Descriptors
The full set of descriptors was filtered out in order to discard both those poorly correlated to the activity descriptors, as well as the highly correlated pairs of descriptors.
To begin with, the correlation of the descriptors with the activity (values: 0—for inactive and 1—for active structures of the set) was studied (
Table S5) in order to find the most correlated descriptors and make up an initial interpretation for those correlations.
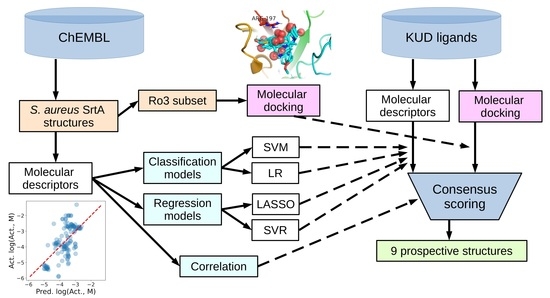
Reasonable interpretations were obtained for the influence of the estimated physicochemical properties of a molecule on its predicted activity. For instance, an increase in lipophilicity (MolLogP) above the mean value of 3.33 adversary affects activity, as do the properties closely related to the general size of a molecule: NumAtoms, MolWt and MolMR when increased based on their mean values of 45.0, 357.2 and 96.0, respectively. Therefore, a large and lipophilic molecule is not beneficial for activity. To extend the above further, the increase of the polar surface (TPSA) and the number of heteroatoms (NumHeteroatoms) leads to higher chances for a molecule to be active for the molecule set studied. Interestingly, there is an asymmetry regarding the influence of changing the number of hydrogen bond donors and acceptors. Thus, an increase of the number of hydrogen bond donors, NHOHCount and NumHDonors, above the mean numbers of 1.8 and 1.8 lowers the chances of a molecule to be active. Meanwhile, an increase of the number of hydrogen bond acceptors, NOCount and NumHAcceptors, above the mean values of 5.0 and 4.3 beneficially affects the probability of a molecule being active. Consequently, an active molecule should contain a significant polar surface represented mainly with acceptors of hydrogen bonds. This observation might be interpreted in terms of the structural requirements that arise due to the binding site constitution. It is well-established that Arg197 is critical to the binding and functioning of SrtA, and it has been also determined that it acts more like a hydrogen bond donor than as a charged species [
56]. It is also corroborated by the not necessarily charged nature of the natural LPXTG-containing ligands. We therefore hypothesize that, in order to coordinate the charged and hydrogen donor Arg197 residue, a ligand should be more like a dipolar aprotic solvent, i.e., it should contain more hydrogen bond acceptors and as few as possible hydrogen bond donors clearly placed in certain positions. To further support this hypothesis, descriptors that reflect the articulated partial atomic charge, MaxPartialCharge, MinPartialCharge and MaxAbsPartialCharge, negatively correlate with the predicted activity.
Another observation is an increase of the number of aromatic rings, NumAromaticRings, and the number of chiral centers, NumChiralCenters, above the mean values of 2.0 and 1.3 is not beneficial for activity. Interestingly, the number of aliphatic rings, NumAliphaticRings, appears to be not significant to determine activity. Excessive conformational flexibility with NumRotatableBonds above the mean value of 5.4 is also not favored for activity.
It is instructive to note that the mean values of descriptors in the molecules set are well within the requirements for Lipinski’s “Ro5” [
27,
28], Ghose [
38] and Veber [
39] filters for druglike molecules. However, as we are interested in fragment-sized lead-like compounds, the “rule of three” (Ro3) [
25,
26] is a more appropriate filter. Its requirements are stretched as judged by the mean descriptor values for the set (
Table S6).
The next step was to exclude the mutually correlated descriptors in order to build more reliable predictive models. To that end, the correlation between the descriptor values on the molecule set was studied. Firstly, a strong correlation was observed between TPSA, NHOHCount, NOCount and NumHeteroatoms. A significant correlation was present between NHOHCount and Kappa1. Another rather predictable correlation was between NumHAcceptors and TPSA, NOCount and NumHeteroatoms. The same was for the NumHDonors and NHOHCount pair. In order to maximize the interpretability, a combination of the descriptors, capturing the general number of heteroatoms, such as TPSA (or, alternatively, NumHeteroatoms), and the hydrogen bond donor/acceptor differentiating descriptors, such as NumHDonors and NumHAcceptors, was decided to be used for further research.
Secondly, the topological indices studied in the work generally established good correlations with each other and with different descriptors. On the one hand, it once again confirmed the usefulness and efficacy of the topological index concept, which made it possible to establish decent quantitative correlations with the properties hardly expressed in terms of the simplest structural descriptors. On the other hand, in cases where the topological descriptors correlated with the more readily interpretable physicochemical or structural descriptors, the latter should be preferred for building the quantitative model to facilitate interpretation of the model. Perhaps a single exception is the Balaban topological connectivity index (BalabanJ), which does not correlate to other indices; however, its correlation to activity is also negligible. As for descriptors of the group Chi, they produce appreciable correlations with the descriptors depicting the general molecule size, such as NumAtoms, MolWt and MolMR. The same holds for Kappa* and BertzCT descriptors. In addition, Chi0 and HallKierAlpha also reasonably correlate to FractionCSP3. This analysis suggests that topological indices should be excluded from the descriptor set to build quantitative models for not bringing additional information in favor of more readily interpretable counterparts.
Finally, descriptors reflecting the general molecule size, such as NumAtoms, MolWt and MolMR, predictably form mutual correlations, so only a single representative, NumAtoms, was retained for the final descriptor set.
Based on the above analysis, 12 descriptors were chosen to be used in building quantitative models (
Table S5). It should be noted that the ratio of the number of endpoint data and the number of descriptors, 12–190 (118 actives and 78 inactives), reasonably corresponds to good practices of QSAR modeling [
57].
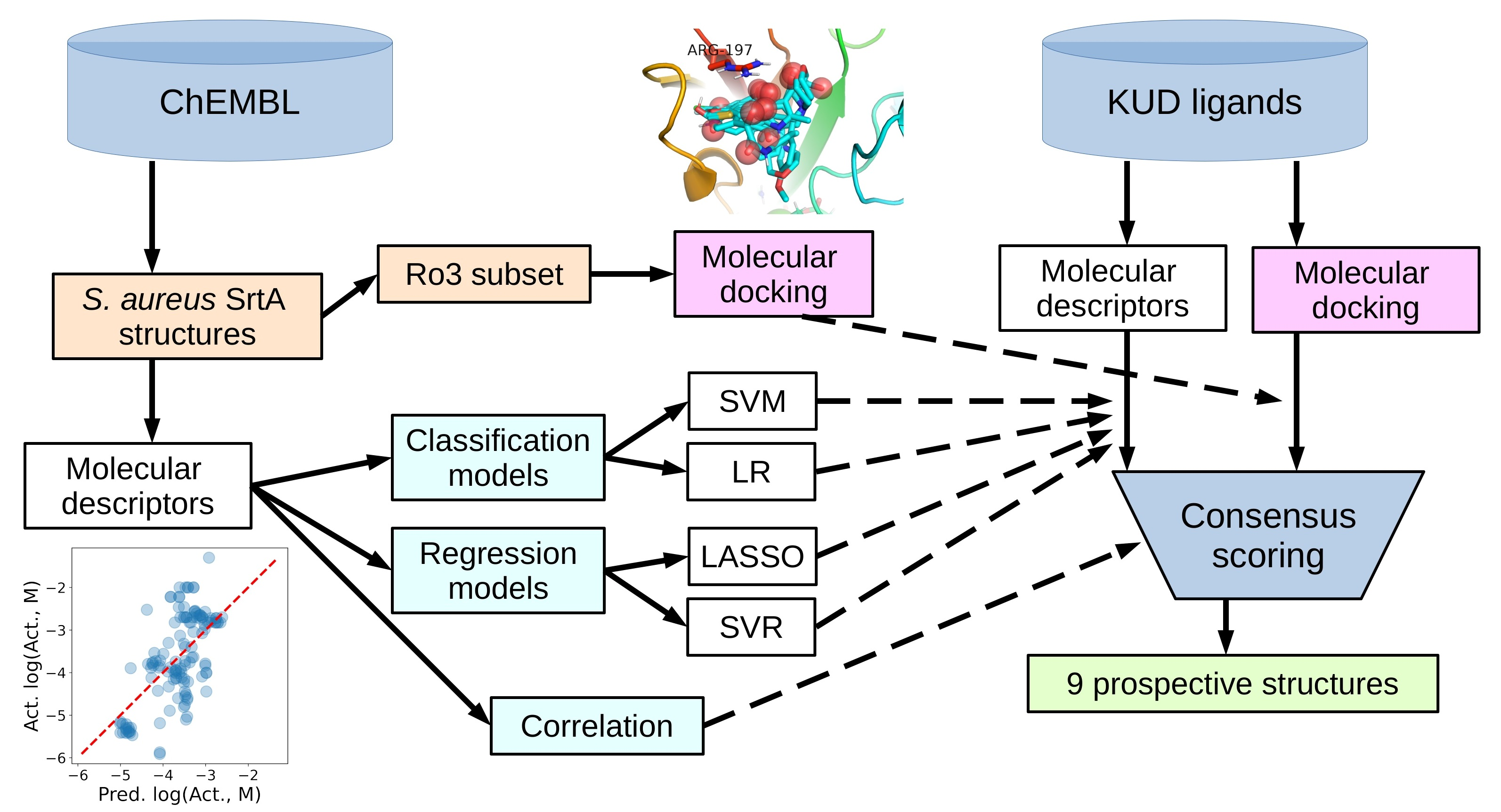
Classification Models on Training Set
Two classification methods were studied in the work for the reasons described in the Materials and Methods: support vector machine (SVM) and logistic regression (LR), both with the L1 regularization of the model coefficients. The main benefit of L1 regularization is that it enables automatic feature (descriptor) selection, based on the significance for the model. At each regularization parameter value, only the coefficients at the most significant descriptors receive nonzero values. The larger the L1 penalty, the fewer descriptors enter the model. This results in a rougher model that is less accurate for the training data but, at the same time, is less prone to be overfitted.
We built three different models for each of the classification methods used, SVM or LR. The first one is a model with high regularization constant, containing only one descriptor, which reveals the most significant descriptor on the set and sets the lower limit to the model accuracy. The second one is built with a mild regularization constant, so that all descriptors that enter the model contribute to the prediction, which sets the upper limit in the accuracy as attainable for the specific molecule and descriptor sets. The third model, built with intermediate values of the regularization constant, is an optimal model containing only a subset of the initial descriptor set and combining reasonable accuracy and robustness.
Using the SVM method, a one-descriptor model contained only the MolLogP descriptor was the most discriminative among the others in the molecule set. Similar to the correlation analysis above, it enters in the model with a negative sign, meaning, generally, the more lipophilicity, the less likely a molecule is active. It is an unusual behavior that reflects the peculiarities of the active/inactive dataset molecules. The visual analysis of the MolLogP distribution among the active and inactive subsets supports the simple distinction (
Figure 2) relative to the mean MolLogP value of 3.34, although the dispersion in each class is also appreciable. The area under the curve (AUC) for the receiver operator characteristic (ROC) for the one-descriptor SVM model is appreciable at 0.71 (grey in
Figure 3), which also supports the discriminating power of the MolLogP descriptor for the molecule set studied. The confusion matrix parameters for this model are TP = 78, TN = 49, FN = 40 and FP = 23.
A mildly regularized model with all 12 descriptors (chosen at the stage of the initial correlation analysis) results in a ROC AUC value of 0.83 (
Figure 3 green) and sets the accuracy limit for the combination of the molecule set and SVM model type. This model is significantly better for early enrichment (left bottom corner of the ROC curve in
Figure 3) compared to the one-descriptor model, despite the AUC increase not being dramatic. The confusion matrix parameters for this model are TP = 106, TN = 43, FN = 12 and FP = 29.
The third model, built with intermediate values for L1 regularization of the parameters, uses effectively only nine out of 12 descriptors. It combines the AUC value, 0.81, close to the full 12-descriptor model, good early enrichment properties and, potentially, the higher robustness (more generalization) via the exclusion of less significant descriptors (
Table S7). The confusion matrix parameters for this model are TP = 103, TN = 40, FN = 15 and FP = 32. The signs of the coefficients at the descriptors for the SVM model and, hence, the interpretation of their influence are similar to those obtained at the study of the correlation of the descriptor value with binary activity. The leave-one-out (LOO) estimation of the AUC for this model is 0.76, close to the value for the whole training set. Finally, this nine-descriptor model was used to predict the activity for the molecules of the prospective ligand list.
Logistic regression (LR) was chosen as the second type of classification model. The setup, like the SVM model training experiment, was used to obtain three models with different levels of the regularization factor and the final number of descriptors used in a model. The results for the LR models were very close to the SVM models. Thus, the one-descriptor model contained only the MolLogP descriptor. The same three descriptors were excluded at the intermediate regularization constant value (
Table S7). All the signs of the coefficients at the descriptors were also the same. Additionally, the statistical performance was the same (
Figure 3, right). The confusion matrix parameters for this model are TP = 78, TN = 49, FN = 40 and FP = 23. Similarly, the nine-descriptor model was used for further predictions on the prospective molecule list. The confusion matrix parameters for this model are TP = 103, TN = 39, FN = 15 and FP = 33. The confusion matrices for the 12-descriptor models for SVM and LR methods coincided. The LOO estimation of the AUC for the nine-descriptor modes is 0.75. The overall similarity in the quality of the prediction in the training set should be attributed not to the model peculiarities but, rather, to the quality of the input data. Therefore, the more flexible models would result in overfit and not in the accuracy gain. Thus, we expect a decent generalization was achieved by the finally chosen models, which lays the foundations for robust activity predictions for dissimilar molecules of the prospective molecule list.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

 5R
5R 5R
5R 5S
5S 5R
5R 5S
5S 7R
7R 7S
7S

 2R
2R 2S
2S 2S
2S 2R
2R 2S
2S 2S
2S 5R
5R 5S
5S 5R
5R 5S
5S 5R
5R 5S
5S 5R
5R 5S
5S 5R
5R 5S
5S 5R
5R 5S
5S 5R
5R 5S
5S 7R
7R 7S
7S 7R
7R 7S
7S 2R
2R 2S
2S 2R
2R 2S
2S 2R
2R 2S
2S