
Spin Hamiltonians in Magnets: Theories and Computations

 ,
,  and
and {kind=link}
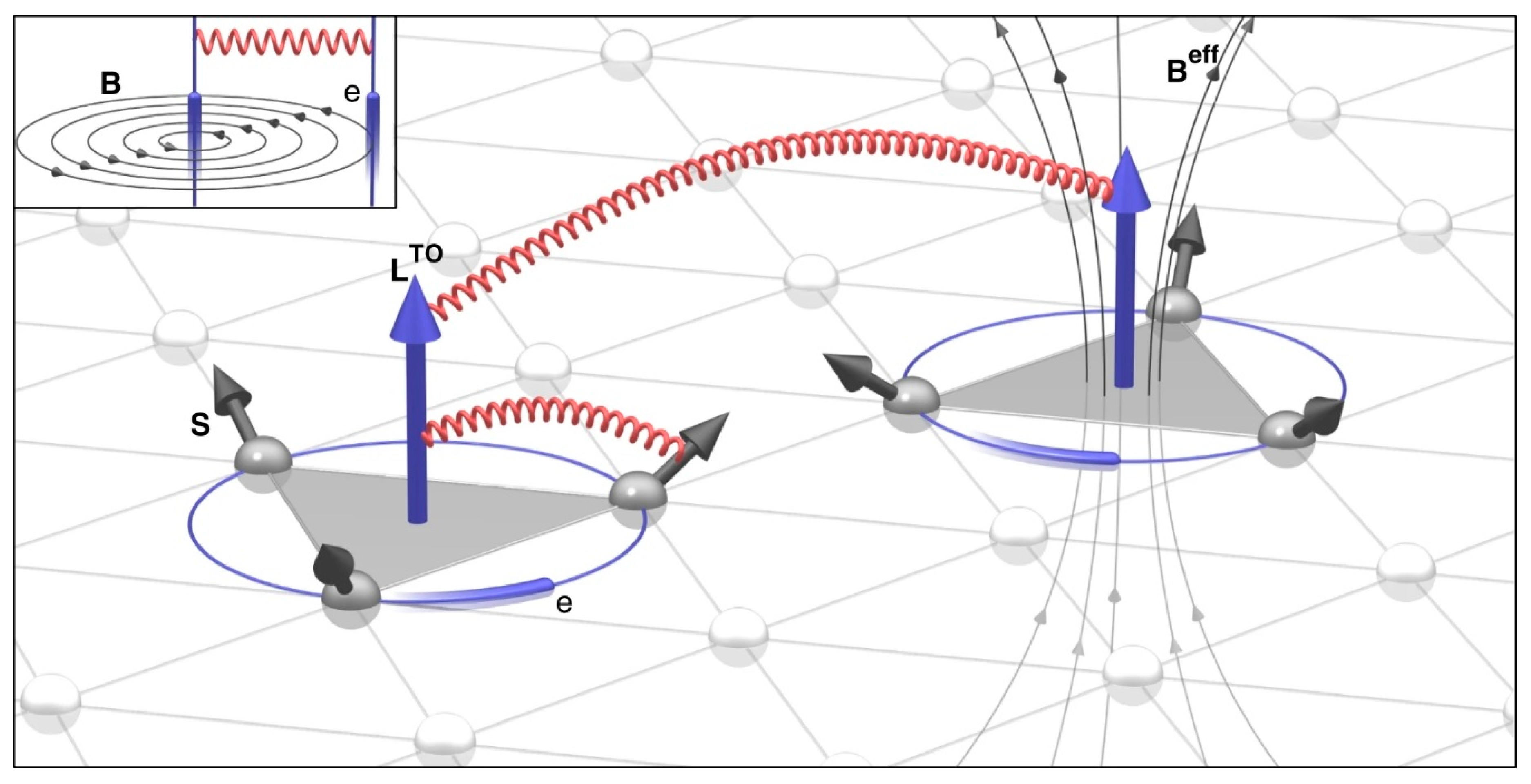{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Effective Spin Hamiltonian Models
2.1. Atomic Magnetic Moments
2.2. Heisenberg Model
2.3. The J Matrices and Single-Ion Anisotropy
2.4. Fourth-Order Interactions without SOC
2.5. Chiral Magnetic Interactions Beyond DMI
2.6. Expansions of Magnetic Interactions
3. Methods of Computing the Parameters of Effective Spin Hamiltonian Models
3.1. Energy-Mapping Analysis
3.1.1. Four-State Method
3.1.2. Direct Least Squares Fitting
3.1.3. Methods Based on Expansions and Selecting Important Terms
3.2. Green’s Function Method Based on Magnetic-Force Linear Response Theory
3.3. More Discussions on Calculating Spin Interaction Parameters
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Baibich, M.N.; Broto, J.M.; Fert, A.; Vandau, F.N.; Petroff, F.; Eitenne, P.; Creuzet, G.; Friederich, A.; Chazelas, J. Giant magnetoresistance of (001)Fe/(001) Cr agnetic superlattices. Phys. Rev. Lett. 1988, 61, 2472–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binasch, G.; Grunberg, P.; Saurenbach, F.; Zinn, W. Enhanced magnetoresistance in layered magnetic-structures with antiferromagnetic interlayer exchange. Phys. Rev. B 1989, 39, 4828–4830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julliere, M. Tunneling between ferromagnetic-films. Phys. Lett. A 1975, 54, 225–226. [Google Scholar] [CrossRef]
- Butler, W.H.; Zhang, X.G.; Schulthess, T.C.; MacLaren, J.M. Spin-dependent tunneling conductance of Fe vertical bar MgO vertical bar Fe sandwiches. Phys. Rev. B 2001, 63, 054416. [Google Scholar] [CrossRef] [Green Version]
- Mathon, J.; Umerski, A. Theory of tunneling magnetoresistance of an epitaxial Fe/MgO/Fe(001) junction. Phys. Rev. B 2001, 63, 220403. [Google Scholar] [CrossRef] [Green Version]
- Bowen, M.; Cros, V.; Petroff, F.; Fert, A.; Boubeta, C.M.; Costa-Kramer, J.L.; Anguita, J.V.; Cebollada, A.; Briones, F.; de Teresa, J.M.; et al. Large magnetoresistance in Fe/MgO/FeCo(001) epitaxial tunnel junctions on GaAs(001). Appl. Phys. Lett. 2001, 79, 1655–1657. [Google Scholar] [CrossRef] [Green Version]
- Yuasa, S.; Nagahama, T.; Fukushima, A.; Suzuki, Y.; Ando, K. Giant room-temperature magnetoresistance in single-crystal Fe/MgO/Fe magnetic tunnel junctions. Nat. Mater. 2004, 3, 868–871. [Google Scholar] [CrossRef]
- Parkin, S.S.P.; Kaiser, C.; Panchula, A.; Rice, P.M.; Hughes, B.; Samant, M.; Yang, S.H. Giant tunnelling magnetoresistance at room temperature with MgO (100) tunnel barriers. Nat. Mater. 2004, 3, 862–867. [Google Scholar] [CrossRef]
- Ikeda, S.; Hayakawa, J.; Ashizawa, Y.; Lee, Y.M.; Miura, K.; Hasegawa, H.; Tsunoda, M.; Matsukura, F.; Ohno, H. Tunnel magnetoresistance of 604% at 300 K by suppression of Ta diffusion in CoFeB/MgO/CoFeB pseudo-spin-valves annealed at high temperature. Appl. Phys. Lett. 2008, 93, 082508. [Google Scholar] [CrossRef]
