Patti, G.J.; Siuzdak, G. An Accelerated Workflow for Untargeted Metabolomics Using the METLIN Database. Nat. Biotechnol. 2012, 30, 826–828. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Comprehensive Analysis of Lipids in Biological Systems by Liquid Chromatography-Mass Spectrometry. Trends Anal. Chem. 2014, 61, 192–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, R.C.; Barkley, R.M.; Zemski Berry, K.; Hankin, J.; Harrison, K.; Johnson, C.; Krank, J.; McAnoy, A.; Uhlson, C.; Zarini, S. Electrospray Ionization and Tandem Mass Spectrometry of Eicosanoids. Anal. Biochem. 2005, 346, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.A.; Milani, C.; Mancabelli, L.; van Sinderen, D.; Ventura, M. MEGAnnotator: A User-Friendly Pipeline for Microbial Genomes Assembly and Annotation. FEMS Microbiol. Lett. 2016, 363, fnw049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, A.E.; Mau, B.; Perna, N.T. ProgressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic | Target | MIC (µg/mL) | MIC Ratio (R15.1 to wt) | ||
|---|---|---|---|---|---|
| wt | R8.1 | R15.5 | |||
| NAI-112 | 16 | 64 | 128 | 8 | |
| Vancomycin | Lipid II | 0.5 | 1 | 2 | 4 |
| NAI-107 * | Lipid II | 0.125 | 0.25 | 0.25 | 2 |
| Ramoplanin * | Lipid II | 0.06 | 0.25 | 0.25 | 4 |
| Erythromycin | Protein synthesis | 0.125 | 0.125 | 0.125 | 1 |
| Ciprofloxacin | DNA replication | 0.12 | 0.12 | 0.12 | 1 |
| Rifampicin | Transcription | 0.004 | 0.004 | 0.004 | 1 |
| Mutant Strain | Genome Position (nt) a | CDS (Locus) a | Type of Mutation | Nucleotide Change | Amino Acid Change | Function |
|---|---|---|---|---|---|---|
| R8.1 R15.3 R15.4 R15.5 | 27431 | SAFDA_0019 | SNP | G to A | Cys598Tyr | WalK (two-component sensor histidine kinase) |
| 587945 | SAFDA_0507 to SAFDA_0508 | SNP | C to A b | Non-coding | Poly(glycerol-phosphate) alpha-glucosyltransferase –GTP cyclohydrolase | |
| 1340002 | SAFDA_1245 to SAFDA_1246 | SNP | G to A | Non-coding | Glycine betaine transporter—aconitate hydratase | |
| 1357466 | SAFDA_1256 | SNP | T to C b | Synonymous | Transposase | |
| 1721972 | SAFDA_1584 | SNP | A to G | Synonymous | Truncated transposase | |
| 1721999 | SNP | T to C | Synonymous | |||
| R8.1 R15.3 R15.5 | 1722017 | SNP | T to C | Synonymous | ||
| 523949 | SAFDA_r0010 to SAFDA_0460 | SNP | T to G b | Non-coding | 5S ribosomal RNA—GntR family transcriptional regulator | |
| R15.3 R15.4 R15.5 | 1523918 | SAFDA_1386 | SNP | G to A | Synonymous | Hypothetical protein |
| 1523955 | SNP | A to G | Synonymous | |||
| R8.1 R15.4 | 1523670 | SNP | A to T b | Asp145Glu | ||
| R15.3 R15.5 | 1523691 | SNP | A to T | Asn138Lys | ||
| 1481440 | SAFDA_1346 to SAFDA_1347 | SNP | A to T | Non-coding | Penicillin-binding protein 2—hypothetical protein | |
| 1481441 | DEL | -G | ||||
| R15.3 R15.4 | 512393 | SAFDA_0459 to SAFDA_r0004 | SNP | A to T | Non-coding | Lysyl-tRNA synthetase—5S ribosomal RNA |
| R15.4 R15.5 | 965417 | SAFDA_0882 to SAFDA_0883 | SNP | C to A b | Non-coding | Hypothetical protein—Na+ alanine symporter |
| R8.1 R15.4 | 751022 | SAFDA_0672 | SNP | C to A | Ala451Ser | Di-/tripeptide ABC transporter |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tocchetti, A.; Iorio, M.; Hamid, Z.; Armirotti, A.; Reggiani, A.; Donadio, S. Understanding the Mechanism of Action of NAI-112, a Lanthipeptide with Potent Antinociceptive Activity. Molecules 2021, 26, 6764. https://doi.org/10.3390/molecules26226764
Tocchetti A, Iorio M, Hamid Z, Armirotti A, Reggiani A, Donadio S. Understanding the Mechanism of Action of NAI-112, a Lanthipeptide with Potent Antinociceptive Activity. Molecules. 2021; 26(22):6764. https://doi.org/10.3390/molecules26226764
Chicago/Turabian StyleTocchetti, Arianna, Marianna Iorio, Zeeshan Hamid, Andrea Armirotti, Angelo Reggiani, and Stefano Donadio. 2021. "Understanding the Mechanism of Action of NAI-112, a Lanthipeptide with Potent Antinociceptive Activity" Molecules 26, no. 22: 6764. https://doi.org/10.3390/molecules26226764
APA StyleTocchetti, A., Iorio, M., Hamid, Z., Armirotti, A., Reggiani, A., & Donadio, S. (2021). Understanding the Mechanism of Action of NAI-112, a Lanthipeptide with Potent Antinociceptive Activity. Molecules, 26(22), 6764. https://doi.org/10.3390/molecules26226764