
Identification of a Novel Delta Opioid Receptor Agonist Chemotype with Potential Negative Allosteric Modulator Capabilities

 ,
, 
Abstract
:1. Introduction
2. Results
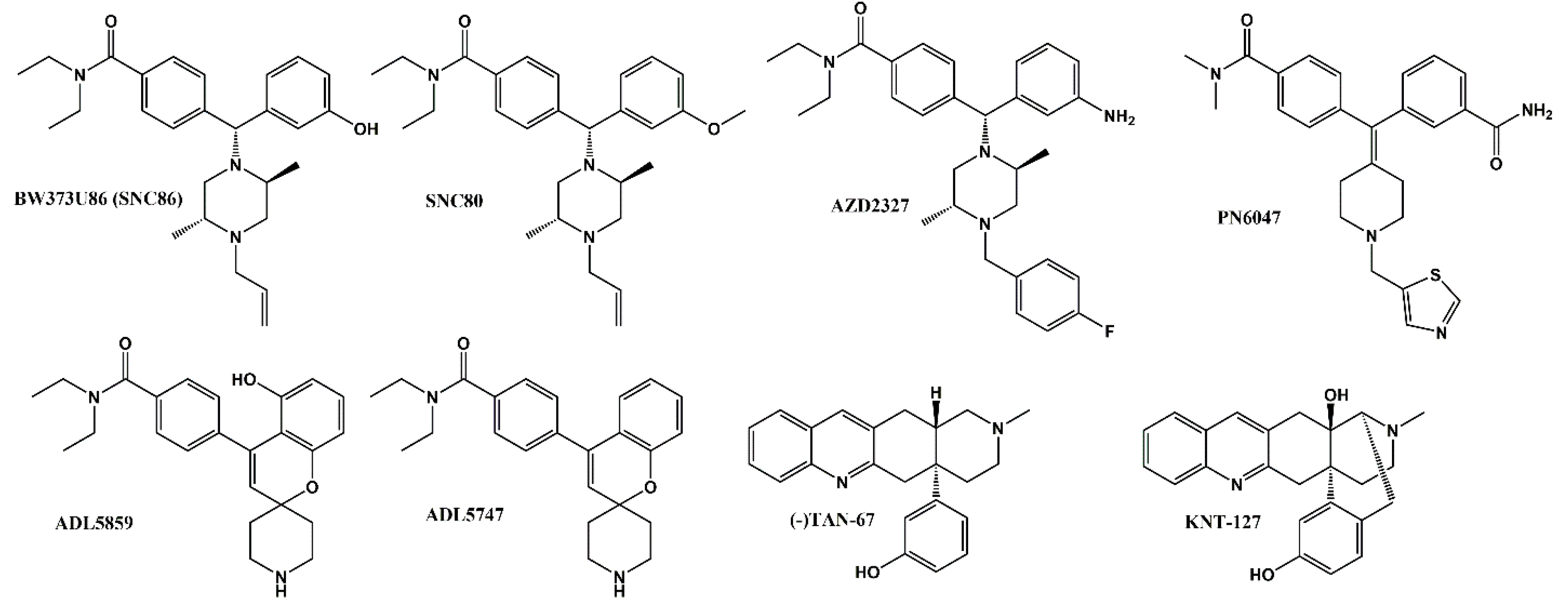
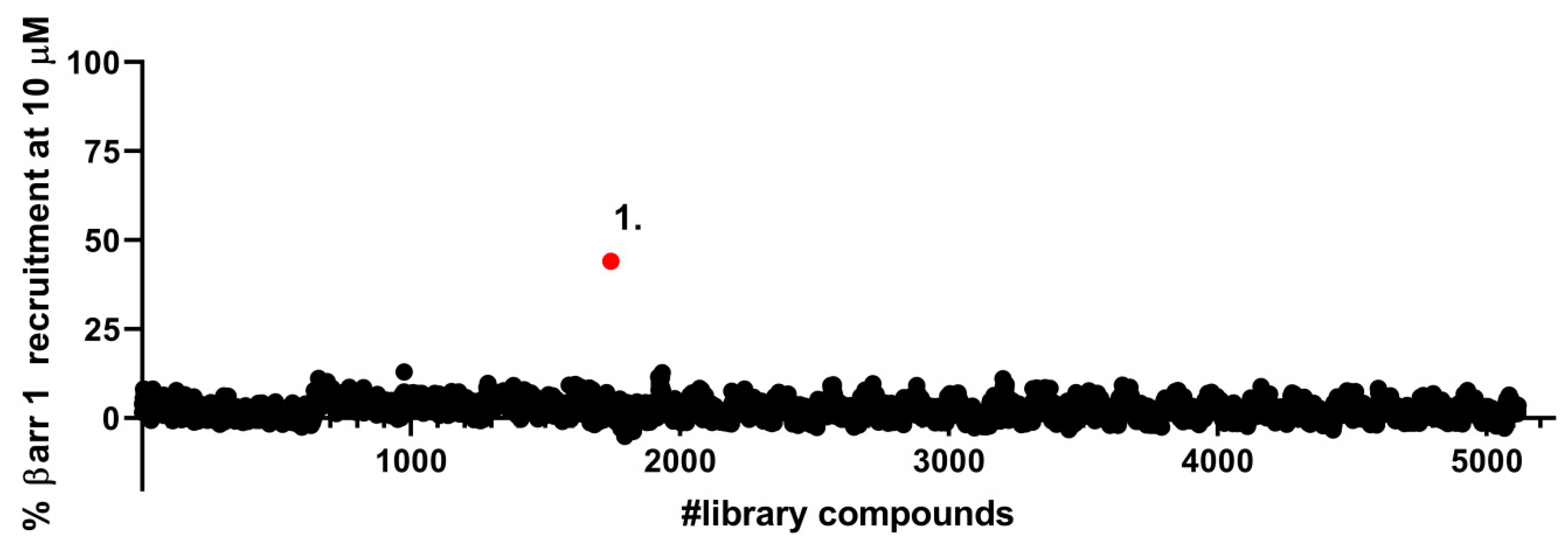
2.1. Identification of a Novel δOR Agonist with Sub-Maximal β-Arrestin Recruitment Efficacy
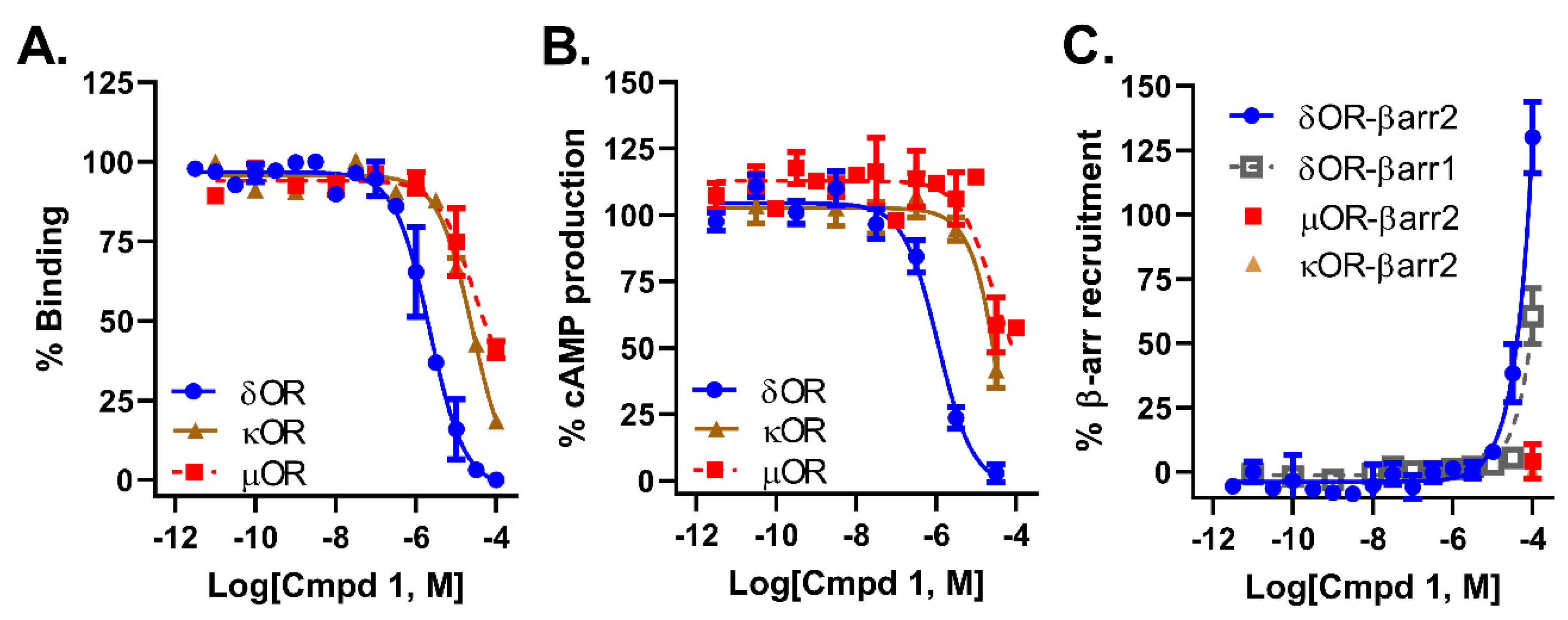
2.2. Compound 1 Displays 10-Fold Selectivity over µOR and κOR
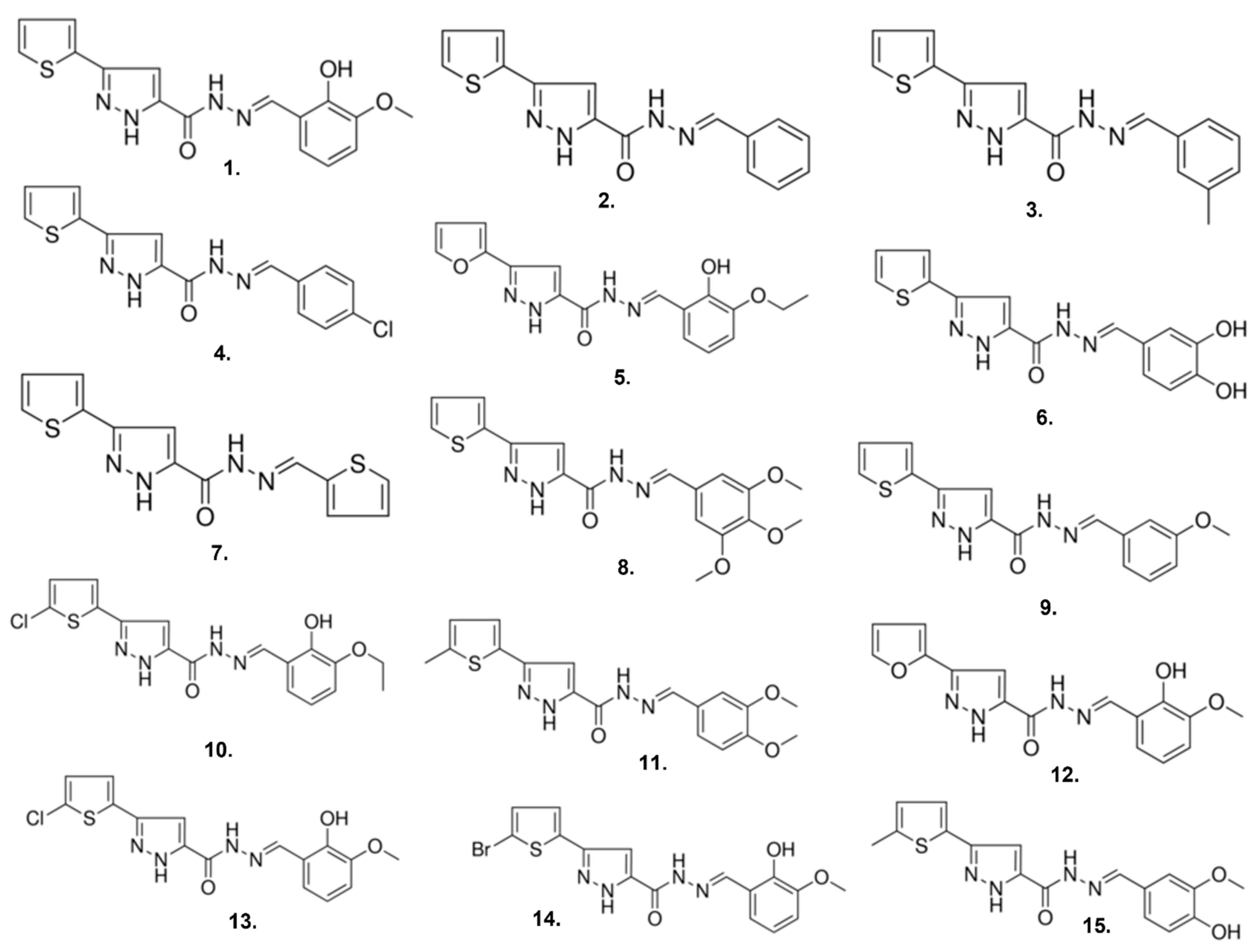
2.3. Compound 1 Derivatives Exhibit Lower δOR Potency
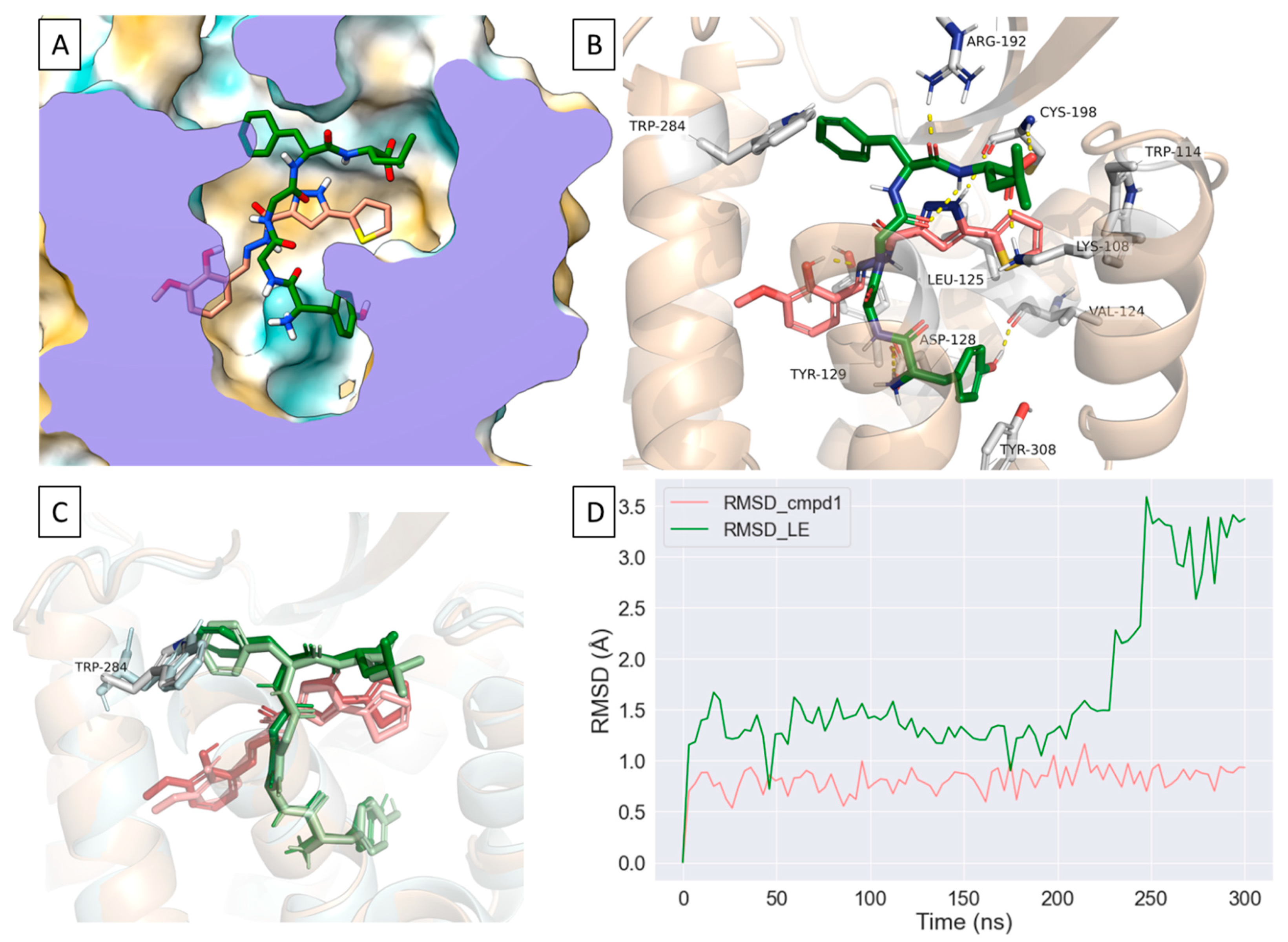
2.4. Compound 1 Engages Amino Acid Residues That Form the Orthosteric Binding Pocket
2.5. Compound 1 Can Occupy an Allosteric Space alongside Leu-Enkephalin
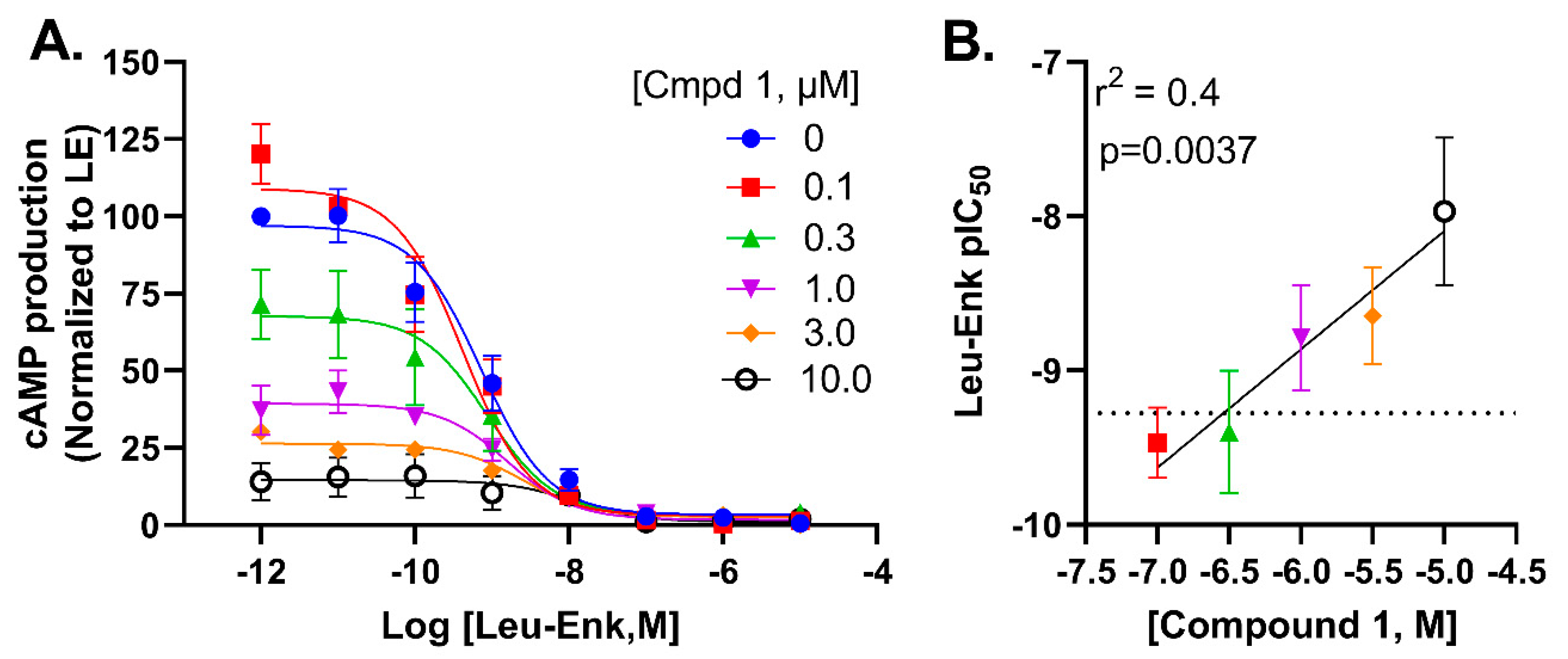
2.6. Compound 1 Potentially Negatively Modulates Potency of Leu-Enkephalin through an Allosteric Mechanism
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Library Screen
4.3. Radioligand Binding Assay
4.4. Cellular Signaling Assays
4.5. Assessment of Allosteric Modulation
4.6. Data and Statistical Analysis
4.7. Receptor and Ligand Preparation for Molecular Modeling
4.8. Ligand Docking Using Glide
4.9. Molecular Dynamics Simulations of Compound 1 at δOR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Pradhan, A.A.; Befort, K.; Nozaki, C.; Gaveriaux-Ruff, C.; Kieffer, B.L. The delta opioid receptor: An evolving target for the treatment of brain disorders. Trends Pharm. Sci. 2011, 32, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Van Rijn, R.M.; Defriel, J.N.; Whistler, J.L. Pharmacological traits of delta opioid receptors: Pitfalls or opportunities? Psychopharmacology 2013, 228, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.J.; Keith, D.E., Jr.; Morrison, H.; Magendzo, K.; Edwards, R.H. Cloning of a delta opioid receptor by functional expression. Science 1992, 258, 1952–1955. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, B.L.; Befort, K.; Gaveriaux-Ruff, C.; Hirth, C.G. The delta-opioid receptor: Isolation of a cDNA by expression cloning and pharmacological characterization. Proc. Natl. Acad. Sci. USA 1992, 89, 12048–12052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagase, H.; Saitoh, A. Research and development of kappa opioid receptor agonists and delta opioid receptor agonists. Pharm. Ther. 2020, 205, 107427. [Google Scholar] [CrossRef]
- Le Bourdonnec, B.; Windh, R.T.; Ajello, C.W.; Leister, L.K.; Gu, M.; Chu, G.H.; Tuthill, P.A.; Barker, W.M.; Koblish, M.; Wiant, D.D.; et al. Potent, orally bioavailable delta opioid receptor agonists for the treatment of pain: Discovery of N,N-diethyl-4-(5-hydroxyspiro[chromene-2,4′-piperidine]-4-yl)benzamide (ADL5859). J. Med. Chem. 2008, 51, 5893–5896. [Google Scholar] [CrossRef]
- Le Bourdonnec, B.; Windh, R.T.; Leister, L.K.; Zhou, Q.J.; Ajello, C.W.; Gu, M.; Chu, G.H.; Tuthill, P.A.; Barker, W.M.; Koblish, M.; et al. Spirocyclic delta opioid receptor agonists for the treatment of pain: Discovery of N,N-diethyl-3-hydroxy-4-(spiro[chromene-2,4′-piperidine]-4-yl) benzamide (ADL5747). J. Med. Chem. 2009, 52, 5685–5702. [Google Scholar] [CrossRef] [PubMed]
- Hudzik, T.J.; Maciag, C.; Smith, M.A.; Caccese, R.; Pietras, M.R.; Bui, K.H.; Coupal, M.; Adam, L.; Payza, K.; Griffin, A.; et al. Preclinical pharmacology of AZD2327: A highly selective agonist of the delta-opioid receptor. J. Pharmacol. Exp. Ther. 2011, 338, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Calderon, S.N.; Rothman, R.B.; Porreca, F.; Flippen-Anderson, J.L.; McNutt, R.W.; Xu, H.; Smith, L.E.; Bilsky, E.J.; Davis, P.; Rice, K.C. Probes for narcotic receptor mediated phenomena. 19. Synthesis of (+)-4-[(alpha R)-alpha-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide (SNC 80): A highly selective, nonpeptide delta opioid receptor agonist. J. Med. Chem. 1994, 37, 2125–2128. [Google Scholar] [CrossRef]
- Chang, K.J.; Rigdon, G.C.; Howard, J.L.; McNutt, R.W. A novel, potent and selective nonpeptidic delta opioid receptor agonist BW373U86. J. Pharmacol. Exp. Ther. 1993, 267, 852–857. [Google Scholar]
- Broom, D.C.; Jutkiewicz, E.M.; Folk, J.E.; Traynor, J.R.; Rice, K.C.; Woods, J.H. Convulsant activity of a non-peptidic delta-opioid receptor agonist is not required for its antidepressant-like effects in Sprague-Dawley rats. Psychopharmacology 2002, 164, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Vicente-Sanchez, A.; Dripps, I.J.; Tipton, A.F.; Akbari, H.; Akbari, A.; Jutkiewicz, E.M.; Pradhan, A.A. Tolerance to high-internalizing delta opioid receptor agonist is critically mediated by arrestin 2. Br. J. Pharmacol. 2018, 175, 3050–3059. [Google Scholar] [CrossRef]
- Blaine, A.T.; Palant, S.; Yuan, J.; Van Rijn, R.M. Role of β-arrestin Isoforms in Delta Opioid Receptor Agonist-Induced Seizures. FASEB J. 2021, 35. [Google Scholar] [CrossRef]
- Chiang, T.; Sansuk, K.; van Rijn, R.M. Beta-arrestin 2 dependence of delta opioid receptor agonists is correlated with alcohol intake. Br. J. Pharmacol. 2016, 173, 323–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, M.J.; Chiang, T.; Mukadam, A.A.; Mulia, G.E.; Gutridge, A.M.; Lin, A.; Chester, J.A.; van Rijn, R.M. beta-Arrestin-dependent ERK signaling reduces anxiety-like and conditioned fear-related behaviors in mice. Sci. Signal. 2021, 14, eaba0245. [Google Scholar] [CrossRef] [PubMed]
- Crombie, A.; Arezzo, J.; Cowan, C.; DeWire, S.; Gowen-McDonald, W.; Hawkins, M.; Jutkiewicz, E.; Kramer, M.; Koblish, M.; Lark, M.; et al. TRV250: A novel G protein-biased ligand at the delta receptor for the potential treatment of migraine. Postgrad. Med. 2015, 127 (Suppl. 1), S61. [Google Scholar]