- Ralph, D.C.; Stiles, M.D. Spin transfer torques. J. Magn. Magn. Mater. 2008, 320, 1190–1216. [Google Scholar] [CrossRef]
- Jonietz, F.; Muehlbauer, S.; Pfleiderer, C.; Neubauer, A.; Muenzer, W.; Bauer, A.; Adams, T.; Georgii, R.; Boeni, P.; Duine, R.A.; et al. Spin Transfer Torques in MnSi at Ultralow Current Densities. Science 2010, 330, 1648–1651. [Google Scholar] [CrossRef] [Green Version]
- Pai, C.-F.; Liu, L.; Li, Y.; Tseng, H.W.; Ralph, D.C.; Buhrman, R.A. Spin transfer torque devices utilizing the giant spin Hall effect of tungsten. Appl. Phys. Lett. 2012, 101, 122404. [Google Scholar] [CrossRef]
- Mellnik, A.R.; Lee, J.S.; Richardella, A.; Grab, J.L.; Mintun, P.J.; Fischer, M.H.; Vaezi, A.; Manchon, A.; Kim, E.A.; Samarth, N.; et al. Spin-transfer torque generated by a topological insulator. Nature 2014, 511, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.F.; Anderson, P.W. Theory of Spin Glasses. J. Phys. F-Met. Phys. 1975, 5, 965–974. [Google Scholar] [CrossRef]
- Binder, K.; Young, A.P. Spin-glasses-experimental facts, theoretical concepts, and open questions. Rev. Mod. Phys. 1986, 58, 801–976. [Google Scholar] [CrossRef]
- Bramwell, S.T.; Gingras, M.J.P. Spin ice state in frustrated magnetic pyrochlore materials. Science 2001, 294, 1495–1501. [Google Scholar] [CrossRef] [Green Version]
- Castelnovo, C.; Moessner, R.; Sondhi, S.L. Magnetic monopoles in spin ice. Nature 2008, 451, 42–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, P.W. Resonating valence bonds: A new kind of insulator? Mater. Res. Bull. 1973, 8, 153–160. [Google Scholar] [CrossRef]
- Shimizu, Y.; Miyagawa, K.; Kanoda, K.; Maesato, M.; Saito, G. Spin liquid state in an organic Mott insulator with a triangular lattice. Phys. Rev. Lett. 2003, 91, 107001. [Google Scholar] [CrossRef] [Green Version]
- Balents, L. Spin liquids in frustrated magnets. Nature 2010, 464, 199–208. [Google Scholar] [CrossRef]
- Yan, S.; Huse, D.A.; White, S.R. Spin-Liquid Ground State of the S = 1/2 Kagome Heisenberg Antiferromagnet. Science 2011, 332, 1173–1176. [Google Scholar] [CrossRef] [Green Version]
- Han, T.-H.; Helton, J.S.; Chu, S.; Nocera, D.G.; Rodriguez-Rivera, J.A.; Broholm, C.; Lee, Y.S. Fractionalized excitations in the spin-liquid state of a kagome-lattice antiferromagnet. Nature 2012, 492, 406–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skyrme, T.H.R. A unifield field theory of mesons and baryons. Nucl. Phys. 1962, 31, 556–569. [Google Scholar] [CrossRef]
- Fert, A.; Cros, V.; Sampaio, J. Skyrmions on the track. Nat. Nanotechnol. 2013, 8, 152–156. [Google Scholar] [CrossRef]
- Muehlbauer, S.; Binz, B.; Jonietz, F.; Pfleiderer, C.; Rosch, A.; Neubauer, A.; Georgii, R.; Boeni, P. Skyrmion Lattice in a Chiral Magnet. Science 2009, 323, 915–919. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, A.; Pfleiderer, C.; Binz, B.; Rosch, A.; Ritz, R.; Niklowitz, P.G.; Boeni, P. Topological Hall Effect in the A Phase of MnSi. Phys. Rev. Lett. 2009, 102, 186602. [Google Scholar] [CrossRef] [Green Version]
- Pappas, C.; Lelievre-Berna, E.; Falus, P.; Bentley, P.M.; Moskvin, E.; Grigoriev, S.; Fouquet, P.; Farago, B. Chiral Paramagnetic Skyrmion-like Phase in MnSi. Phys. Rev. Lett. 2009, 102, 197202. [Google Scholar] [CrossRef] [Green Version]
- Roessler, U.K.; Bogdanov, A.N.; Pfleiderer, C. Spontaneous skyrmion ground states in magnetic metals. Nature 2006, 442, 797–801. [Google Scholar] [CrossRef] [Green Version]
- Fujishiro, Y.; Kanazawa, N.; Tokura, Y. Engineering skyrmions and emergent monopoles in topological spin crystals. Appl. Phys. Lett. 2020, 116, 090501. [Google Scholar] [CrossRef]
- Hastings, W.K. Monte-carlo sampling methods using markov chains and their applications. Biometrika 1970, 57, 97–109. [Google Scholar] [CrossRef]
- Ma, P.-W.; Woo, C.H.; Dudarev, S.L. Large-scale simulation of the spin-lattice dynamics in ferromagnetic iron. Phys. Rev. B 2008, 78, 024434. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.-W.; Dudarev, S.L.; Woo, C.H. Spin-lattice-electron dynamics simulations of magnetic materials. Phys. Rev. B 2012, 85, 184301. [Google Scholar] [CrossRef] [Green Version]
- Omelyan, I.P.; Mryglod, I.M.; Folk, R. Algorithm for molecular dynamics simulations of spin liquids. Phys. Rev. Lett. 2001, 86, 898–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Vleck, J.H. Theory of the variations in paramagnetic anisotropy among different salts of the iron group. Phys. Rev. 1932, 41, 208–215. [Google Scholar] [CrossRef]
- Ballhausen, C.J. Introduction to Ligand Field Theory; McGraw-Hill: New York, NY, USA, 1962. [Google Scholar]
- Griffith, J.S. The Theory of Transition-Metal Ions; Cambridge University Press: Cambridge, UK, 1964. [Google Scholar]
- Mattis, D.C. The Theory of Magnetism Made Simple: An. Introduction to Physical Concepts and to Some Useful Mathematical Methods; World Scientific Publishing Company: Singapore, 2006. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. B 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, J. Electron correlations in narrow energy bands. Proc. R. Soc. Lond. Ser. A-Math. Phys. Sci. 1963, 276, 238–257. [Google Scholar]
- Liechtenstein, A.I.; Anisimov, V.I.; Zaanen, J. Density-functional theory and strong-interactions-orbital ordering in mott-hubbard insulators. Phys. Rev. B 1995, 52, R5467–R5470. [Google Scholar] [CrossRef] [Green Version]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Himmetoglu, B.; Floris, A.; de Gironcoli, S.; Cococcioni, M. Hubbard-Corrected DFT Energy Functionals: The LDA + U Description of Correlated Systems. Int. J. Quantum Chem. 2014, 114, 14–49. [Google Scholar] [CrossRef] [Green Version]
- The VASP Manual—Vaspwiki. Available online: https://www.vasp.at/wiki/index.php/The_VASP_Manual (accessed on 3 February 2021).
- Dederichs, P.H.; Blugel, S.; Zeller, R.; Akai, H. Ground-states of constrained systems-application to cerium impurities. Phys. Rev. Lett. 1984, 53, 2512–2515. [Google Scholar] [CrossRef]
- Hybertsen, M.S.; Schluter, M.; Christensen, N.E. Calculation of coulomb-interaction parameters for la2cuo4 using a constrained-density-functional approach. Phys. Rev. B 1989, 39, 9028–9041. [Google Scholar] [CrossRef]
- Gunnarsson, O.; Andersen, O.K.; Jepsen, O.; Zaanen, J. Density-functional calculation of the parameters in the anderson model-application to Mn in CdTe. Phys. Rev. B 1989, 39, 1708–1722. [Google Scholar] [CrossRef] [Green Version]
- Springer, M.; Aryasetiawan, F. Frequency-dependent screened interaction in Ni within the random-phase approximation. Phys. Rev. B 1998, 57, 4364–4368. [Google Scholar] [CrossRef]
- Kotani, T. Ab initio random-phase-approximation calculation of the frequency-dependent effective interaction between 3d electrons: Ni, Fe, and MnO. J. Phys. Condens. Matter 2000, 12, 2413–2422. [Google Scholar] [CrossRef]
- Aryasetiawan, F.; Imada, M.; Georges, A.; Kotliar, G.; Biermann, S.; Lichtenstein, A.I. Frequency-dependent local interactions and low-energy effective models from electronic structure calculations. Phys. Rev. B 2004, 70, 195104. [Google Scholar] [CrossRef] [Green Version]
- Metzner, W.; Vollhardt, D. Correlated lattice fermions in dinfinity dimensions. Phys. Rev. Lett. 1989, 62, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Georges, A.; Kotliar, G. Hubbard-model in infinite dimensions. Phys. Rev. B 1992, 45, 6479–6483. [Google Scholar] [CrossRef] [PubMed]
- Georges, A.; Kotliar, G.; Krauth, W.; Rozenberg, M.J. Dynamical mean-field theory of strongly correlated fermion systems and the limit of infinite dimensions. Rev. Mod. Phys. 1996, 68, 13–125. [Google Scholar] [CrossRef] [Green Version]
- Pavarini, E.; Koch, E.; Vollhardt, D.; Lichtenstein, A. The LDA+ DMFT approach to strongly correlated materials. Reihe Modeling Simul. 2011, 1, 2–13. [Google Scholar]
- Embedded DMFT Functional Tutorials. Available online: http://hauleweb.rutgers.edu/tutorials/ (accessed on 3 February 2021).
- Yang, W.T.; Zhang, Y.K.; Ayers, P.W. Degenerate ground states and a fractional number of electrons in density and reduced density matrix functional theory. Phys. Rev. Lett. 2000, 84, 5172–5175. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Dewhurst, J.K.; Lathiotakis, N.N.; Gross, E.K.U. Reduced density matrix functional for many-electron systems. Phys. Rev. B 2008, 78, 201103. [Google Scholar] [CrossRef] [Green Version]
- Roos, B.O.; Taylor, P.R.; Siegbahn, P.E.M. A complete active space scf method (casscf) using a density-matrix formulated super-ci approach. Chem. Phys. 1980, 48, 157–173. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Almlof, J.; Heiberg, A.; Roos, B.O. The complete active space scf (casscf) method in a newton-raphson formulation with application to the HNO molecule. J. Chem. Phys. 1981, 74, 2384–2396. [Google Scholar] [CrossRef]
- Knowles, P.J.; Sexton, G.J.; Handy, N.C. Studies using the casscf wavefunction. Chem. Phys. 1982, 72, 337–347. [Google Scholar] [CrossRef]
- Snyder, J.W., Jr.; Parrish, R.M.; Martinez, T.J. alpha-CASSCF: An Efficient, Empirical Correction for SA-CASSCF To Closely Approximate MS-CASPT2 Potential Energy Surfaces. J. Phys. Chem. Lett. 2017, 8, 2432–2437. [Google Scholar] [CrossRef] [PubMed]
- Roos, B.O.; Linse, P.; Siegbahn, P.E.M.; Blomberg, M.R.A. A simple method for the evaluation of the 2nd-order perturbation energy from external double-excitations with a casscf reference wavefunction. Chem. Phys. 1982, 66, 1–2, 197–207. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.A.; Roos, B.O.; Sadlej, A.J.; Wolinski, K. Second-order perturbation theory with a CASSCF reference function. J. Phys. Chem. 1990, 94, 5483–5488. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.A.; Roos, B.O. 2nd-order perturbation-theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar] [CrossRef]
- Werner, H.J. Third-order multireference perturbation theory—The CASPT3 method. Mol. Phys. 1996, 89, 645–661. [Google Scholar] [CrossRef]
- Miralles, J.; Daudey, J.P.; Caballol, R. Variational calculation of small energy differences—The singlet-triplet gap in Cu2Cl6 2. Chem. Phys. Lett. 1992, 198, 555–562. [Google Scholar] [CrossRef]
- Miralles, J.; Castell, O.; Caballol, R.; Malrieu, J.P. Specific ci calculation of energy differences—transition energies and bond-energies. Chem. Phys. 1993, 172, 33–43. [Google Scholar] [CrossRef]
- Malrieu, J.P.; Daudey, J.P.; Caballol, R. Multireference self-consistent size-consistent singles and doubles configuration-interaction for ground and excited-states. J. Chem. Phys. 1994, 101, 8908–8921. [Google Scholar] [CrossRef]
- Malrieu, J.P.; Caballol, R.; Calzado, C.J.; de Graaf, C.; Guihery, N. Magnetic Interactions in Molecules and Highly Correlated Materials: Physical Content, Analytical Derivation, and Rigorous Extraction of Magnetic Hamiltonians. Chem. Rev. 2014, 114, 429–492. [Google Scholar] [CrossRef] [PubMed]
- Heitler, W.; London, F. Wechselwirkung neutraler Atome und homöopolare Bindung nach der Quantenmechanik. Z. Für Phys. 1927, 44, 6–7, 455–472. [Google Scholar] [CrossRef]
- Levine, D.S.; Head-Gordon, M. Clarifying the quantum mechanical origin of the covalent chemical bond. Nat. Commun. 2020, 11, 4893. [Google Scholar] [CrossRef]
- White, R.M.; White, R.M.; Bayne, B. Quantum Theory of Magnetism; Springer: Berlin/Heidelberg, Germany, 1983. [Google Scholar]
- Hay, P.J.; Thibeault, J.C.; Hoffmann, R. Orbital interactions in metal dimer complexes. J. Am. Chem. Soc. 1975, 97, 4884–4899. [Google Scholar] [CrossRef]
- Novak, P.; Chaplygin, I.; Seifert, G.; Gemming, S.; Laskowski, R. Ab-initio calculation of exchange interactions in YMnO(3). Comput. Mater. Sci. 2008, 44, 79–81. [Google Scholar] [CrossRef]
- Zener, C. Interaction between the d-shells in the transition metals. 2. ferromagnetic compounds of manganese with perovskite structure. Phys. Rev. 1951, 82, 403–405. [Google Scholar] [CrossRef]
- Anderson, P.W.; Hasegawa, H. Considerations on double exchange. Phys. Rev. 1955, 100, 675–681. [Google Scholar] [CrossRef]
- Anderson, P.W. Antiferromagnetism—Theory of superexchange interaction. Phys. Rev. 1950, 79, 350–356. [Google Scholar] [CrossRef]
- Kramers, H. L’interaction entre les atomes magnétogènes dans un cristal paramagnétique. Physica 1934, 1, 182–192. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Loeb, A.L. Theory of ionic ordering, crystal distortion, and magnetic exchange due to covalent forces in spinels. Phys. Rev. 1955, 98, 391–408. [Google Scholar] [CrossRef]
- Goodenough, J.B. Theory of the role of covalence in the perovskite-type manganites la,m(ii) mno3. Phys. Rev. 1955, 100, 564–573. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, J. Superexchange interaction and symmetry properties of electron orbitals. J. Phys. Chem. Solids 1959, 10, 87–98. [Google Scholar] [CrossRef]
- Goodenough, J.B. Magnetism and the Chemical Bond; Interscience Publishers: New York, NY, USA, 1963; Volume 1. [Google Scholar]
- Gong, C.; Li, L.; Li, Z.; Ji, H.; Stern, A.; Xia, Y.; Cao, T.; Bao, W.; Wang, C.; Wang, Y.; et al. Discovery of intrinsic ferromagnetism in two-dimensional van der Waals crystals. Nature 2017, 546, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ni, J.Y.; Li, X.Y.; Koo, H.J.; Whangbo, M.H.; Feng, J.S.; Xiang, H.J. Intralayer ferromagnetism between S = 5/2 ions in MnBi2Te4: Role of empty Bi p states. Phys. Rev. B 2020, 101, 201408. [Google Scholar] [CrossRef]
- Oles, A.M.; Horsch, P.; Feiner, L.F.; Khaliullin, G. Spin-orbital entanglement and violation of the Goodenough-Kanamori rules. Phys. Rev. Lett. 2006, 96, 147205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whangbo, M.H.; Koo, H.J.; Dai, D. Spin exchange interactions and magnetic structures of extended magnetic solids with localized spins: Theoretical descriptions on formal, quantitative and qualitative levels. J. Solid State Chem. 2003, 176, 417–481. [Google Scholar] [CrossRef]
- Ruderman, M.A.; Kittel, C. Indirect exchange coupling of nuclear magnetic moments by conduction electrons. Phys. Rev. 1954, 96, 99–102. [Google Scholar] [CrossRef]
- Zener, C. Interaction between the d-shells in the transition metals. Phys. Rev. 1951, 81, 440–444. [Google Scholar] [CrossRef]
- Kasuya, T. A Theory of metallic ferromagnetism and antiferromagnetism on zeners model. Prog. Theor. Phys. 1956, 16, 45–57. [Google Scholar] [CrossRef]
- Yosida, K. Magnetic properties of cu-mn alloys. Phys. Rev. 1957, 106, 893–898. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.-Q.; Zhang, P.; Zhao, L.; Guo, M.; Tang, J. Single-Molecule Magnet Behavior Enhanced by Synergic Effect of Single-Ion Anisotropy and Magnetic Interactions. Inorg. Chem. 2017, 56, 7882–7889. [Google Scholar] [CrossRef]
- Degennes, P.G. Effects of double exchange in magnetic crystals. Phys. Rev. 1960, 118, 141–154. [Google Scholar] [CrossRef]
- Gordon, E.E.; Derakhshan, S.; Thompson, C.M.; Whangho, M.-H. Spin-Density Wave as a Superposition of Two Magnetic States of Opposite Chirality and Its Implications. Inorg. Chem. 2018, 57, 9782–9785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, H.-J.; PN, R.S.; Orlandi, F.; Sundaresan, A.; Whangbo, M.-H. On Ferro- and Antiferro-Spin-Density Waves Describing the Incommensurate Magnetic Structure of NaYNiWO6. Inorg. Chem. 2020, 59, 17856–17859. [Google Scholar] [CrossRef]
- Xiang, H.J.; Whangbo, M.H. Density-functional characterization of the multiferroicity in spin spiral chain cuprates. Phys. Rev. Lett. 2007, 99, 257203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzyaloshinsky, I. A thermodynamic theory of “weak” ferromagnetism of antiferromagnetics. J. Phys. Chem. Solids 1958, 4, 241–255. [Google Scholar] [CrossRef]
- Moriya, T. Anisotropic superexchange interaction and weak ferromagnetism. Phys. Rev. 1960, 120, 91–98. [Google Scholar] [CrossRef]
- Moriya, T. New mechanism of anisotropic superexchange interaction. Phys. Rev. Lett. 1960, 4, 228–230. [Google Scholar] [CrossRef]
- Xu, C.; Xu, B.; Dupe, B.; Bellaiche, L. Magnetic interactions in BiFeO3: A first-principles study. Phys. Rev. B 2019, 99, 104420. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Feng, J.; Prokhorenko, S.; Nahas, Y.; Xiang, H.; Bellaiche, L. Topological spin texture in Janus monolayers of the chromium trihalides Cr(I, X)(3). Phys. Rev. B 2020, 101, 060404. [Google Scholar] [CrossRef] [Green Version]
- Spirin, D.V. Magnetization of two-layer ferromagnets with easy-plane and/or easy-axis single-site anisotropy. J. Magn. Magn. Mater. 2003, 264, 121–129. [Google Scholar] [CrossRef]
- Onufrieva, F.P. Magnetic property anomalies of ferromagnets with mono-ionic anisotropy. Fiz. Tverd. Tela 1984, 26, 3435–3437. [Google Scholar]
- Zadrozny, J.M.; Xiao, D.J.; Atanasov, M.; Long, G.J.; Grandjean, F.; Neese, F.; Long, J.R. Magnetic blocking in a linear iron(I) complex. Nat. Chem. 2013, 5, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, Y.-C.; Liu, J.-L.; Vieru, V.; Ungur, L.; Jia, J.-H.; Chibotaru, L.F.; Lan, Y.; Wernsdorfer, W.; Gao, S.; et al. A Stable Pentagonal Bipyramidal Dy(III) Single-Ion Magnet with a Record Magnetization Reversal Barrier over 1000 K. J. Am. Chem. Soc. 2016, 138, 5441–5450. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.-F.; Wang, Z.; Ouyang, Z.-W.; Yin, B.; Zheng, Z.; Zheng, Y.-Z. Large Easy-Plane Magnetic Anisotropy in a Three-Coordinate Cobalt(II) Complex Li(THF)(4) Co(NPh2)(3). Chem. A Eur. J. 2016, 22, 14821–14825. [Google Scholar] [CrossRef]
- Antal, A.; Lazarovits, B.; Udvardi, L.; Szunyogh, L.; Ujfalussy, B.; Weinberger, P. First-principles calculations of spin interactions and the magnetic ground states of Cr trimers on Au(111). Phys. Rev. B 2008, 77, 174429. [Google Scholar] [CrossRef] [Green Version]
- Bak, P.; Jensen, M.H. Theory of helical magnetic-structures and phase-transitions in mnsi and fege. J. Phys. C Solid State Phys. 1980, 13, L881–L885. [Google Scholar] [CrossRef]
- Yu, X.Z.; Kanazawa, N.; Onose, Y.; Kimoto, K.; Zhang, W.Z.; Ishiwata, S.; Matsui, Y.; Tokura, Y. Near room-temperature formation of a skyrmion crystal in thin-films of the helimagnet FeGe. Nat. Mater. 2011, 10, 106–109. [Google Scholar] [CrossRef]
- Huang, S.X.; Chien, C.L. Extended Skyrmion Phase in Epitaxial FeGe(111) Thin Films. Phys. Rev. Lett. 2012, 108, 267201. [Google Scholar] [CrossRef] [Green Version]
- Weber, T.; Waizner, J.; Tucker, G.S.; Georgii, R.; Kugler, M.; Bauer, A.; Pfleiderer, C.; Garst, M.; Boeni, P. Field dependence of nonreciprocal magnons in chiral MnSi. Phys. Rev. B 2018, 97, 224403. [Google Scholar] [CrossRef] [Green Version]
- Lado, J.L.; Fernandez-Rossier, J. On the origin of magnetic anisotropy in two dimensional CrI3. 2d Materials 2017, 4, 035002. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Feng, J.; Xiang, H.; Bellaiche, L. Interplay between Kitaev interaction and single ion anisotropy in ferromagnetic CrI3 and CrGeTe3 monolayers. Npj Comput. Mater. 2018, 4, 57. [Google Scholar] [CrossRef]
- Kitaev, A. Anyons in an exactly solved model and beyond. Ann. Phys. 2006, 321, 2–111. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Bridges, C.A.; Yan, J.Q.; Aczel, A.A.; Li, L.; Stone, M.B.; Granroth, G.E.; Lumsden, M.D.; Yiu, Y.; Knolle, J.; et al. Proximate Kitaev quantum spin liquid behaviour in a honeycomb magnet. Nat. Mater. 2016, 15, 733–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, S.H.; Do, S.H.; Choi, K.Y.; Kwon, Y.S.; Wolter, A.U.B.; Nishimoto, S.; van den Brink, J.; Buechner, B. Evidence for a Field-Induced Quantum Spin Liquid in alpha-RuCl3. Phys. Rev. Lett. 2017, 119, 037201. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Ran, K.; Li, T.; Wang, J.; Wang, P.; Liu, B.; Liu, Z.-X.; Normand, B.; Wen, J.; Yu, W. Gapless Spin Excitations in the Field-Induced Quantum Spin Liquid Phase of alpha-RuCl3. Phys. Rev. Lett. 2017, 119, 227208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, G.; Qi, T.F.; Li, L.; Terzic, J.; Cao, V.S.; Yuan, S.J.; Tovar, M.; Murthy, G.; Kaul, R.K. Evolution of magnetism in the single-crystal honeycomb iridates (Na1-xLix)(2)IrO3. Phys. Rev. B 2013, 88, 220414. [Google Scholar] [CrossRef] [Green Version]
- Manni, S.; Choi, S.; Mazin, I.I.; Coldea, R.; Altmeyer, M.; Jeschke, H.O.; Valenti, R.; Gegenwart, P. Effect of isoelectronic doping on the honeycomb-lattice iridate A(2)IrO(3). Phys. Rev. B 2014, 89, 245113. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Feng, J.; Kawamura, M.; Yamaji, Y.; Nahas, Y.; Prokhorenko, S.; Qi, Y.; Xiang, H.; Bellaiche, L. Possible Kitaev Quantum Spin Liquid State in 2D Materials with S = 3/2. Phys. Rev. Lett. 2020, 124, 087205. [Google Scholar] [CrossRef] [Green Version]
- Harris, E.A.; Owen, J. Biquadratic exchange between Mn2+ ions in MgO. Phys. Rev. Lett. 1963, 11, 9. [Google Scholar] [CrossRef]
- Rodbell, D.S.; Harris, E.A.; Owen, J.; Jacobs, I.S. Biquadratic exchange and behavior of some antiferromagnetic substances. Phys. Rev. Lett. 1963, 11, 10. [Google Scholar] [CrossRef]
- Fedorova, N.S.; Ederer, C.; Spaldin, N.A.; Scaramucci, A. Biquadratic and ring exchange interactions in orthorhombic perovskite manganites. Phys. Rev. B 2015, 91, 165122. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.-F.; Cao, H.-Y.; Xie, Y.; Hou, Y.-S.; Chen, S.; Xiang, H.; Gong, X.-G. Giant biquadratic interaction-induced magnetic anisotropy in the iron-based superconductor A(x)Fe(2-y)Se(2). Phys. Rev. B 2016, 93, 024511. [Google Scholar] [CrossRef] [Green Version]
- Kartsev, A.; Augustin, M.; Evans, R.F.L.; Novoselov, K.S.; Santos, E.J.G. Biquadratic exchange interactions in two-dimensional magnets. Npj Comput. Mater. 2020, 6, 150. [Google Scholar] [CrossRef]
- Li, X.-Y.; Lou, F.; Gong, X.-G.; Xiang, H. Constructing realistic effective spin Hamiltonians with machine learning approaches. New J. Phys. 2020, 22, 053036. [Google Scholar] [CrossRef]
- Ivanov, N.B.; Ummethum, J.; Schnack, J. Phase diagram of the alternating-spin Heisenberg chain with extra isotropic three-body exchange interactions. Eur. Phys. J. B 2014, 87, 226. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Haldar, S.; von Malottki, S.; Heinze, S. Role of higher-order exchange interactions for skyrmion stability. Nat. Commun. 2020, 11, 4756. [Google Scholar] [CrossRef]
- Grytsiuk, S.; Hanke, J.P.; Hoffmann, M.; Bouaziz, J.; Gomonay, O.; Bihlmayer, G.; Lounis, S.; Mokrousov, Y.; Bluegel, S. Topological-chiral magnetic interactions driven by emergent orbital magnetism. Nat. Commun. 2020, 11, 511. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Weischenberg, J.; Dupe, B.; Freimuth, F.; Ferriani, P.; Mokrousov, Y.; Heinze, S. Topological orbital magnetization and emergent Hall effect of an atomic-scale spin lattice at a surface. Phys. Rev. B 2015, 92, 020401. [Google Scholar] [CrossRef] [Green Version]
- Hanke, J.P.; Freimuth, F.; Nandy, A.K.; Zhang, H.; Bluegel, S.; Mokrousov, Y. Role of Berry phase theory for describing orbital magnetism: From magnetic heterostructures to topological orbital ferromagnets. Phys. Rev. B 2016, 94, 121114. [Google Scholar] [CrossRef] [Green Version]
- Dias, M.D.S.; Bouaziz, J.; Bouhassoune, M.; Bluegel, S.; Lounis, S. Chirality-driven orbital magnetic moments as a new probe for topological magnetic structures. Nat. Commun. 2016, 7, 13613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanke, J.-P.; Freimuth, F.; Bluegel, S.; Mokrousov, Y. Prototypical topological orbital ferromagnet gamma-FeMn. Sci. Rep. 2017, 7, 41078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, M.D.S.; Lounis, S. Insights into the orbital magnetism of noncollinear magnetic systems. Proc. SPIE 2017, 10357, 103572A. [Google Scholar]
- Lux, F.R.; Freimuth, F.; Bluegel, S.; Mokrousov, Y. Engineering chiral and topological orbital magnetism of domain walls and skyrmions. Commun. Phys. 2018, 1, 60. [Google Scholar] [CrossRef]
- Tanigaki, T.; Shibata, K.; Kanazawa, N.; Yu, X.; Onose, Y.; Park, H.S.; Shindo, D.; Tokura, Y. Real-Space Observation of Short-Period Cubic Lattice of Skyrmions in MnGe. Nano Lett. 2015, 15, 5438–5442. [Google Scholar] [CrossRef] [Green Version]
- Brinker, S.; Dias, M.D.S.; Lounis, S. The chiral biquadratic pair interaction. New J. Phys. 2019, 21, 083015. [Google Scholar] [CrossRef]
- Laszloffy, A.; Rozsa, L.; Palotas, K.; Udvardi, L.; Szunyogh, L. Magnetic structure of monatomic Fe chains on Re(0001): Emergence of chiral multispin interactions. Phys. Rev. B 2019, 99, 184430. [Google Scholar] [CrossRef] [Green Version]
- Parihari, D.; Pati, S.K. Effect of chiral interactions in frustrated magnetic chains. Phys. Rev. B 2004, 70, 180403. [Google Scholar] [CrossRef]
- Kostyrko, T.; Bulka, B.R. Canonical perturbation theory for inhomogeneous systems of interacting fermions. Phys. Rev. B 2011, 84, 035123. [Google Scholar] [CrossRef]
- Mankovsky, S.; Polesya, S.; Ebert, H. Extension of the standard Heisenberg Hamiltonian to multispin exchange interactions. Phys. Rev. B 2020, 101, 174401. [Google Scholar] [CrossRef]
- Wen, X.G.; Wilczek, F.; Zee, A. Chiral spin states and superconductivity. Phys. Rev. B 1989, 39, 11413–11423. [Google Scholar] [CrossRef] [Green Version]
- Bauer, B.; Cincio, L.; Keller, B.P.; Dolfi, M.; Vidal, G.; Trebst, S.; Ludwig, A.W.W. Chiral spin liquid and emergent anyons in a Kagome lattice Mott insulator. Nat. Commun. 2014, 5, 5137. [Google Scholar] [CrossRef] [Green Version]
- Drautz, R.; Fahnle, M. Spin-cluster expansion: Parametrization of the general adiabatic magnetic energy surface with ab initio accuracy. Phys. Rev. B 2004, 69, 104404. [Google Scholar] [CrossRef]
- Singer, R.; Faehnle, M. Construction of basis functions for the spin-cluster expansion of the magnetic energy on the atomic scale in rotationally invariant systems. J. Math. Phys. 2006, 47, 113503. [Google Scholar] [CrossRef]
- Singer, R.; Dietermann, F.; Faehnle, M. Spin Interactions in bcc and fcc Fe beyond the Heisenberg Model. Phys. Rev. Lett. 2011, 107, 017204. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, P.; Habas, M.P.; Dovesi, R.; Moreira, I.D.; Illas, F. Magnetic coupling in the weak ferromagnet CuF2. Phys. Rev. B 1999, 59, 1016–1023. [Google Scholar] [CrossRef] [Green Version]
- Illas, F.; Moreira, I.D.R.; de Graaf, C.; Barone, V. Magnetic coupling in biradicals, binuclear complexes and wide-gap insulators: A survey of ab initio wave function and density functional theory approaches. Theor. Chem. Acc. 2000, 104, 265–272. [Google Scholar] [CrossRef]
- Xiang, H.; Lee, C.; Koo, H.-J.; Gong, X.; Whangbo, M.-H. Magnetic properties and energy-mapping analysis. Dalton Trans. 2013, 42, 823–853. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.J.; Kan, E.J.; Wei, S.-H.; Whangbo, M.H.; Gong, X.G. Predicting the spin-lattice order of frustrated systems from first principles. Phys. Rev. B 2011, 84, 224429. [Google Scholar] [CrossRef] [Green Version]
- Weingart, C.; Spaldin, N.; Bousquet, E. Noncollinear magnetism and single-ion anisotropy in multiferroic perovskites. Phys. Rev. B 2012, 86, 094413. [Google Scholar] [CrossRef] [Green Version]
- Liechtenstein, A.I.; Katsnelson, M.I.; Antropov, V.P.; Gubanov, V.A. Local spin-density functional-approach to the theory of exchange interactions in ferromagnetic metals and alloys. J. Magn. Magn. Mater. 1987, 67, 65–74. [Google Scholar] [CrossRef]
- Katsnelson, M.I.; Lichtenstein, A.I. First-principles calculations of magnetic interactions in correlated systems. Phys. Rev. B 2000, 61, 8906–8912. [Google Scholar] [CrossRef] [Green Version]
- Katsnelson, M.I.; Kvashnin, Y.O.; Mazurenko, V.V.; Lichtenstein, A.I. Correlated band theory of spin and orbital contributions to Dzyaloshinskii-Moriya interactions. Phys. Rev. B 2010, 82, 100403. [Google Scholar] [CrossRef] [Green Version]
- Lounis, S.; Dederichs, P.H. Mapping the magnetic exchange interactions from first principles: Anisotropy anomaly and application to Fe, Ni, and Co. Phys. Rev. B 2010, 82, 180404. [Google Scholar] [CrossRef] [Green Version]
- Szilva, A.; Costa, M.; Bergman, A.; Szunyogh, L.; Nordstrom, L.; Eriksson, O. Interatomic Exchange Interactions for Finite-Temperature Magnetism and Nonequilibrium Spin Dynamics. Phys. Rev. Lett. 2013, 111, 127204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korotin, D.M.; Mazurenko, V.V.; Anisimov, V.I.; Streltsov, S.V. Calculation of exchange constants of the Heisenberg model in plane-wave-based methods using the Green’s function approach. Phys. Rev. B 2015, 91, 224405. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Helbig, N.; Verstraete, M.J.; Bousquet, E. TB2J: A Python Package for Computing Magnetic Interaction Parameters. