
The Development of BTK Inhibitors: A Five-Year Update





Abstract
:1. Introduction
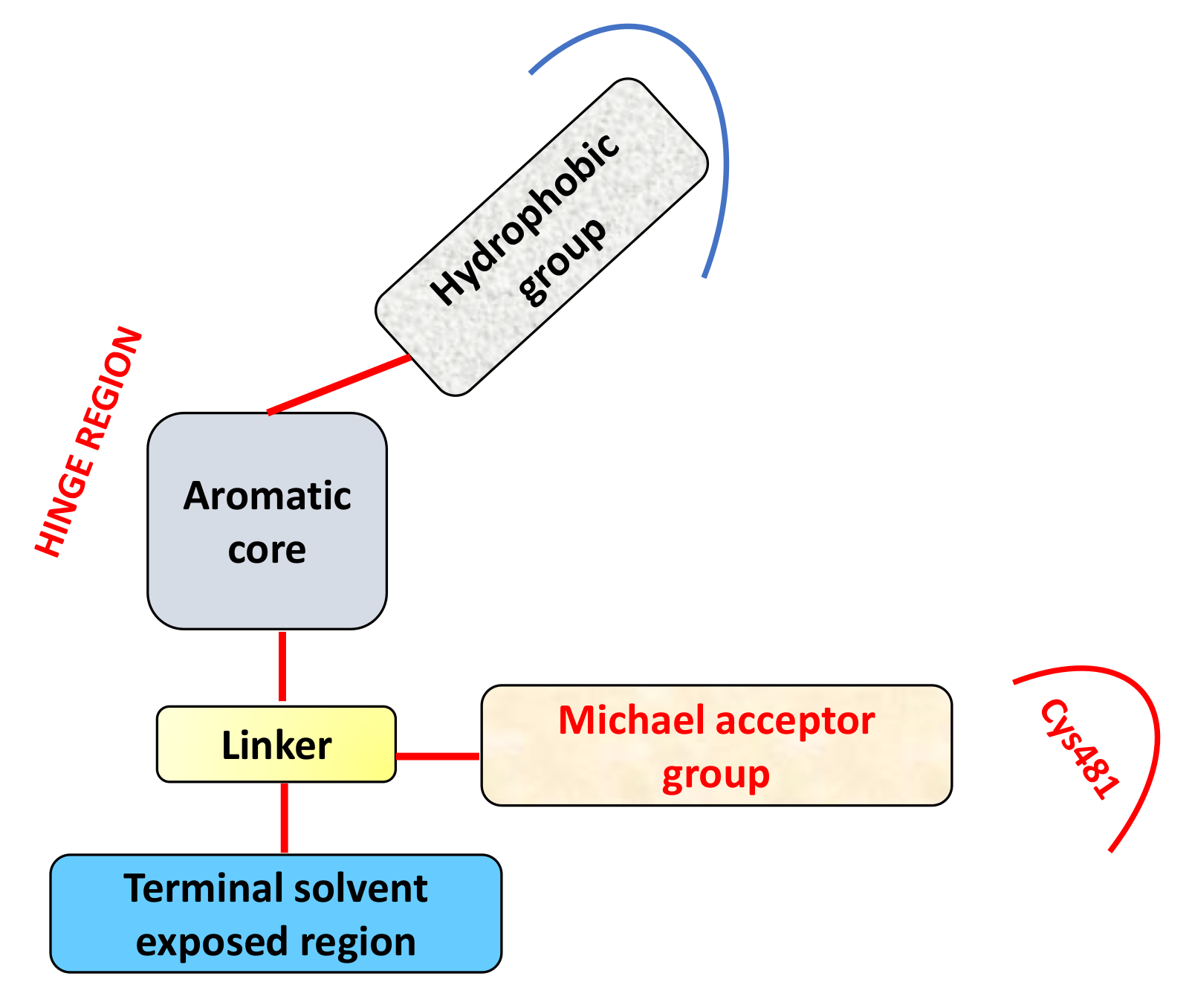
2. BTKIs: Their Classification
3. Irreversible BTKIs on the Market
4. BTKIs in Clinical and Pre-Clinical Evaluations
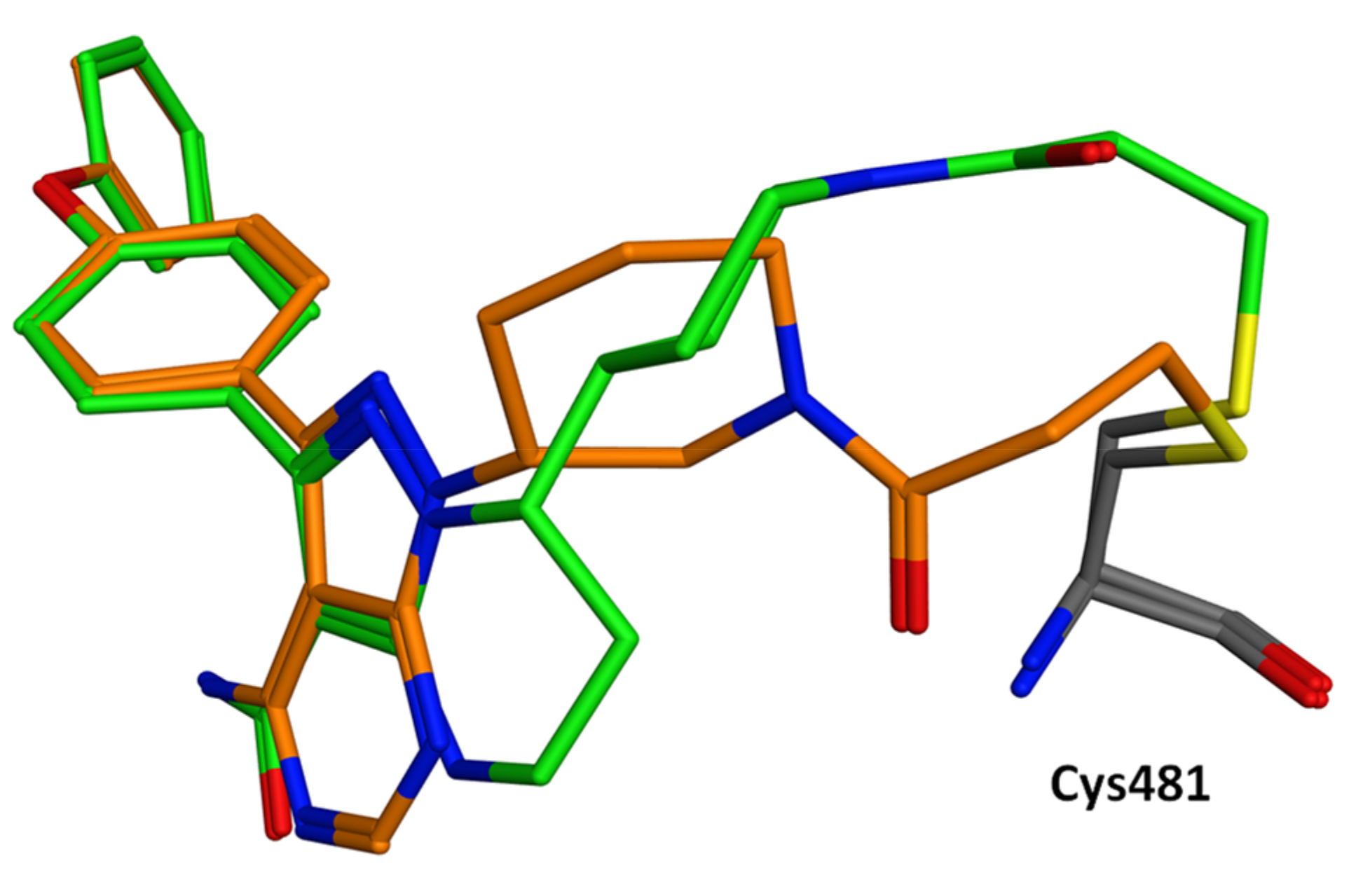
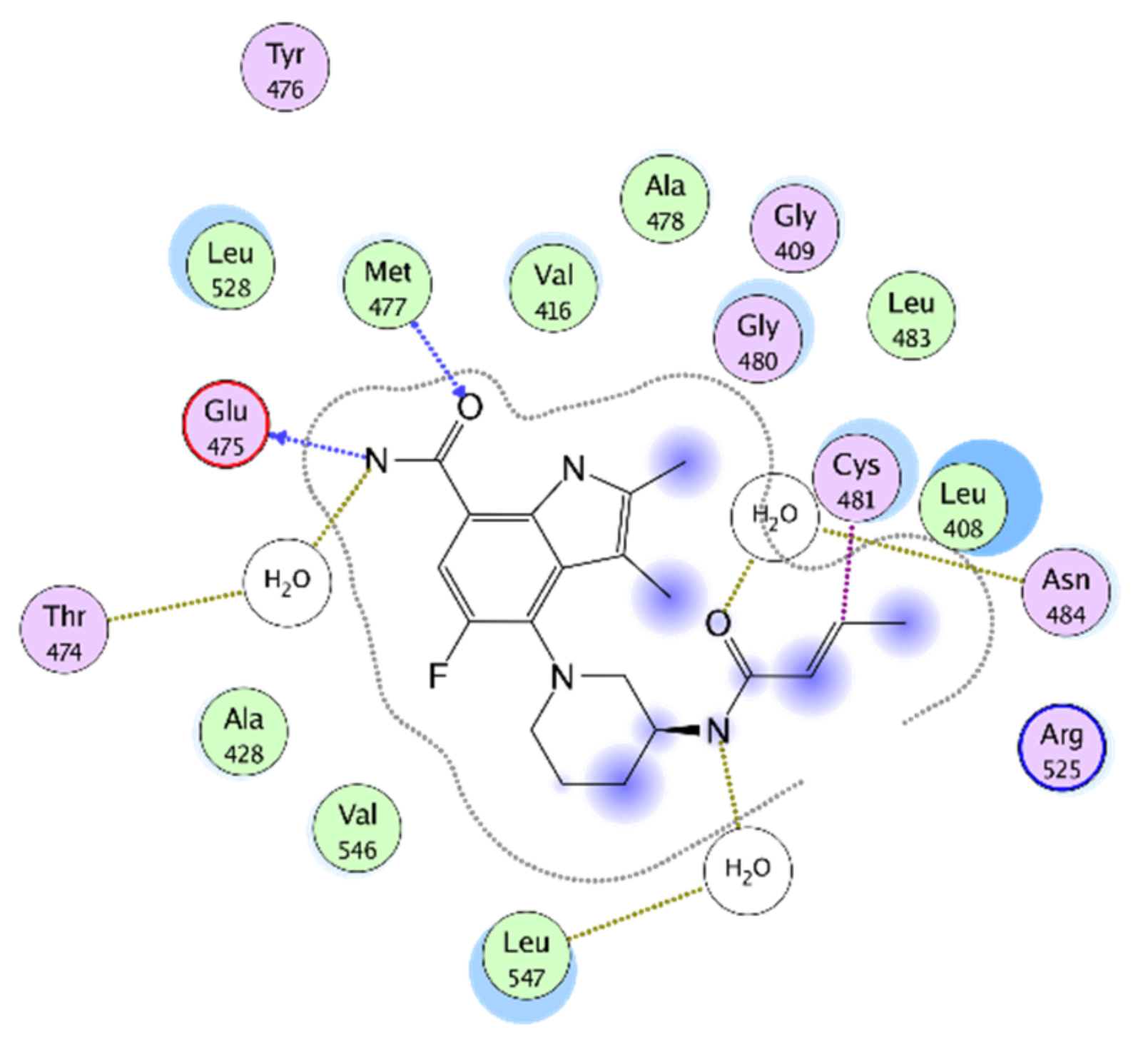
4.1. Irreversible BTKIs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Company | Chemical Structure | IC50 or pIC50 Values on BTK | Clinical Phases | Ref |
|---|---|---|---|---|---|
| Evobrutinib | Merk |  | 37.9 μM | Phase III | [29,30] |
| Spebrutinib | Avila Therapeutics/ Celegene |  | 0.5 μM | Pre-clinical studies | [35] |
| Remibrutinib | Novartis |  | 1.3 nM | Phase II | [36,37] |
| Tolebrutinib | Sanofi/Principia Biopharma |  | 0.4–0.7 nM | Phase III | [38,39] |
| Olmutinib | Hamni Pharmaceuticals |  | 1.0 nM | Phase II | [13,40] |
| Branebrutinib | Bristol-Myers Squibb |  | 0.1 nM | Phase I | [41,42] |
| TAK-020 | Takeda |  | pIC50 > 8.7 | Phase I | [43,44] |
| Elsubrutinib | AbbVie |  | 0.18 μM | Phase II | [45,46] |
| Rilzabrutinib | Sanofi |  | 3.1 nM | Phase III | [4,7,14,47,48] |
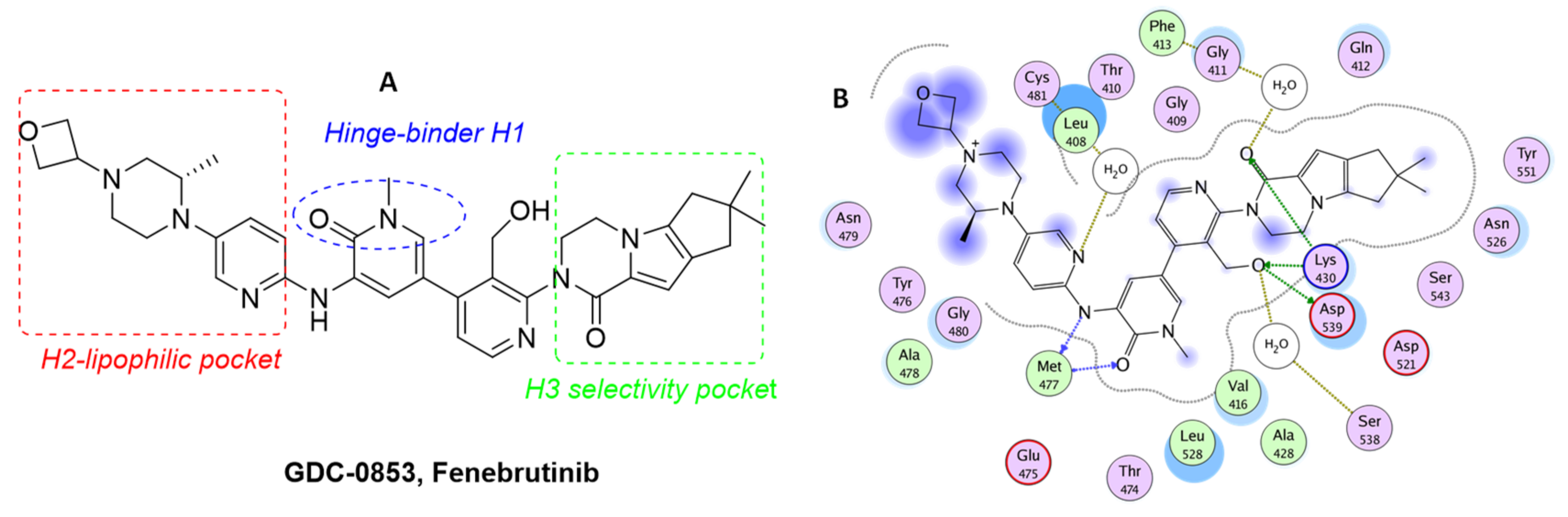
4.2. Reversible BTKIs
| Name | Company | Chemical Structure | Clinical Phases | Ref |
|---|---|---|---|---|
| Vecabrutinib | SNSS |  | Phase II | [13] |
| GNE-431 | Genentech |  | Pre-clinical investigation | [49,50] |
| RN-486 | Roche |  | Pre-clinical investigation | [4] |
| BMS-935177 | Bristol Meyers Squibb |  | Phase II | [53,54,55,56,57] |
| BMS-986142 | Bristol Meyers Squibb |  | Phase II | [53,54,55,56,57] |
| CGI-1746 | CGI/ Genentech |  | Pre-clinical investigation | [58] |
| GDC-0834 | Gilead/ Genetech |  | Pre-clinical investigation | [25,59] |
| G-744 | Gilead/ Genetech |  | Pre-clinical investigation | [60,61] |
| G-278 | Gilead/ Genetech |  | Pre-clinical investigation, now abandoned | [62] |
| Fenebrutinib | Genetech |  | Phase III | [63] |
5. Recent Advances in BTKIs
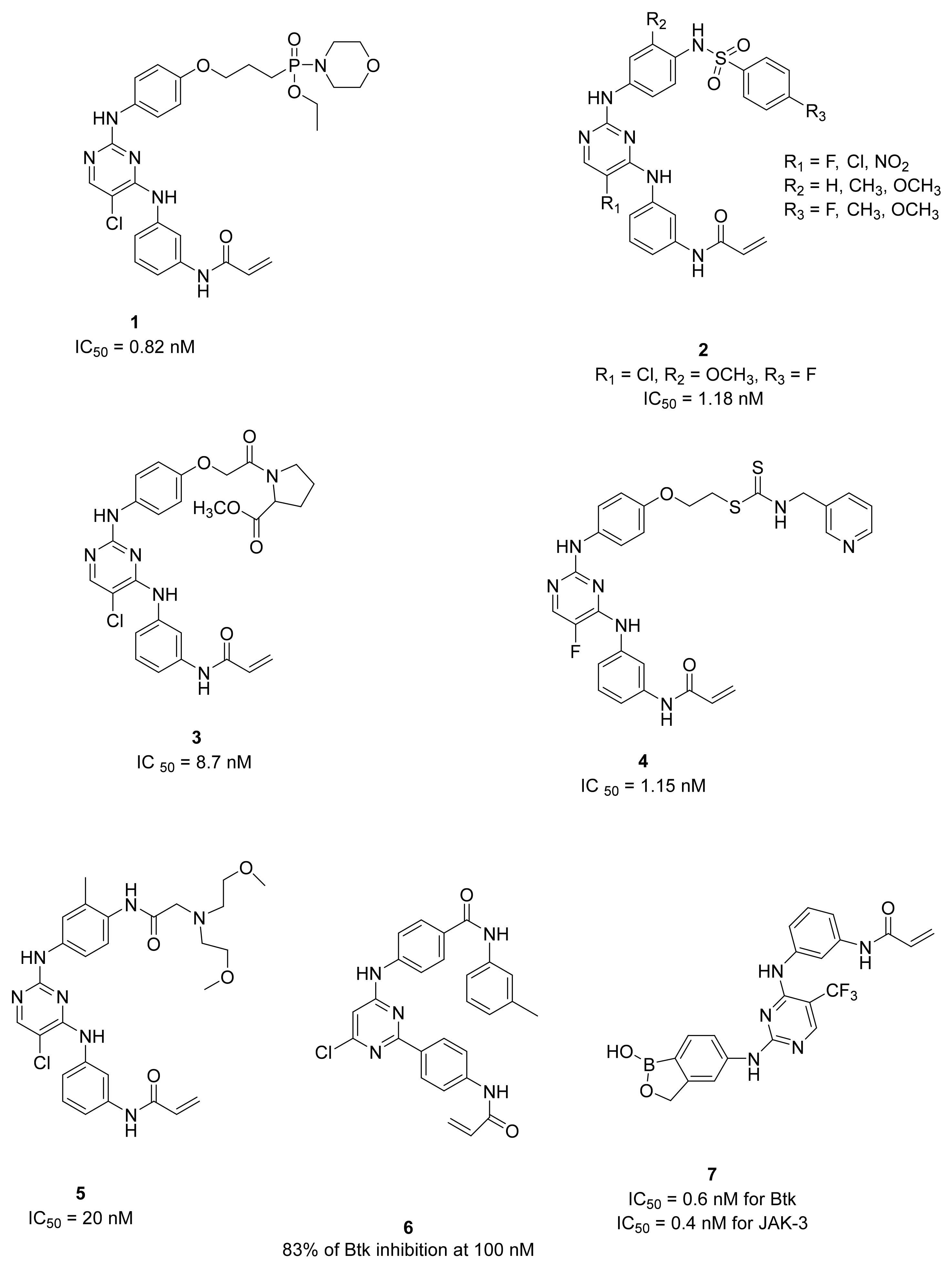
5.1. Diphenylaminopyrimidines (DPPYs)
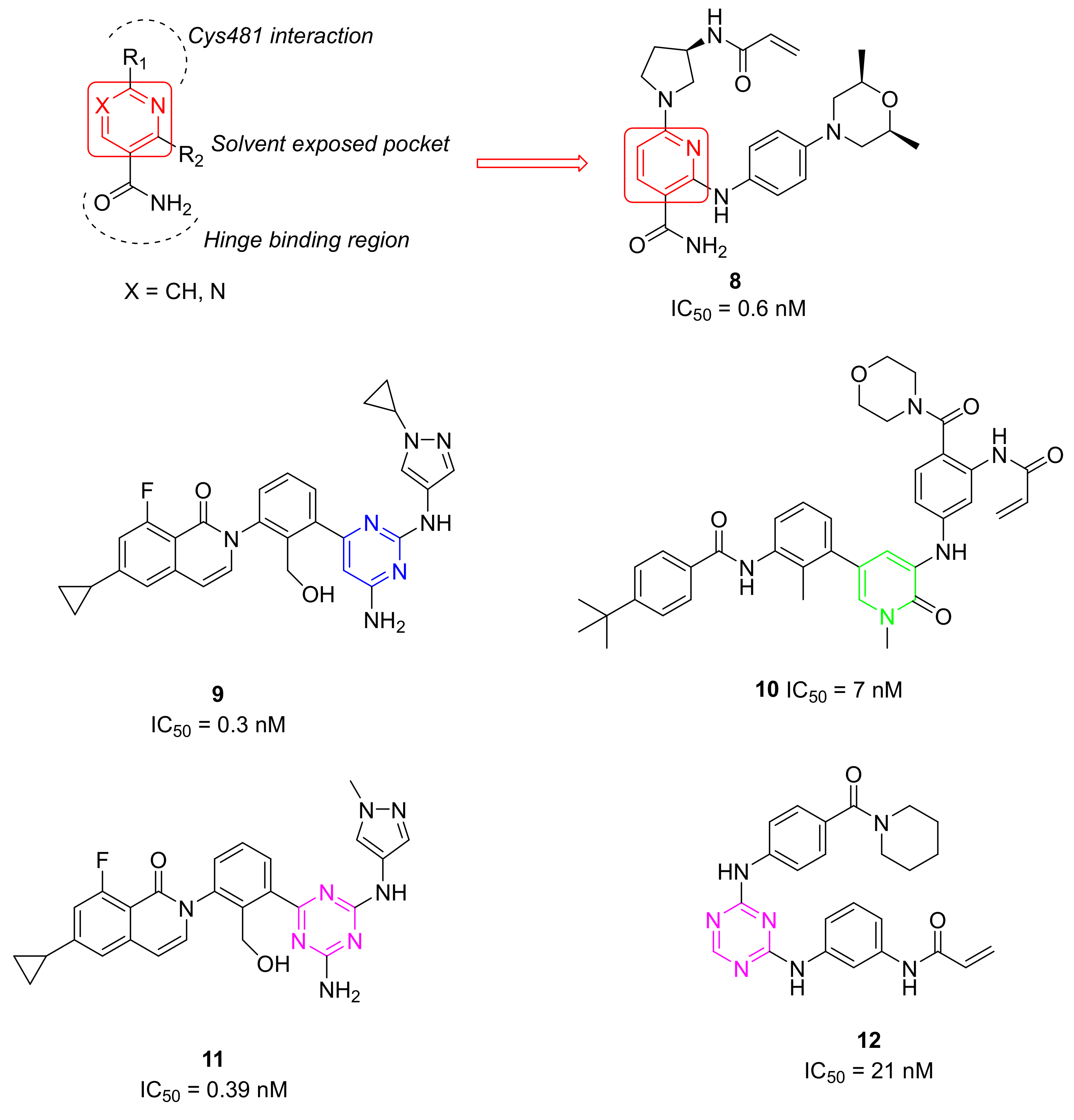
5.2. Other Pyrimidine and Pyridine Derivatives
5.3. Pyridinone Derivatives
5.4. 1,3,5-Triazine Derivatives
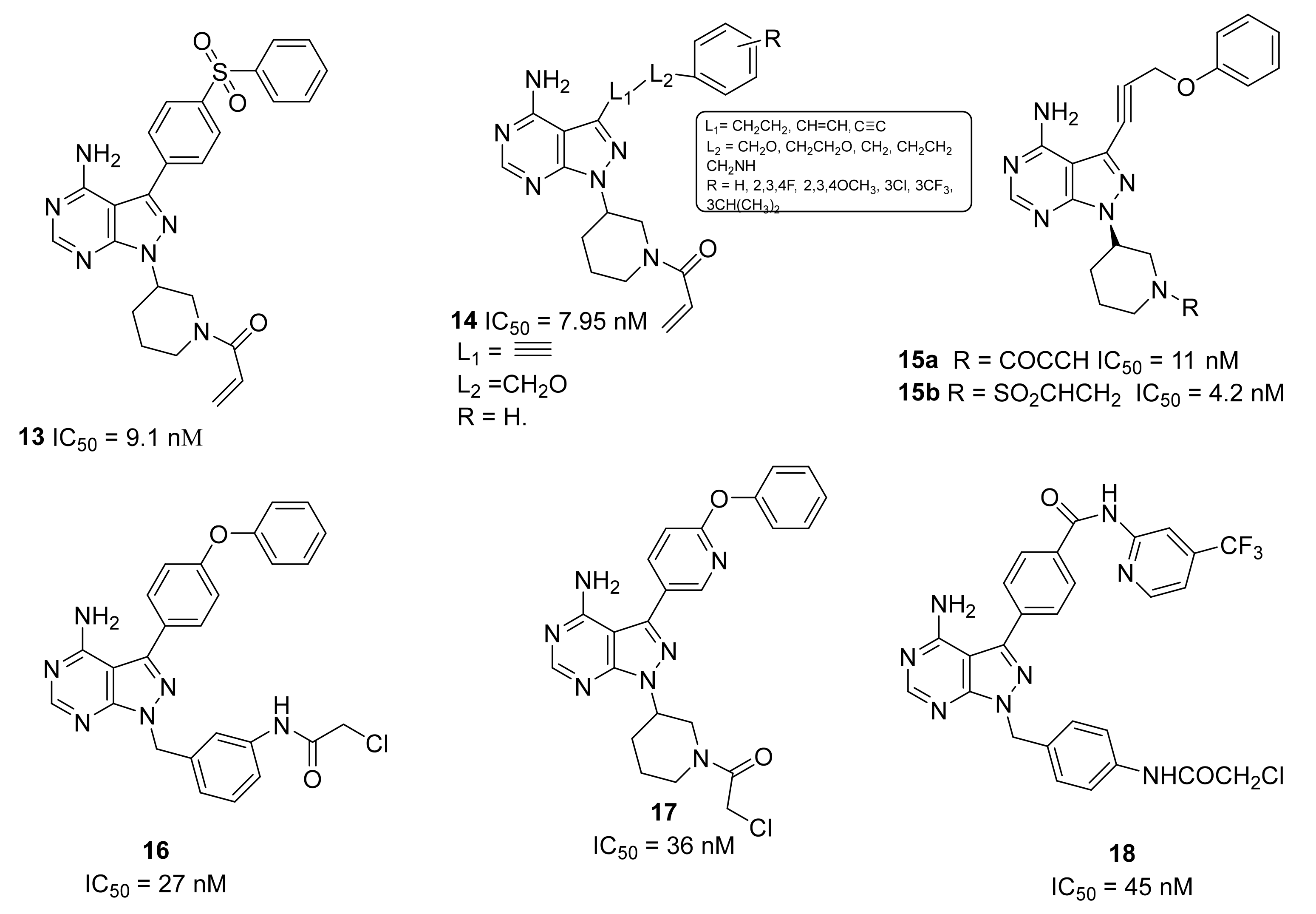
5.5. Pyrazolo-Pyrimidine Derivatives
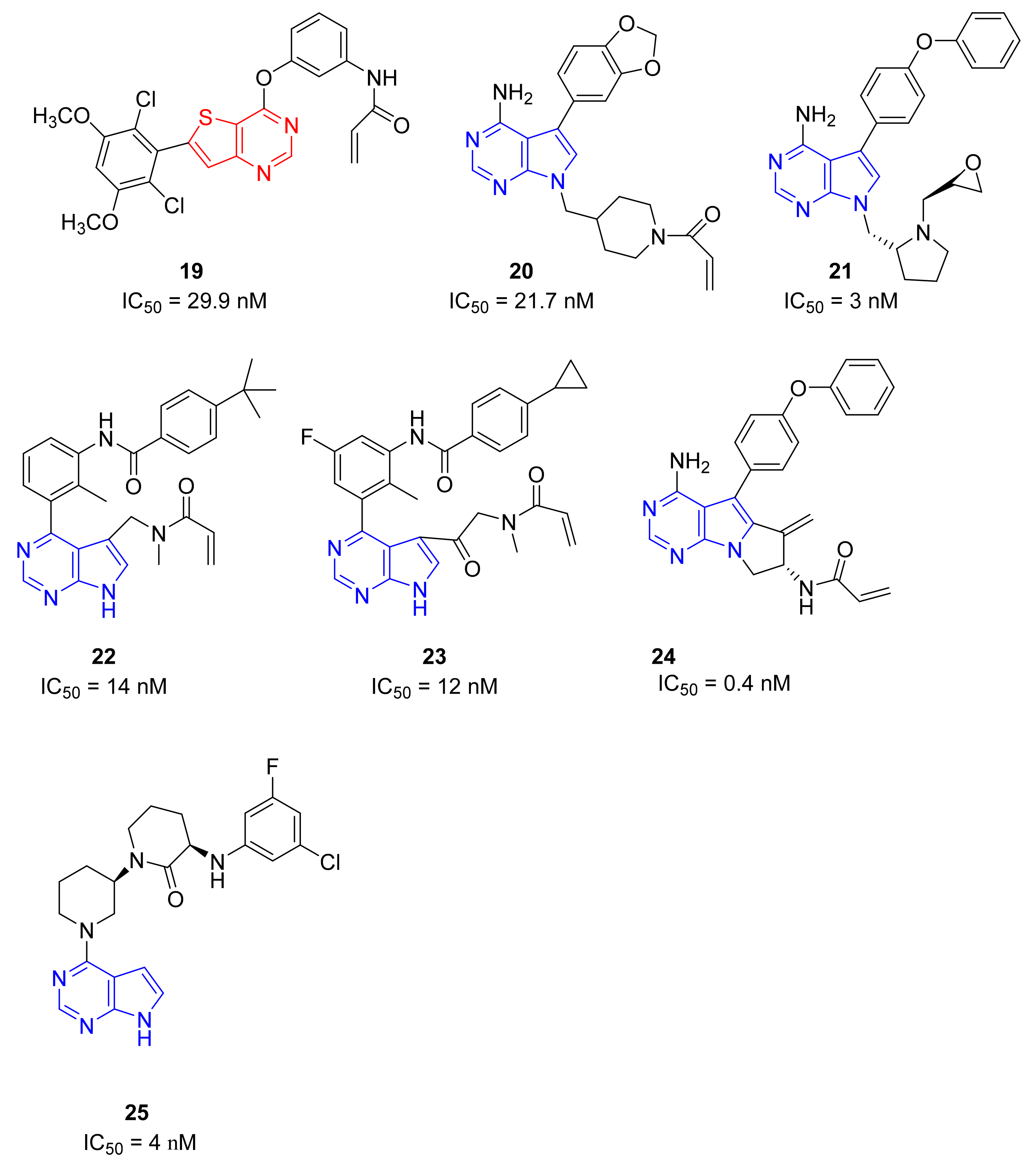
5.6. Tieno-Pyrimidine Derivatives
5.7. Pyrrolo[2,3-d]pyrimidine Derivatives
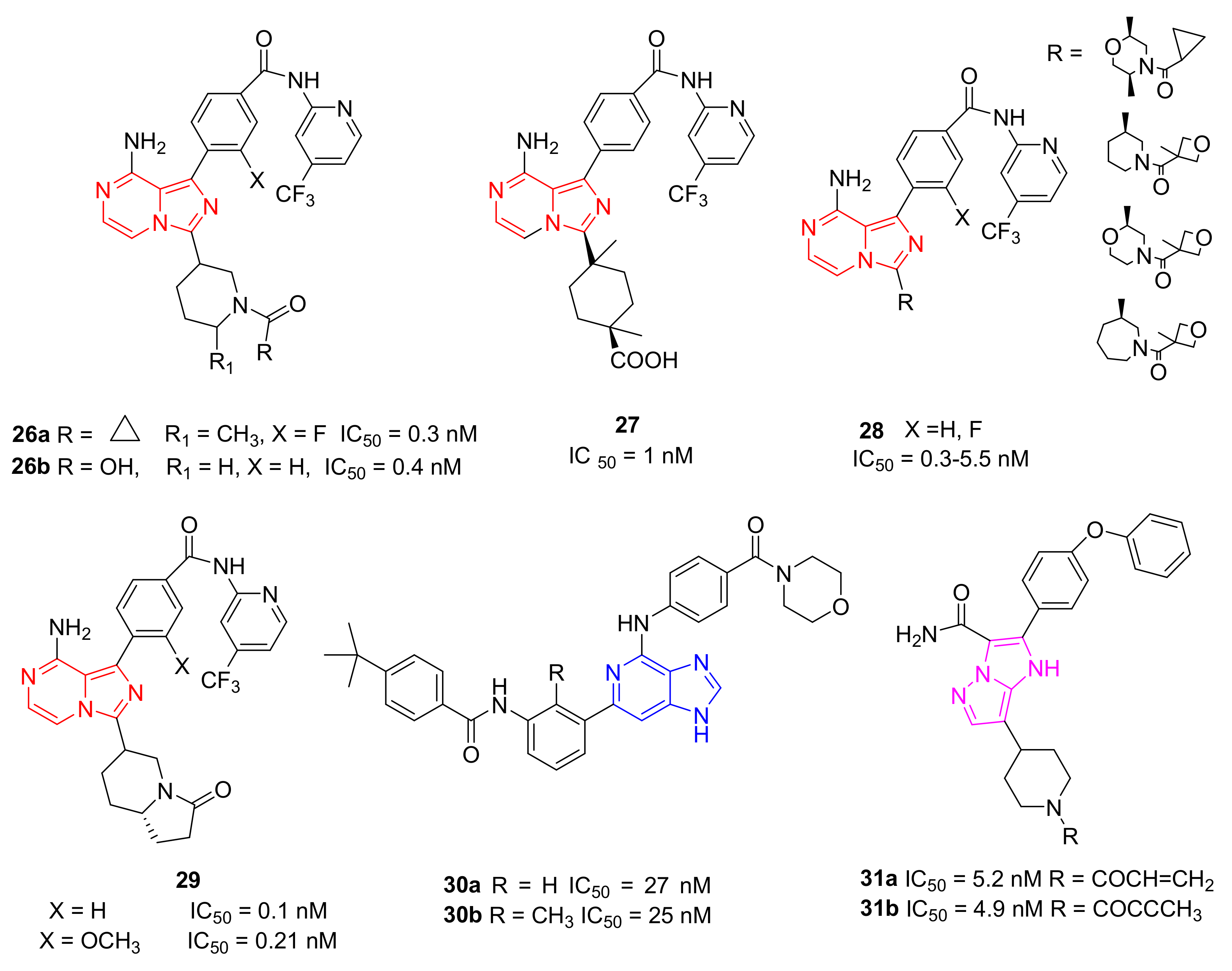
5.8. Imidazo-Pyrazine, Imidazo-Pyridine and Imidazo-Pyrazole Derivatives
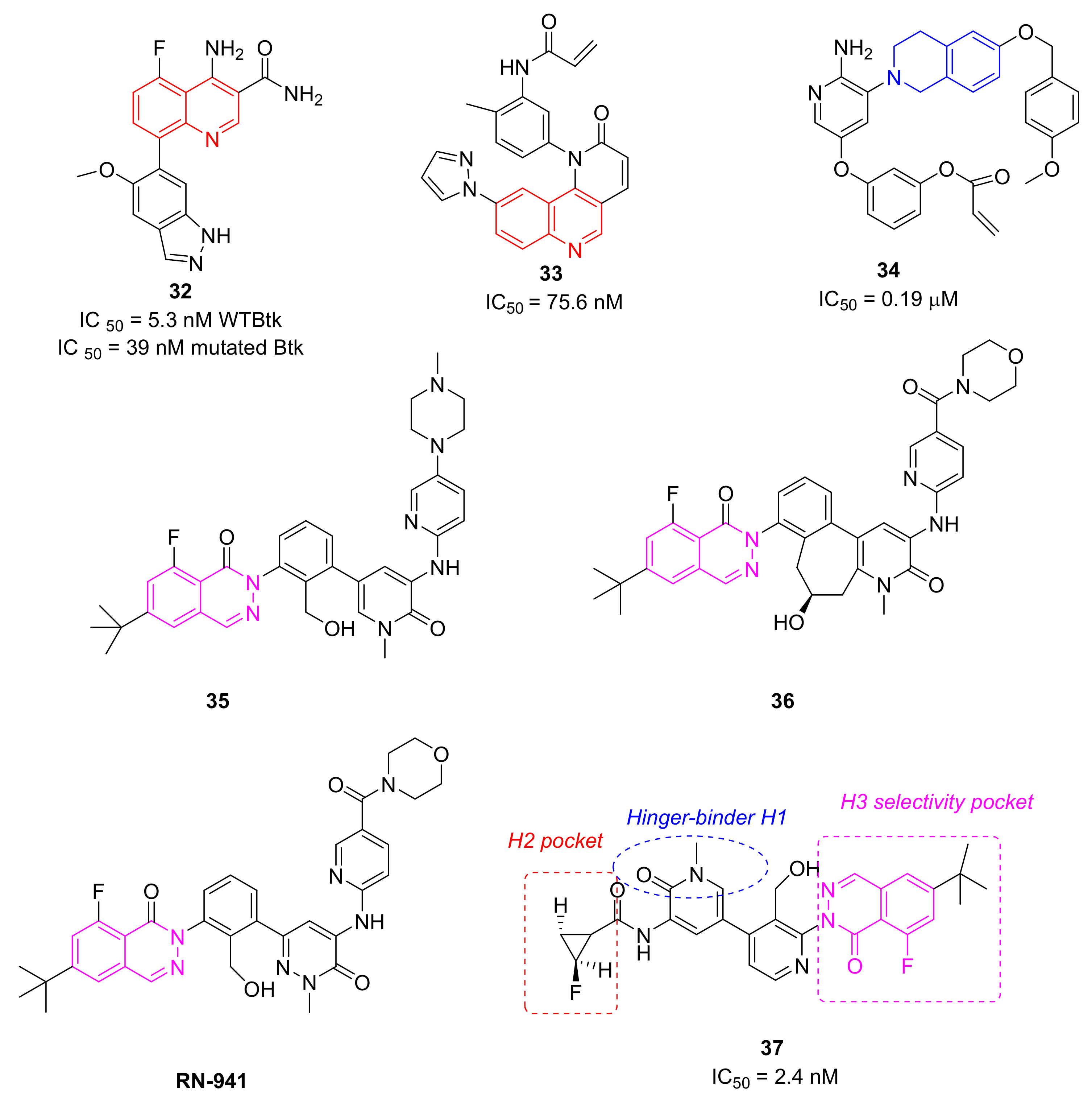
5.9. Quinoline and Isoquinoline Derivatives
5.10. Phthalazine Derivatives
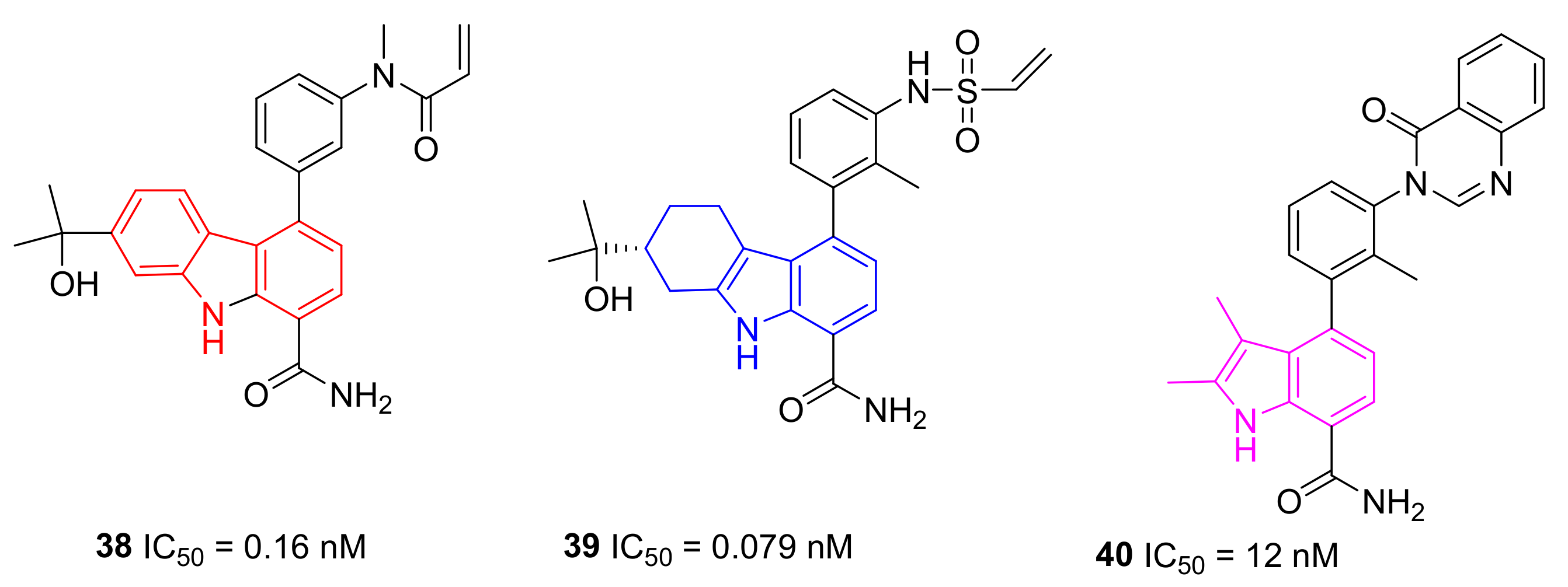
5.11. Carbazole, Tetrahydrocarbazole, and Indole Derivatives
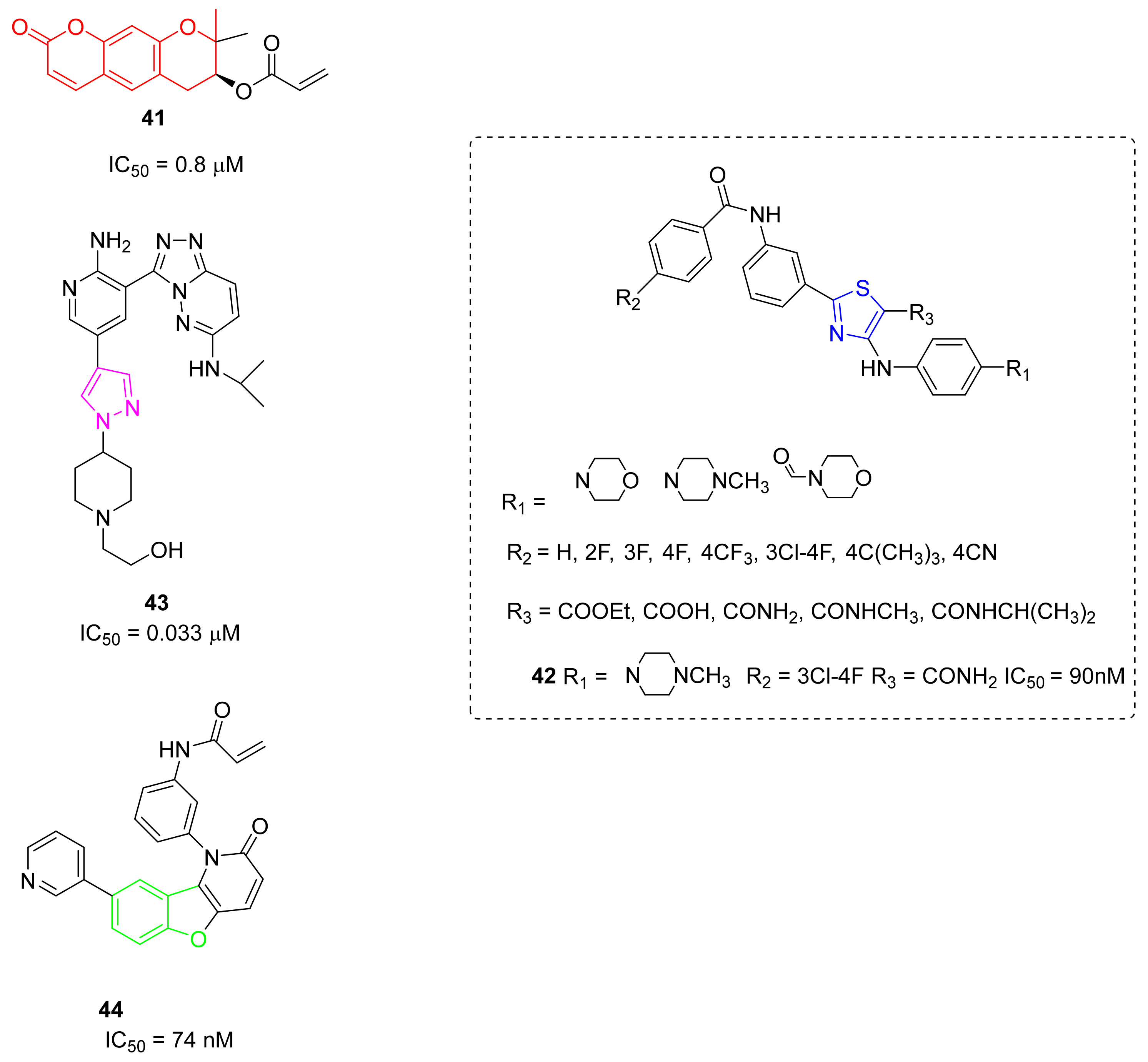
5.12. Miscellaneus Compounds
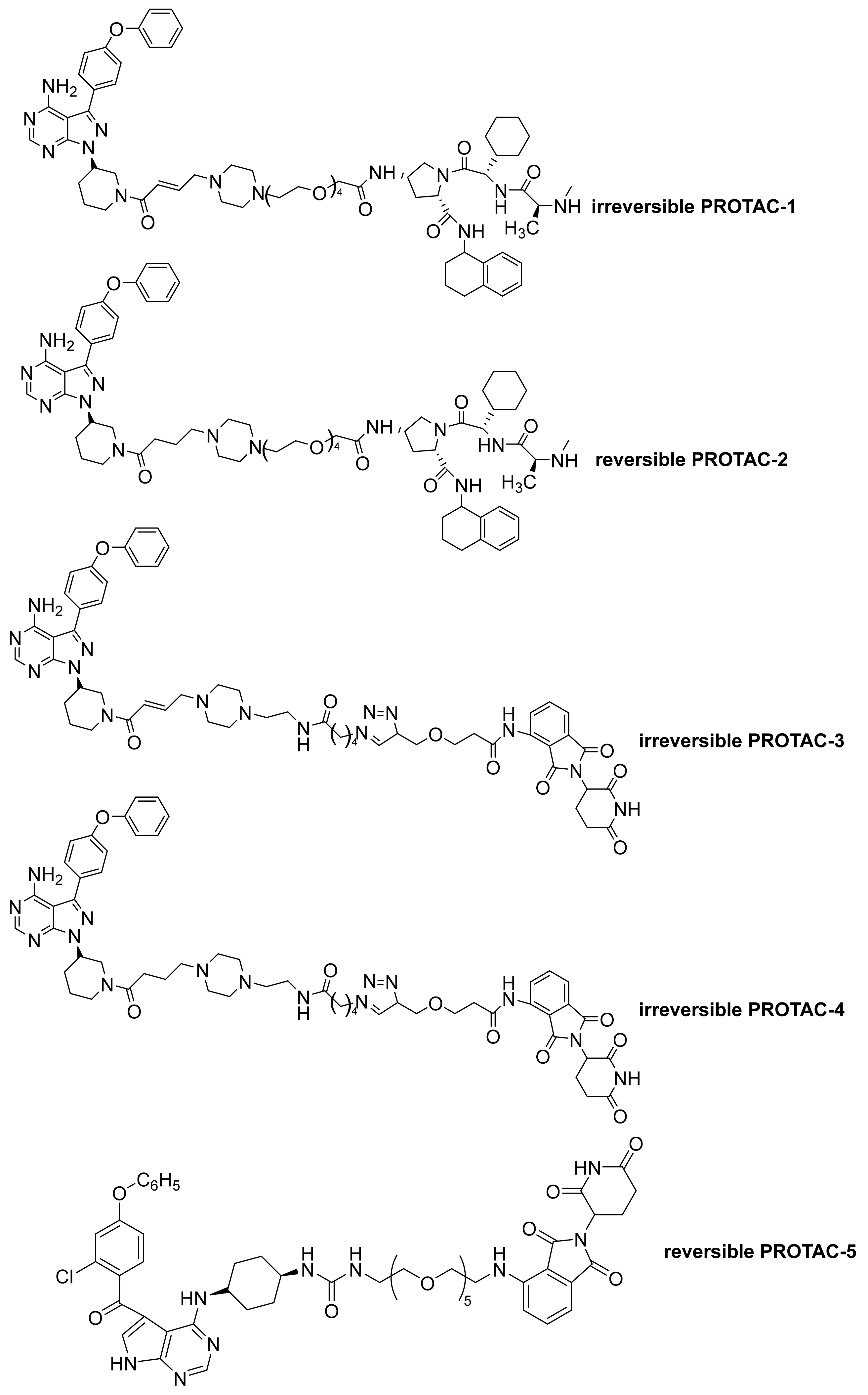
6. PROTAC Molecules
7. Future Directions
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BCR | B-cell receptor |
| BTK | Bruton’s kinase |
| BTKIs | BTK inhibitors |
| CLL | lymphocytic leukemia |
| CIA | collagen-induced arthritis |
| DPPY | diphenylaminopyrimidine |
| EGFR | epidermal growth factor receptor |
| FAK | focal adgesion kinase |
| FBDD | fragment-based drug discovery |
| cGVHD | chronic graft versus host disease |
| MZL | marginal zone lymphoma |
| MCL | mantle cell lymphoma |
| MS | multiple scherosis |
| NSCLC | non-small cell lung cancer |
| PMDA | Pharmaceuticals and Medical Devices Agency |
| PH | pleckstrin homology domain |
| Pho-DPYYs | diphenylpyrimidine derivatives |
| PLCγ2 | phospholipase γ2 |
| PK | pharmacokinetic |
| PROTAC | proteolysis-targeting chimera compounds |
| PRR | proline-rich regions |
| RA | rheumatoid arthritis |
| RMS | Relapsing Multiple Sclerosis |
| ROS | reactive oxygen species |
| SBD | structure-based design |
| SLL | small lymphocytic lymphoma |
| SH2/SH3 | Src homology 2/3 domain |
| SLE | Systemic Lupus Erythematosus |
| TK | tyrosine kinases |
| WM | Waldenström’s macroglobulinemia |
| WT | wild type |
References
- Carnero Contentti, E.; Correale, J. Bruton’s tyrosine kinase inhibitors: A promising emerging treatment option for multiple sclerosis. Expert Opin. Emerg. Drugs 2020, 25, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2013, 369, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bilal, M.; Raza, A.; Khan, M.I.; Mehmood, S.; Hayat, U.; Hassan, S.T.S.; Iqbal, H.M.N. Tyrosine kinase inhibitors and their unique therapeutic potentialities to combat cancer. Int. J. Biol. Macromol. 2020, 168, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Tian, D.; Ren, X.; Ding, S.; Jia, M.; Xin, M.; Thareja, S. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: A mini-review. Eur. J. Med. Chem. 2018, 151, 315–326. [Google Scholar] [CrossRef]