- Conibear, A.E.; Asghar, J.; Hill, R.; Henderson, G.; Borbely, E.; Tekus, V.; Helyes, Z.; Palandri, J.; Bailey, C.; Starke, I.; et al. A Novel G Protein-Biased Agonist at the delta Opioid Receptor with Analgesic Efficacy in Models of Chronic Pain. J. Pharmacol. Exp. Ther. 2020, 372, 224–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagase, H.; Nemoto, T.; Matsubara, A.; Saito, M.; Yamamoto, N.; Osa, Y.; Hirayama, S.; Nakajima, M.; Nakao, K.; Mochizuki, H.; et al. Design and synthesis of KNT-127, a delta-opioid receptor agonist effective by systemic administration. Bioorg. Med. Chem. Lett. 2010, 20, 6302–6305. [Google Scholar] [CrossRef]
- Nagase, H.; Wakita, H.; Kawai, K.; Endoh, T.; Matsura, H.; Tanaka, C.; Takezawa, Y. Syntheses of non-peptidid delta opioid agonists and their structure activity relationships. Jpn. J. Pharmacol. 1994, 64. [Google Scholar] [CrossRef]
- Gutridge, A.M.; Robins, M.T.; Cassell, R.J.; Uprety, R.; Mores, K.L.; Ko, M.J.; Pasternak, G.W.; Majumdar, S.; van Rijn, R.M. G protein-biased kratom-alkaloids and synthetic carfentanil-amide opioids as potential treatments for alcohol use disorder. Br. J. Pharmacol. 2020, 177, 1497–1513. [Google Scholar] [CrossRef]
- Cassell, R.J.; Sharma, K.K.; Su, H.; Cummins, B.R.; Cui, H.; Mores, K.L.; Blaine, A.T.; Altman, R.A.; van Rijn, R.M. The Meta-Position of Phe(4) in Leu-Enkephalin Regulates Potency, Selectivity, Functional Activity, and Signaling Bias at the Delta and Mu Opioid Receptors. Molecules 2019, 24, 4542. [Google Scholar] [CrossRef] [Green Version]
- Claff, T.; Yu, J.; Blais, V.; Patel, N.; Martin, C.; Wu, L.; Han, G.W.; Holleran, B.J.; Van der Poorten, O.; White, K.L.; et al. Elucidating the active delta-opioid receptor crystal structure with peptide and small-molecule agonists. Sci. Adv. 2019, 5, eaax9115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Y.; Yeatman, H.R.; Provasi, D.; Alt, A.; Christopoulos, A.; Canals, M.; Filizola, M. Proposed Mode of Binding and Action of Positive Allosteric Modulators at Opioid Receptors. ACS Chem. Biol. 2016, 11, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, A.; Changeux, J.P.; Catterall, W.A.; Fabbro, D.; Burris, T.P.; Cidlowski, J.A.; Olsen, R.W.; Peters, J.A.; Neubig, R.R.; Pin, J.P.; et al. International Union of Basic and Clinical Pharmacology. XC. multisite pharmacology: Recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol. Rev. 2014, 66, 918–947. [Google Scholar] [CrossRef] [Green Version]
- Kelly, E. Efficacy and ligand bias at the mu-opioid receptor. Br. J. Pharmacol. 2013, 169, 1430–1446. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Wang, H.; Li, C.; Zhang, J.Z.H.; Ji, C. MolGpka: A Web Server for Small Molecule pKa Prediction Using a Graph-Convolutional Neural Network. J. Chem. Inf. Model. 2021, 61, 3159–3165. [Google Scholar] [CrossRef]
- Stanczyk, M.A.; Livingston, K.E.; Chang, L.; Weinberg, Z.Y.; Puthenveedu, M.A.; Traynor, J.R. The delta-opioid receptor positive allosteric modulator BMS 986187 is a G-protein-biased allosteric agonist. Br. J. Pharmacol. 2019, 176, 1649–1663. [Google Scholar] [CrossRef]
- Burford, N.T.; Clark, M.J.; Wehrman, T.S.; Gerritz, S.W.; Banks, M.; O’Connell, J.; Traynor, J.R.; Alt, A. Discovery of positive allosteric modulators and silent allosteric modulators of the mu-opioid receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 10830–10835. [Google Scholar] [CrossRef] [Green Version]
- Burford, N.T.; Wehrman, T.; Bassoni, D.; O’Connell, J.; Banks, M.; Zhang, L.; Alt, A. Identification of selective agonists and positive allosteric modulators for micro- and delta-opioid receptors from a single high-throughput screen. J. Biomol. Screen. 2014, 19, 1255–1265. [Google Scholar] [CrossRef] [Green Version]
- Kathmann, M.; Flau, K.; Redmer, A.; Trankle, C.; Schlicker, E. Cannabidiol is an allosteric modulator at mu- and delta-opioid receptors. Naunyn. Schmiedebergs. Arch. Pharmacol. 2006, 372, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.D.; Jara, Z.P.; Harford, T.; Saha, P.P.; Pardhi, T.R.; Desnoyer, R.; Karnik, S.S. Novel allosteric ligands of the angiotensin receptor AT1R as autoantibody blockers. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hubner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165–1175.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.T.; Pitis, P.; Liu, G.; Yuan, C.; Gotchev, D.; Cowan, C.L.; Rominger, D.H.; Koblish, M.; Dewire, S.M.; Crombie, A.L.; et al. Structure-activity relationships and discovery of a G protein biased mu opioid receptor ligand, [(3-methoxythiophen-2-yl)methyl]({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan- 9-yl]ethyl})amine (TRV130), for the treatment of acute severe pain. J. Med. Chem. 2013, 56, 8019–8031. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.P.; Carroll, F.I.; et al. Structure of the human kappa-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef]
- Zheng, Z.; Huang, X.P.; Mangano, T.J.; Zou, R.; Chen, X.; Zaidi, S.A.; Roth, B.L.; Stevens, R.C.; Katritch, V. Structure-Based Discovery of New Antagonist and Biased Agonist Chemotypes for the Kappa Opioid Receptor. J. Med. Chem. 2017, 60, 3070–3081. [Google Scholar] [CrossRef] [Green Version]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef]
- Stein, R.M.; Kang, H.J.; McCorvy, J.D.; Glatfelter, G.C.; Jones, A.J.; Che, T.; Slocum, S.; Huang, X.P.; Savych, O.; Moroz, Y.S.; et al. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 2020, 579, 609–614. [Google Scholar] [CrossRef]
- Cassell, R.J.; Mores, K.L.; Zerfas, B.L.; Mahmoud, A.H.; Lill, M.A.; Trader, D.J.; van Rijn, R.M. Rubiscolins are naturally occurring G protein-biased delta opioid receptor peptides. Eur. Neuropsychopharmacol. 2019, 29, 450–456. [Google Scholar] [CrossRef]
- Creed, S.M.; Gutridge, A.M.; Argade, M.D.; Hennessy, M.R.; Friesen, J.B.; Pauli, G.F.; van Rijn, R.M.; Riley, A.P. Isolation and Pharmacological Characterization of Six Opioidergic Picralima nitida Alkaloids. J. Nat. Prod. 2021, 84, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug. Des. 2006, 67, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.; Sondergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory. Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Sondergaard, C.R.; Olsson, M.H.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory. Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided. Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK(a) prediction and protonation state generation for drug-like molecules. J. Comput. Aided. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug. Des. 2007, 69, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ramsey, S.; Provasi, D.; El Daibani, A.; Appourchaux, K.; Chakraborty, S.; Kapoor, A.; Che, T.; Majumdar, S.; Filizola, M. Predicted Mode of Binding to and Allosteric Modulation of the mu-Opioid Receptor by Kratom’s Alkaloids with Reported Antinociception In Vivo. Biochemistry 2021, 60, 1420–1429. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | δOR | µOR | κOR |
|---|---|---|---|
| Affinity (pKi ± SEM) | 5.94 ± 0.16 | <5 | <5 |
| cAMP Potency (pIC50 ± SEM) | 6.01 ± 0.09 | <5 | <5 |
| β-ARR2 potency (pEC50) | <5 | ND | ND |
| β-ARR1 potency (pEC50) | <5 | - | - |
| Compound | Sigma Catalog Number | pIC50 ± SEM |
|---|---|---|
| 1 | R995045 | 6.0 ± 0.1 |
| 2 | R563412 | 4.9 ± 0.1 |
| 3 | R723622 | 5.1 ± 0.2 |
| 4 | R443638 | 4.9± 0.2 |
| 5 | R442488 | 5.0 ± 0.1 |
| 6 | R910759 | 4.9 ± 0.2 |
| 7 | R994944 | ND |
| 8 | R817031 | 5.0 ± 0.1 |
| 9 | R563420 | 4.8 ± 0.1 |
| 10 | R729426 | 5.1 ± 0.2 |
| 11 | R731501 | 5.4 ± 0.1 |
| 12 | R455865 | 5.1 ± 0.2 |
| 13 | R728691 | 5.1 ± 0.1 |
| 14 | R729639 | 5.0 ± 0.1 |
| 15 | L262382 | 5.0 ± 0.4 |
| Leu5-enkephalin | - | 9.1 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meqbil, Y.J.; Su, H.; Cassell, R.J.; Mores, K.L.; Gutridge, A.M.; Cummins, B.R.; Chen, L.; van Rijn, R.M. Identification of a Novel Delta Opioid Receptor Agonist Chemotype with Potential Negative Allosteric Modulator Capabilities. Molecules 2021, 26, 7236. https://doi.org/10.3390/molecules26237236
Meqbil YJ, Su H, Cassell RJ, Mores KL, Gutridge AM, Cummins BR, Chen L, van Rijn RM. Identification of a Novel Delta Opioid Receptor Agonist Chemotype with Potential Negative Allosteric Modulator Capabilities. Molecules. 2021; 26(23):7236. https://doi.org/10.3390/molecules26237236
Chicago/Turabian StyleMeqbil, Yazan J., Hongyu Su, Robert J. Cassell, Kendall L. Mores, Anna M. Gutridge, Benjamin R. Cummins, Lan Chen, and Richard M. van Rijn. 2021. "Identification of a Novel Delta Opioid Receptor Agonist Chemotype with Potential Negative Allosteric Modulator Capabilities" Molecules 26, no. 23: 7236. https://doi.org/10.3390/molecules26237236
APA StyleMeqbil, Y. J., Su, H., Cassell, R. J., Mores, K. L., Gutridge, A. M., Cummins, B. R., Chen, L., & van Rijn, R. M. (2021). Identification of a Novel Delta Opioid Receptor Agonist Chemotype with Potential Negative Allosteric Modulator Capabilities. Molecules, 26(23), 7236. https://doi.org/10.3390/molecules26237236