- Jongstra-Bilen, J.; Cano, A.P.; Hasija, M.; Xiao, H.; Smith, C.I.E.; Cybulsky, M. Dual Functions of Bruton’s Tyrosine Kinase and Tec Kinase during Fcγ Receptor-Induced Signaling and Phagocytosis. J. Immunol. 2008, 181, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Hata, D.; Kawakami, Y.; Inagaki, N.; Lantz, C.S.; Kitamura, T.; Khan, W.N.; Maeda-Yamamoto, M.; Miura, T.; Han, W.; Hartman, S.E.; et al. Involvement of Bruton’s Tyrosine Kinase in FcεRI-dependent Mast Cell Degranulation and Cytokine Production. J. Exp. Med. 1998, 187, 1235–1247. [Google Scholar] [CrossRef]
- Brullo, C.; Villa, C.; Tasso, B.; Russo, E.; Spallarossa, A. Btk Inhibitors: A Medicinal Chemistry and Drug Delivery Perspective. Int. J. Mol. Sci. 2021, 22, 7641. [Google Scholar] [CrossRef]
- Vetrie, D.; Vořechovský, I.; Sideras, P.; Holland, J.; Davies, A.; Flinter, F.A.; Hammarström, L.; Kinnon, C.; Levinsky, R.J.; Bobrow, M.; et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 1993, 361, 226–233. [Google Scholar] [CrossRef]
- Manji, F.; Puckrin, R.; Stewart, D.A. Novel synthetic drugs for the treatment of non-Hodgkin lymphoma. Expert Opin. Pharmacother. 2021, 22, 1417–1427. [Google Scholar] [CrossRef]
- Wen, T.; Wang, J.; Shi, Y.; Qian, H.; Liu, P. Inhibitors targeting Bruton’s tyrosine kinase in cancers: Drug development advances. Leukemia 2021, 35, 312–332. [Google Scholar] [CrossRef]
- Tinworth, C.P.; Lithgow, H.; Dittus, L.; Bassi, Z.I.; Hughes, S.E.; Muelbaier, M.; Dai, H.; Smith, I.E.D.; Kerr, W.J.; Burley, G.A.; et al. PROTAC-Mediated Degradation of Bruton’s Tyrosine Kinase Is Inhibited by Covalent Binding. ACS Chem. Biol. 2019, 14, 342–347. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Shu, Y.; Lin, J.; Chen, Z.; Xie, Q.; Bao, Y.; Lu, L.; Sun, N.; Wang, Y. Discovery of novel BTK PROTACs for B-Cell lymphomas. Eur. J. Med. Chem. 2021, 225, 113820. [Google Scholar] [CrossRef]
- Feng, Y.; Duan, W.; Cu, X.; Xin, M. Bruton’s tyrosine kinase (BTK) inhibitors in treating cancer: A patent review. Expert. Opin. Ther. Pat. 2019, 29, 217–241. [Google Scholar] [CrossRef]
- Dolgin, E. BTK blockers make headway in multiple sclerosis. Nat. Biotechnol. 2021, 39, 3–5. [Google Scholar] [CrossRef]
- Zhang, D.; Gong, H.; Meng, F. Recent Advances in BTK Inhibitors for the Treatment of Inflammatory and Autoimmune Diseases. Molecules 2021, 26, 4907. [Google Scholar] [CrossRef]
- Dhillon, S. Tirabrutinib: First approval. Drugs 2020, 80, 835–840. [Google Scholar] [CrossRef]
- Estupiñán, H.Y.; Berglöf, A.; Zain, R.; Smith, C.I.E. Comparative Analysis of BTK Inhibitors and Mechanisms Underlying Adverse Effects. Front. Cell Dev. Biol. 2021, 9, 331. [Google Scholar] [CrossRef]
- Dhillon, S. Orelabrutinib: First Approval. Drugs 2021, 81, 503–507. [Google Scholar] [CrossRef]
- Chong, E.A.; Roeker, L.E.; Shadman, M.; Davids, M.S.; Schuster, S.J.; Mato, A.R. BTK Inhibitors in Cancer Patients with COVID-19: “The Winner Will be the One Who Controls That Chaos” (Napoleon Bonaparte). Clin. Cancer Res. 2020, 26, 3514–3516. [Google Scholar] [CrossRef]
- Acalabrutinib Study with Best Supportive Care in Participants Hospitalized with COVID-19. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04497948 (accessed on 16 June 2021).
- Sibaud, V.; Beylot-Barry, M.; Protin, C.; Vigarios, E.; Recher, C.; Ysebaert, L. Dermatological Toxicities of Bruton’s Tyrosine Kinase nhibitors. Am. J. Clin. Dermatol. 2020, 21, 799–812. [Google Scholar] [CrossRef]
- Woyach, J.A.; Furman, R.R.; Liu, T.M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.; Steggerda, S.M.; Versele, M.; et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chen, C.; Wang, D.; Zhang, J.; Zhang, T. Emerging small-molecule inhibitors of the Bruton’s tyrosine kinase (BTK): Current development. Eur. J. Med. Chem. 2021, 217, 113329. [Google Scholar] [CrossRef]
- Young, W.B.; Barbosa, J.; Blomgren, P.; Bremer, M.C.; Crawford, J.J.; Dambach, D.; Eigenbrot, C.; Gallion, S.; Johnson, A.R.; Kropf, J.E.; et al. Discovery of highly potent and selective Bruton’s tyrosine kinase inhibitors: Pyridazinone analogs with improved metabolic stability. Bioorg. Med. Chem. Lett. 2016, 26, 575–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, W.B.; Barbosa, J.; Blomgren, P.; Bremer, M.C.; Crawford, J.; Dambach, D.; Gallion, S.; Hymowitz, S.G.; Kropf, J.E.; Lee, S.H.; et al. Potent and selective Bruton’s tyrosine kinase inhibitors: Discovery of GDC-0834. Bioorg. Med. Chem. Lett. 2015, 25, 1333–1337. [Google Scholar] [CrossRef] [Green Version]
- Aw, A.; Brown, J.R. Current Status of Bruton’s Tyrosine Kinase Inhibitor Development and Use in B-Cell Malignancies. Drugs Aging 2017, 34, 509–527. [Google Scholar] [CrossRef]
- Noy, A.; De Vos, S.; Thieblemont, C.; Martin, P.; Flowers, C.R.; Morschhauser, F.; Collins, G.P.; Ma, S.; Coleman, M.; Peles, S.; et al. Targeting Bruton tyrosine kinase with ibrutinib in relapsed/refractory marginal zone lymphoma. Blood 2017, 129, 2224–2232. [Google Scholar] [CrossRef]
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing Irreversible Inhibitors of the Protein Kinase Cysteinome. Chem. Biol. 2013, 20, 146–159. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, R.D.; Qiu, H.; Askew, B.C.; Bender, A.T.; Brugger, N.; Camps, M.; Dhanabal, M.; Dutt, V.; Eichhorn, T.; Gardberg, A.S.; et al. Discovery of Evobrutinib: An Oral, Potent, and Highly Selective, Covalent Bruton’s Tyrosine Kinase (BTK) Inhibitor for the Treatment of Immunological Diseases. J. Med. Chem. 2019, 62, 7643–7655. [Google Scholar] [CrossRef] [Green Version]
- Haselmayer, P.; Camps, M.; Liu-Bujalski, L.; Nguyen, N.; Morandi, F.; Head, J.; O’Mahony, A.; Zimmerli, S.C.; Bruns, L.; Bender, A.T.; et al. Efficacy and Pharmacodynamic Modeling of the BTK Inhibitor Evobrutinib in Autoimmune Disease Models. J. Immunol. 2019, 202, 2888–2906. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Li, R.; Ning, X.; Zhao, X.; Jin, Y.; Yin, Y. Synthesis and anti-tumor efficacy of novel 2,4-diarylaminopyrimidine derivatives bearing N-(3-pyridinylmethyl) urea moiety as anaplastic lymphoma kinase inhibitors. Eur. J. Med. Chem. 2019, 178, 141–153. [Google Scholar] [CrossRef]
- Brullo, C. New Insights on Fak and Fak Inhibitors. Curr. Med. Chem. 2021, 28, 3318–3338. [Google Scholar] [CrossRef] [PubMed]
- Song, A.; Zhang, J.; Ge, Y.; Wang, C.; Meng, Q.; Tang, Z.; Peng, J.; Liu, K.; Li, Y.; Ma, X. C-2(E)-4-(Styryl)aniline substituted diphenylpyrimidine derivatives (Sty-DPPYs) as specific kinase inhibitors targeting clinical resistance related EGFR T790M mutant. Bioorg. Med. Chem. 2017, 25, 2724–2729. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Jin, Y.; Ge, Y.; Wang, C.; Zhang, J.; Tang, Z.; Peng, J.; Liu, K.; Li, Y.; Ma, X. Synthesis and biological evaluation of azole-diphenylpyrimidine derivatives (AzDPPYs) as potent T790M mutant form of epidermal growth factor receptor inhibitors. Bioorg. Med. Chem. 2016, 24, 5505–5512. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Qiu, M.; Luo, Q.; Zhou, S.; Zhang, L.; Yan, X.; Yuan, L.; Zhang, Y. Combination of FAK Inhibitor and BTK Inhibitor for Treating a Disease. PCT Int. Appl. Patent WO 2020259553, 30 December 2020. [Google Scholar]
- Angst, D.; Gessier, F.; Vulpetti, A. Preparation of Novel Amino Pyrimidine Derivatives as Bruton’s Tyrosine Kinase Inhibitors. PCT Int. Appl. Patent WO 2015079417, 4 June 2015. [Google Scholar]
- Angst, D.; Gessier, F.; Janser, P.; Vulpetti, A.; Wälchli, R.; Beerli, C.; Littlewood-Evans, A.; Dawson, J.; Nuesslein-Hildesheim, B.; Wieczorek, G.; et al. Discovery of LOU064 (Remibrutinib), a Potent and Highly Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase. J. Med. Chem. 2020, 63, 5102–5118. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Qian, C.; He, Q.; Huang, Y.; Ma, Z.; Qin, S.; Ye, C.; Zhong, X. Preparation of Dihydroimidazopyridine Derivatives for Treatment of Bruton’s Tyrosine Kinase Related Diseases. Faming Zhuanli Shenqing Patent CN 105753863, 13 July 2016. [Google Scholar]
- Goldstein, D.; Owens, T.D. Preparation of 1H-imidazo[4,5-c]pyridin-2(3H)-one Derivatives as Tyrosine Kinase Inhibitors. PCT Int. Appl. Patent WO 2016196840, 8 December 2016. [Google Scholar]
- Roskoski, R. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2018, 139, 395–411. [Google Scholar] [CrossRef]
- Watterson, S.H.; Liu, Q.; Bertrand, M.B.; Batt, D.G.; Li, L.; Pattoli, M.A.; Skala, S.; Cheng, L.; Obermeier, M.T.; Moore, R.; et al. Discovery of Branebrutinib (BMS-986195): A Strategy for Identifying a Highly Potent and Selective Covalent Inhibitor Providing Rapid in Vivo Inactivation of Bruton’s Tyrosine Kinase (BTK). J. Med. Chem. 2019, 62, 3228–3250. [Google Scholar] [CrossRef] [Green Version]
- Catlett, I.M.; Nowak, M.; Kundu, S.; Zheng, N.; Liu, A.; He, B.; Girgis, I.G.; Grasela, D.M. Safety, pharmacokinetics and pharmacodynamics of branebrutinib (BMS-986195), a covalent, irreversible inhibitor of Bruton’s tyrosine kinase: Randomised phase I, placebo-controlled trial in healthy participants. Br. J. Clin. Pharmacol. 2020, 86, 1849–1859. [Google Scholar] [CrossRef]
- Sabat, M.; Dougan, D.R.; Knight, B.; Lawson, J.D.; Scorah, N.; Smith, C.R.; Taylor, E.R.; Vu, P.; Wyrick, C.; Wang, H.; et al. Discovery of the Bruton’s Tyrosine Kinase Inhibitor Clinical Candidate TAK-020 (S)-5-(1-((1-Acryloylpyrrolidin-3-yl)oxy)isoquinolin-3-yl)-2,4-dihydro-3H-1,2,4-triazol-3-one, by Fragment-Based Drug Design. J. Med. Chem. 2021, 64, 12893–12902. [Google Scholar] [CrossRef]
- Esfandiari, E.; Chen, M.; Smithson, G.; Blair, D.; Faessel, H.; Wagner, J.; Mclean, L.; Fedyk, E.R. A Phase I, Randomized, Double-Blind, Placebo-Controlled, Single-Dose and Multiple-Rising-Dose Study of the BTK Inhibitor TAK-020 in Healthy Subjects. Clin. Transl. Sci. 2021, 14, 820–828. [Google Scholar] [CrossRef]
- Bonafoux, D.; Davis, H.M.; Frank, K.E.; Friedman, M.M.; Herold, J.M.; Hoemann, M.Z.; Huntley, R.; Osuma, A.; Sheppard, G.; Somal, G.K.; et al. Preparation of Heterocycle Carboxamides as Bruton’s Tyrosine Kinase (Btk) Inhibitors. PCT Int. Appl. Patent WO 2014210255, 31 December 2014. [Google Scholar]
- Goess, C.; Harris, C.M.; Murdock, S.; McCarthy, R.W.; Sampson, E.; Twomey, R.; Mathieu, S.; Mario, R.; Perham, M.; Goedken, E.R.; et al. ABBV-105, a selective and irreversible inhibitor of Bruton’s tyrosine kinase, is efficacious in multiple preclinical models of inflammation. Mod. Rheumatol. 2018, 29, 510–522. [Google Scholar] [CrossRef]
- Smith, P.F.; Krishnarajah, J.; Nunn, P.A.; Hill, R.J.; Karr, D.; Tam, D.; Masjedizadeh, M.; Gourlay, S.G. SAT0232 A Phase 1 Clinical Trial of PRN1008, an Oral, Reversible, Covalent BTK Inhibitor Demonstrates Clinical Safety and Therapeutic Levels of BTK Occupancy Without Sustained Systemic Exposure. Ann. Rheum. Dis. 2015, 74, 742. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.F.; Krishnarajah, J.; Nunn, P.A.; Hill, R.J.; Karr, D.; Tam, D.; Masjedizadeh, M.; Funk, J.O.; Gourlay, S.G. A phase I trial of PRN1008, a novel reversible covalent inhibitor of Bruton’s tyrosine kinase, in healthy volunteers. Br. J. Clin. Pharmacol. 2017, 83, 2367–2376. [Google Scholar] [CrossRef]
- Crawford, J.J.; Ortwine, D.F.; Young, W.B. Bicyclic Piperazine Compounds as BTK Inhibitors and Their Preparation. PCT Int. Appl. Patent WO 2013067260, 10 May 2013. [Google Scholar]
- Johnson, A.R.; Kohli, P.B.; Katewa, A.; Gogol, E.; Belmont, L.D.; Choy, R.; Penuel, E.; Burton, L.; Eigenbrot, C.; Yu, C.; et al. Battling Btk Mutants with Noncovalent Inhibitors That Overcome Cys481 and Thr474 Mutations. ACS Chem. Biol. 2016, 11, 2897–2907. [Google Scholar] [CrossRef]
- Berthel, S.; Firooznia, F.; Fishlock, D.; Hong, J.B.; Lou, Y.; Lucas, M.; Owens, T.D.; Sarma, K.; Sweeney, Z.K.; Taygerly, J.P.G. Preparation of 5-phenyl-1H-pyridin-2-one, 6-phenyl-2H-pyridazin-3-one, and 5-phenyl-1H-pyrazin-2-one Derivatives as Inhibitors of Bruton’s Tyrosine Kinase. US Patent US 20100222325, 2 September 2010. [Google Scholar]
- Lou, Y.; Han, X.; Kuglstatter, A.; Kondru, R.K.; Sweeney, Z.K.; Soth, M.; McIntosh, J.; Litman, R.; Suh, J.; Kocer, B.; et al. Structure-Based Drug Design of RN486, a Potent and Selective Bruton’s Tyrosine Kinase (BTK) Inhibitor, for the Treatment of Rheumatoid Arthritis. J. Med. Chem. 2014, 58, 512–516. [Google Scholar] [CrossRef]
- Watterson, S.H.; De Lucca, G.V.; Shiuhang, Y.; Langevine, C.M.; Liu, Q.; Batt, D.G.; Bertrand, M.B.; Gong, H.; Dai, J.; Yip, S.; et al. Discovery of 6-Fluoro-5-(R)-(3-(S)-(8-fluoro-1-methyl-2,4-dioxo-1,2-dihydroquinazolin-3(4H)-yl)-2-methylphenyl)-2-(S)-(2-hydroxypropan-2-yl)-2,3,4,9-tetrahydro-1H-carbazole-8-carboxamide (BMS-986142): A Reversible Inhibitor of Bruton’s Tyrosine Kinase (BTK) Conformationally Constrained by Two Locked Atropisomers. J. Med. Chem. 2016, 59, 9173–9200. [Google Scholar] [CrossRef]
- Liu, Q.; Batt, D.G.; Lippy, J.S.; Surti, N.; Tebben, A.J.; Muckelbauer, J.K.; Chen, L.; An, Y.; Chang, C.; Pokross, M.; et al. Design and synthesis of carbazole carboxamides as promising inhibitors of Bruton’s tyrosine kinase (BTK) and Janus kinase 2 (JAK2). Bioorg. Med. Chem. Lett. 2015, 25, 4265–4269. [Google Scholar] [CrossRef]
- De Lucca, G.V.; Shi, Q.; Liu, Q.; Batt, D.G.; Bertrand, M.B.; Rampulla, R.; Mathur, A.; Discenza, L.; D’Arienzo, C.; Dai, J.; et al. Small Molecule Reversible Inhibitors of Bruton’s Tyrosine Kinase (BTK): Structure–Activity Relationships Leading to the Identification of 7-(2-Hydroxypropan-2-yl)-4-[2-methyl-3-(4-oxo-3,4-dihydroquinazolin-3-yl)phenyl]-9H-carbazole-1-carboxamide (BMS-935177). J. Med. Chem. 2016, 59, 7915–7935. [Google Scholar] [CrossRef]
- Ahmad, S.; Batt, D.G.; Liu, Q.; Macor, J.E.; Tino, J.A.; Watterson, S.H.; Nair, S.K.; Maishal, T.K. Preparation of Carbazole Derivatives as Inhibitors of Bruton’s Tyrosine Kinase. PCT Int. Appl. Patent WO 2016065236 A1 20160428, 28 April 2016. [Google Scholar]
- Carrasquillo, R.; Geng, P.; Huang, E.C.; Katipally, K.; Lee, A.; Mudryk, B.; Qian, X.; Razler, T.M.; Wang, J.; Wei, C.S.; et al. Process for Preparing BMS 986142. PCT Int. Appl. Patent WO 2018118830, 28 June 2018. [Google Scholar]
- Di Paolo, J.A.; Huang, T.; Balazs, M.; Barbosa, J.; Barck, K.H.; Bravo, B.J.; Carano, R.A.D.; Darrow, J.; Davies, D.R.; DeForge, L.E.; et al. Specific Btk inhibition suppresses B cell– and myeloid cell–mediated arthritis. Nat. Chem. Biol. 2011, 7, 41–50. [Google Scholar] [CrossRef]
- Blomgren, P.A.; Lee, S.H.; Mitchell, S.A.; Xu, J.; Schmitt, A.C.; Kropf, J.E.; Currie, K.S. Certain Substituted Amides, Method of Making, and Method of Use Thereof. US Patent US 20080153834, 26 June 2008. [Google Scholar]
- Wang, X.; Barbosa, J.; Blomgren, P.; Bremer, M.C.; Chen, J.; Crawford, J.J.; Deng, W.; Dong, L.; Eigenbrot, C.; Gallion, S.; et al. Discovery of Potent and Selective Tricyclic Inhibitors of Bruton’s Tyrosine Kinase with Improved Druglike Properties. ACS Med. Chem. Lett. 2017, 8, 608–613. [Google Scholar] [CrossRef]
- Barbosa, A.J.M.; Blomgren, P.A.; Currie, K.S.; Krishnamoorthy, R.; Kropf, J.E.; Lee, S.H.; Mitchell, S.A.; Ortwine, D.; Schmitt, A.C.; Wang, X. Preparation of Pyridone and Azapyridone Compounds as Btk Inhibitors for Treating Immune Disorders, inflammation, Cancer, and Other Btk-Mediated Diseases. PCT Int. Appl. Patent WO 2011140488, 10 November 2011. [Google Scholar]
- Erickson, R.I.; Schutt, L.K.; Tarrant, J.M.; McDowell, M.; Liu, L.; Johnson, A.R.; Lewin-Koh, S.-C.; Hedehus, M.; Ross, J.; Carano, R.A.D.; et al. Bruton’s Tyrosine Kinase Small Molecule Inhibitors Induce a Distinct Pancreatic Toxicity in Rats. J. Pharmacol. Exp. Ther. 2016, 360, 226–238. [Google Scholar] [CrossRef] [Green Version]
- Crawford, J.J.; Johnson, A.R.; Misner, D.L.; Belmont, L.D.; Castanedo, G.M.; Choy, R.; Coraggio, M.; Dong, L.; Eigenbrot, C.; Erickson, R.; et al. Discovery of GDC-0853: A Potent, Selective, and Noncovalent Bruton’s Tyrosine Kinase Inhibitor in Early Clinical Development. J. Med. Chem. 2018, 61, 2227–2245. [Google Scholar] [CrossRef] [Green Version]
- Alberts, D.S.; Hallum III, A.V.; Stratton-Custis, M.; Garcia, D.J.; Gleason-Guzman, M.; Salmon, S.E.; Santabarbara, P.; Niesor, E.J.; Floret, S.; Bentzen, C.L. Phase I pharmacokinetic trial and correlative in vitro phase II tumor kinetic study of Apomine (SR-45023A), a novel oral biphosphonate anticancer drug. Clin. Cancer Res. 2001, 7, 1246–1250. [Google Scholar]
- Huttunen, K.M.; Rautio, J. Prodrugs—An efficient way to breach delivery and targeting barriers. Curr. Top. Med. Chem. 2011, 11, 2265–2287. [Google Scholar] [CrossRef]
- Ge, Y.; Yang, H.; Wang, C.; Meng, Q.; Li, L.; Sun, H.; Zhen, Y.; Liu, K.; Li, Y.; Ma, X. Design and synthesis of phosphoryl-substituted diphenylpyrimidines (Pho-DPPYs) as potent Bruton’s tyrosine kinase (BTK) inhibitors: Targeted treatment of B lymphoblastic leukemia cell lines. Bioorg. Med. Chem. 2016, 25, 765–772. [Google Scholar] [CrossRef]
- Ma, X.; Ge, Y.; Song, Z.; Huang, S.; Wang, C.; Zhang, J.; Tang, Z.; Liu, K. Phosphoryl Pyrimidine Compound, and Its Composition and Application. Faming Zhuanli Shenqing Patent CN 106565782, 19 April 2017. [Google Scholar]
- Supuran, C.T.; Casini, A.; Scozzafava, A. Protease inhibitors of the sulfonamide type: Anticancer, antiinflammatory, and antiviral agents. Med. Res. Rev. 2003, 23, 535–558. [Google Scholar] [CrossRef]
- Liu, H.; Qu, M.; Xu, L.; Han, X.; Wang, C.; Shu, X.; Yao, J.; Liu, K.; Peng, J.; Li, Y.; et al. Design and synthesis of sulfonamide-substituted diphenylpyrimidines (SFA-DPPYs) as potent Bruton’s tyrosine kinase (BTK) inhibitors with improved activity toward B-cell lymphoblastic leukemia. Eur. J. Med. Chem. 2017, 135, 60–69. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, D.; Wang, L.; Qu, M.; Huang, S.; Wang, C.; Shu, X.; Liu, K. Sulfoamido Pyrimidine Compound as Bruton Tyrosine Kinase Inhibitor and Its Preparation. Faming Zhuanli Shenqing Patent CN 106749042, 31 May 2017. [Google Scholar]
- Wang, C.; Li, S.; Meng, Q.; Sun, X.; Li, H.; Shu, X.; Sun, H.; Liu, K.; Liu, Z.; Ma, X. Novel amino acid-substituted diphenylpyrimidine derivatives as potent BTK inhibitors against B cell lymphoma cell lines. Bioorg. Med. Chem. 2018, 26, 4179–4186. [Google Scholar] [CrossRef]
- Su, Y.; Li, R.; Ning, X.; Lin, Z.; Zhao, X.; Zhou, J.; Liu, J.; Jin, Y.; Yin, Y. Discovery of 2,4-diarylaminopyrimidine derivatives bearing dithiocarbamate moiety as novel FAK inhibitors with antitumor and anti-angiogenesis activities. Eur. J. Med. Chem. 2019, 177, 32–46. [Google Scholar] [CrossRef]
- Zhai, Z.; Li, R.; Bai, X.; Ning, X.; Lin, Z.; Zhao, X.; Jin, Y.; Yin, Y. Design, synthesis and biological evaluation of novel dithiocarbamate-substituted diphenylaminopyrimidine derivatives as BTK inhibitors. Bioorg. Med. Chem. 2019, 27, 4124–4142. [Google Scholar] [CrossRef]
- Zhao, D.; Huang, S.; Qu, M.; Wang, C.; Liu, Z.; Li, Z.; Peng, J.; Liu, K.; Li, Y.; Ma, X.; et al. Structural optimization of diphenylpyrimidine derivatives (DPPYs) as potent Bruton’s tyrosine kinase (BTK) inhibitors with improved activity toward B leukemia cell lines. Eur. J. Med. Chem. 2017, 126, 444–455. [Google Scholar] [CrossRef]
- Li, X.; Shi, B.; Teng, Y.; Cheng, Y.; Yang, H.; Li, J.; Wang, L.; He, S.; You, Q.; Xiang, H. Design, synthesis and biological evaluation of novel 2-phenyl pyrimidine derivatives as potent Bruton’s tyrosine kinase (BTK) inhibitors. MedChemComm 2019, 10, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Shi, W.; Zhao, D.; Wang, Q.; Chang, X.; He, X.; Wang, X.; Gao, Y.; Lu, P.; Zhang, X.; et al. Design and synthesis of boron-containing diphenylpyrimidines as potent BTK and JAK3 dual inhibitors. Bioorg. Med. Chem. 2019, 28, 115236. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.; Liu-Bujalski, L.; Qiu, H.; Mochalkin, I.; Jones, R.; Neagu, C.; Goutopoulos, A.; Grenningloh, R.; Johnson, T.; Sherer, B.; et al. Discovery of a novel series of pyridine and pyrimidine carboxamides as potent and selective covalent inhibitors of Btk. Bioorg. Med. Chem. Lett. 2018, 28, 3419–3424. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, W.; Asami, T.; Irie, T.; Sawa, M. Design and synthesis of novel pyrimidine analogs as highly selective, non-covalent BTK inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 145–151. [Google Scholar] [CrossRef]
- Liang, Q.; Chen, Y.; Yu, K.; Chen, C.; Zhang, S.; Wang, A.; Wang, W.; Wu, H.; Liu, X.; Wang, B.; et al. Discovery of N-(3-(5-((3-acrylamido-4-(morpholine-4-carbonyl)phenyl)amino)-1-methyl-6-oxo-1,6-dihydropyridin-3-yl)-2-methylphenyl)-4-(tert-butyl)benzamide (CHMFL-BTK-01) as a highly selective irreversible Bruton’s tyrosine kinase (BTK) inhibitor. Eur. J. Med. Chem. 2017, 131, 107–125. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Q.; Liang, Q.; Chen, Y.; Chen, C.; Wang, A.; Wu, H.; Yu, K.; Wang, W.; Hu, C.; et al. Novel Irreversible Bruton’s Tyrosine Kinase Inhibitor and Its Preparation. PCT Int. Appl. Patent WO 2017133341, 11 August 2017. [Google Scholar]
- Kawahata, W.; Asami, T.; Kiyoi, T.; Irie, T.; Taniguchi, H.; Asamitsu, Y.; Inoue, T.; Miyake, T.; Sawa, M. Design and Synthesis of Novel Amino-triazine Analogues as Selective Bruton’s Tyrosine Kinase Inhibitors for Treatment of Rheumatoid Arthritis. J. Med. Chem. 2018, 61, 8917–8933. [Google Scholar] [CrossRef]
- Kawahata, W.; Asami, T.; Sawa, M.; Asamitsu, Y.; Irie, T.; Miyake, T.; Kiyoi, T. Preparation of triazine derivatives as BTK inhibitors. PCT Int. Appl. Patent WO 2015012149, 29 January 2015. [Google Scholar]
- Teng, Y.; Lu, X.; Xiao, M.; Li, Z.; Zou, Y.; Ren, S.; Cheng, Y.; Luo, G.; Xiang, H. Discovery of Potent and Highly Selective Covalent Inhibitors of Bruton’s Tyrosine Kinase Bearing Triazine Scaffold. Eur. J. Med. Chem. 2020, 199, 112339. [Google Scholar] [CrossRef]
- Park, H.; Park, C.H.; Kang, S.-T.; Jeon, J.H.; Archary, R.; Lee, J.-Y.; Kim, P.; Jung, H.; Yun, C.-S.; Hwang, J.Y.; et al. Design and Synthesis of Novel Pyrazolo[3,4-d]pyrimidin-1-yl piperidine Derivatives as Bruton’s Tyrosine Kinase Inhibitors. Bull. Korean Chem. Soc. 2017, 38, 278–281. [Google Scholar] [CrossRef]
- Zheng, N.; Pan, J.; Hao, Q.; Li, Y.; Zhou, W. Design, synthesis and biological evaluation of novel 3-substituted pyrazolopyrimidine derivatives as potent Bruton’s tyrosine kinase (BTK) inhibitors. Bioorg. Med. Chem. 2018, 26, 2165–2172. [Google Scholar] [CrossRef]
- Zheng, N.; Hao, Q.; Lin, K.; Pan, J.; Li, Y.; Zhou, W. Synthesis and biological evaluation of novel 1-substituted 3-(3-phenoxyprop-1-yn-1-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amines as potent Bruton’s tyrosine kinase (BTK) inhibitors. Bioorg. Med. Chem. Lett. 2018, 29, 225–229. [Google Scholar] [CrossRef]
- Ran, F.; Liu, Y.; Liu, M.; Zhang, D.; Wang, P.; Dong, J.; Tang, W.; Zhao, G. Discovery of pyrazolopyrimidine derivatives as potent BTK inhibitors with effective anticancer activity in MCL. Bioorg. Chem. 2019, 89, 102943. [Google Scholar] [CrossRef]
- Ran, F.; Liu, Y.; Yu, S.; Guo, K.; Tang, W.; Chen, X.; Zhao, G. Design and synthesis of novel 1-substituted 3-(6-phenoxypyridin-3-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine analogs as selective BTK inhibitors for the treatment of mantle cell lymphoma. Bioorg. Chem. 2019, 94, 103367. [Google Scholar] [CrossRef]
- Ran, F.; Liu, Y.; Chen, X.; Zhuo, H.; Xu, C.; Li, Y.; Duan, X.; Zhao, G. Design and synthesis of novel substituted benzyl pyrrolopyrimidine derivatives as selective BTK inhibitors for treating mantle cell lymphoma. Bioorg. Chem. 2021, 112, 104968. [Google Scholar] [CrossRef]
- Zhang, Q.; Hu, Z.; Shen, Q.; Chen, Y.; Lu, W. Design, Synthesis and Anti-Proliferative Activities of 2,6-Substituted Thieno[3,2-d]pyrimidine Derivatives Containing Electrophilic Warheads. Molecules 2017, 22, 788. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, L.; Yu, J.; Li, H.; He, S.; Tang, W.; Zuo, J.; Lu, W. Discovery of new BTK inhibitors with B cell suppression activity bearing a 4,6-substituted thieno[3,2-d]pyrimidine scaffold. RSC Adv. 2017, 7, 26060–26069. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Lv, W.; Tang, W.; Zhang, Q.; He, S.; Zhang, L.; Li, H.; Lin, Z. Thieno[3,2-d] Pyrimidines as BTK Inhibitor and Preparation Method. Faming Zhuanli Shenqing Patent CN 108727404, 2 November 2018. [Google Scholar]
- He, L.; Pei, H.; Zhang, C.; Shao, M.; Li, D.; Tang, M.; Wang, T.; Chen, X.; Xiang, M.; Chen, L. Design, synthesis and biological evaluation of 7 H -pyrrolo[2,3- d ]pyrimidin-4-amine derivatives as selective Btk inhibitors with improved pharmacokinetic properties for the treatment of rheumatoid arthritis. Eur. J. Med. Chem. 2018, 145, 96–112. [Google Scholar] [CrossRef]
- Zhang, C.; Pei, H.; He, J.; Zhu, J.; Li, W.; Niu, T.; Xiang, M.; Chen, L. Design, synthesis and evaluation of novel 7H-pyrrolo[2,3-d]pyrimidin-4-amine derivatives as potent, selective and reversible Bruton’s tyrosine kinase (BTK) inhibitors for the treatment of rheumatoid arthritis. Eur. J. Med. Chem. 2019, 169, 121–143. [Google Scholar] [CrossRef]
- Pulz, R.; Angst, D.; Dawson, J.; Gessier, F.; Gutmann, S.; Hersperger, R.; Hinniger, A.; Janser, P.; Koch, G.; Revesz, L.; et al. Design of Potent and Selective Covalent Inhibitors of Bruton’s Tyrosine Kinase Targeting an Inactive Conformation. ACS Med. Chem. Lett. 2019, 10, 1467–1472. [Google Scholar] [CrossRef]
- Xue, Y.; Song, P.; Song, Z.; Wang, A.; Tong, L.; Geng, M.; Ding, J.; Liu, Q.; Sun, L.; Xie, H.; et al. Discovery of 4,7-Diamino-5-(4-phenoxyphenyl)-6-methylene-pyrimido[5,4-b]pyrrolizines as Novel Bruton’s Tyrosine Kinase Inhibitors. J. Med. Chem. 2018, 61, 4608–4627. [Google Scholar] [CrossRef]
- Zhang, A.; Ding, J.; Xie, H.; Song, Z.; Xue, Y.; Tong, L.; Geng, M. Preparation of Pyrimido[5,4-b]indolizine or Pyrimido[5,4-b]pyrrolizine Compounds Useful as BTK Kinase Inhibitors for the Treatment of Cancer. PCT Int. Appl. Patent WO 2018095398, 1 June 2018. [Google Scholar]
- Hopkins, B.T.; Bame, E.; Bell, N.; Bohnert, T.; Bowden-Verhoek, J.K.; Bui, M.; Cancilla, M.T.; Conlon, P.; Cullen, P.; Erlanson, D.A.; et al. Utilizing structure based drug design and metabolic soft spot identification to optimize the in vitro potency and in vivo pharmacokinetic properties leading to the discovery of novel reversible Bruton’s tyrosine kinase inhibitors. Bioorg. Med. Chem. 2021, 44, 116275. [Google Scholar] [CrossRef]
- Gao, X.; Wang, J.; Liu, J.; Guiadeen, D.; Krikorian, A.; Boga, S.B.; Alhassan, A.-B.; Selyutin, O.; Yu, W.; Yu, Y.; et al. Discovery of novel BTK inhibitors with carboxylic acids. Bioorg. Med. Chem. Lett. 2016, 27, 1471–1477. [Google Scholar] [CrossRef]
- Boga, S.B.; Alhassan, A.-B.; Liu, J.; Guiadeen, D.; Krikorian, A.; Gao, X.; Wang, J.; Yu, Y.; Anand, R.; Liu, S.; et al. Discovery of 3-morpholino-imidazole[1,5-a]pyrazine BTK inhibitors for rheumatoid arthritis. Bioorg. Med. Chem. Lett. 2017, 27, 3939–3943. [Google Scholar] [CrossRef]
- Liu, J.; Guiadeen, D.; Krikorian, A.; Gao, X.; Wang, J.; Boga, S.B.; Alhassan, A.-B.; Yu, W.; Selyutin, O.; Yu, Y.; et al. Potent, non-covalent reversible BTK inhibitors with 8-amino-imidazo[1,5-a]pyrazine core featuring 3-position bicyclic ring substitutes. Bioorg. Med. Chem. Lett. 2020, 30, 127390. [Google Scholar] [CrossRef]
- Jorda, R.; Krajcovicova, S.; Králová, P.; Soural, M.; Kryštof, V. Scaffold hopping of the SYK inhibitor entospletinib leads to broader targeting of the BCR signalosome. Eur. J. Med. Chem. 2020, 204, 112636. [Google Scholar] [CrossRef]
- Krajčovičová, S.; Jorda, R.; Vanda, D.; Soural, M.; Kryštof, V. 1,4,6-Trisubstituted imidazo[4,5-c]pyridines as inhibitors of Bruton’s tyrosine kinase. Eur. J. Med. Chem. 2020, 211, 113094. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, G.; Zhao, J.; Wang, Y.; Wu, X.; He, X.; Li, W.; Zhang, S.; Yang, S.; Ma, C.; et al. Struc-ture-activity relationship investigation for imidazopyrazole-3-carboxamide derivatives as novel selective inhibitors of Bruton’s tyrosine kinase. Eur. J. Med. Chem. 2021, 225, 113724. [Google Scholar] [CrossRef]
- Yao, X.; Sun, X.; Jin, S.; Yang, L.; Xu, H.; Rao, Y. Discovery of 4-Aminoquinoline-3-carboxamide Derivatives as Potent Reversible Bruton’s Tyrosine Kinase Inhibitors for the Treatment of Rheumatoid Arthritis. J. Med. Chem. 2019, 62, 6561–6574. [Google Scholar] [CrossRef]
- Wang, B.; Deng, Y.; Chen, Y.; Yu, K.; Wang, A.; Liang, Q.; Wang, W.; Chen, C.; Wu, H.; Hu, C.; et al. Structure-activity relationship investigation for benzonaphthyridinone derivatives as novel potent Bruton’s tyrosine kinase (BTK) irreversible inhibitors. Eur. J. Med. Chem. 2017, 137, 545–557. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, J.; Chen, Y.; Wu, H.; Wang, A.; Wang, B.; Hu, C.; Wang, W.; Chen, C. Preparation of Bruton’s Tyrosine Kinase Inhibitors. PCT Int. Appl. Patent WO 2015192658, 23 December 2015. [Google Scholar]
- Lee, E.; Cho, H.; Lee, D.K.; Ha, J.; Choi, B.J.; Jeong, J.H.; Ryu, J.-H.; Kang, J.S.; Jeon, R. Discovery of 5-Phenoxy-2-aminopyridine Derivatives as Potent and Selective Irreversible Inhibitors of Bruton’s Tyrosine Kinase. Int. J. Mol. Sci. 2020, 21, 8006. [Google Scholar] [CrossRef]
- Lou, Y.; Lopez, F.; Jiang, Y.; Han, X.; Brotherton, C.; Billedeau, R.; Gabriel, S.; Gleason, S.; Goldstein, D.M.; Hilgenkamp, R.; et al. Mitigation of reactive metabolite formation for a series of 3-amino-2-pyridone inhibitors of Bruton’s tyrosine kinase (BTK). Bioorg. Med. Chem. Lett. 2016, 27, 632–635. [Google Scholar] [CrossRef]
- Lopez-Tapia, F.; Lou, Y.; Brotherton-Pleiss, C.; Kuglstatter, A.; So, S.-S.; Kondru, R. A potent seven-membered cyclic BTK (Bruton’s tyrosine Kinase) chiral inhibitor conceived by structure-based drug design to lock its bioactive conformation. Bioorg. Med. Chem. Lett. 2019, 29, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.J.; Lee, W.; Johnson, A.R.; Delatorre, K.J.; Chen, J.; Eigenbrot, C.; Heidmann, J.; Kakiuchi-Kiyota, S.; Katewa, A.; Kiefer, J.R.; et al. Stereochemical Differences in Fluorocyclopropyl Amides Enable Tuning of Btk Inhibition and Off-Target Activity. ACS Med. Chem. Lett. 2020, 11, 1588–1597. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Batt, D.G.; Chaudhry, C.; Lippy, J.S.; Pattoli, M.A.; Surti, N.; Xu, S.; Carter, P.H.; Burke, J.R.; Tino, J.A. Conversion of carbazole carboxamide based reversible inhibitors of Bruton’s tyrosine kinase (BTK) into potent, selective irreversible inhibitors in the carbazole, tetrahydrocarbazole, and a new 2,3-dimethylindole series. Bioorg. Med. Chem. Lett. 2018, 28, 3080–3084. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Lee, E.; Kwon, H.A.; Seul, L.; Jeon, H.-J.; Yu, J.H.; Ryu, J.-H.; Jeon, R. Discovery of Tricyclic Pyranochromenone as Novel Bruton’s Tyrosine Kinase Inhibitors with in Vivo Antirheumatic Activity. Int. J. Mol. Sci. 2020, 21, 7919. [Google Scholar] [CrossRef]
- Guo, X.; Yang, D.; Fan, Z.; Zhang, N.; Zhao, B.; Huang, C.; Wang, F.; Ma, R.; Meng, M.; Deng, Y. Discovery and structure-activity relationship of novel diphenylthiazole derivatives as BTK inhibitor with potent activity against B cell lymphoma cell lines. Eur. J. Med. Chem. 2019, 178, 767–781. [Google Scholar] [CrossRef]
- Park, C.H.; Kim, D.; Jung, H.; Jeon, J.H.; Achary, R.; Lee, J.-Y.; Kim, P.; Jung, H.; Hwang, J.Y.; Ryu, D.H.; et al. Design and Synthesis of Novel 3-(2-Aminopyridin-3-yl)-1,2,4-Triazolo[4,3-b]Pyridazine Derivatives as a Reversible Bruton’s Tyrosine Kinase Inhibitors. Bull. Korean Chem. Soc. 2018, 39, 853–857. [Google Scholar] [CrossRef]
- Cho, S.Y.; Kim, P.H.; Kim, H.R.; Lee, J.O.; Ha, J.D.; Jung, H.J.; Yoon, C.S.; Park, J.H.; Hwang, J.Y. Triazolopyridazine Derivative as Bruton’s Tyrosine Kinase Inhibitor, and Method for the Preparation Thereof. Repub. Korean Kongkae Taeho Kongbo Patent KR 2018094194, 23 August 2018. [Google Scholar]
- Liu, L.; Li, X.; Cheng, Y.; Wang, L.; Yang, H.; Li, J.; He, S.; Wu, S.; Yin, Q.; Xiang, H. Optimization of novel benzofuro[3,2-b]pyridin-2(1H)-one derivatives as dual inhibitors of BTK and PI3Kδ. Eur. J. Med. Chem. 2018, 164, 304–316. [Google Scholar] [CrossRef]













| Drug | Molecular Formula | Diseases |
|---|---|---|
| Ibrutinib |  | CLL/SLL WM MZL MCL cGVHD |
| Acalabrutinib |  | CLL SLL MCL |
| Zanubrutinib |  | MCL |
| Tirabrutinib |  | CNS lymphoma WM CLL |
| Orelabrutinib |  | MCL, CLL SLL |
| Chemical Class | Type of Inhibition | Ref |
|---|---|---|
| Diphenylaminopyrimidines | Irreversible BTKIs | [64,65,66,67,68,69,70,71,72,73,74,75,76] |
| Pyridines | Irreversible BTKIs | [77] |
| Pyrimidines | Reversible BTKIs | [78] |
| Pyridinones | Irreversible BTKIs | [79,80] |
| Triazines | Irreversible and reversible BTKIs | [81,82,83] |
| Pyrazolo-pyrimidines | Irreversible BTKIs | [84,85,86,87,88,89] |
| Thieno-pyrimidines | Irreversible BTKIs | [90,91,92] |
| Pyrrolo-pyrimidines | Irreversible BTKIs | [93,94,95,96,97,98] |
| Imidazo-pyrazines | Reversible BTKIs | [99,100,101] |
| Imidazo-pyridines | Reversible BTKIs | [102,103] |
| Imidazo-pyrazoles | Irreversible BTKIs | [104] |
| Quinolines | Irreversible and Reversible BTKIs | [105,106,107] |
| Dihydroisoquinolines | Irreversible BTKIs | [108] |
| Phthalazines | Reversible BTKIs | [109,110,111] |
| Carbazoles | Irreversible BTKIs | [112] |
| Tetrahydrocarbazole | Irreversible BTKIs | [112] |
| Indoles | Irreversible BTKIs | [112] |
| Pyrano-chromenone | Irreversible BTKIs | [113] |
| Thiazoles | Reversible BTKIs | [114] |
| Pyrazoles | Reversible BTKIs | [115,116] |
| benzofuranes | Irreversible BTKIs | [117] |
| PROTAC | Irreversible and reversible BTKIs | [11,12] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tasso, B.; Spallarossa, A.; Russo, E.; Brullo, C. The Development of BTK Inhibitors: A Five-Year Update. Molecules 2021, 26, 7411. https://doi.org/10.3390/molecules26237411
Tasso B, Spallarossa A, Russo E, Brullo C. The Development of BTK Inhibitors: A Five-Year Update. Molecules. 2021; 26(23):7411. https://doi.org/10.3390/molecules26237411
Chicago/Turabian StyleTasso, Bruno, Andrea Spallarossa, Eleonora Russo, and Chiara Brullo. 2021. "The Development of BTK Inhibitors: A Five-Year Update" Molecules 26, no. 23: 7411. https://doi.org/10.3390/molecules26237411
APA StyleTasso, B., Spallarossa, A., Russo, E., & Brullo, C. (2021). The Development of BTK Inhibitors: A Five-Year Update. Molecules, 26(23), 7411. https://doi.org/10.3390/molecules26237411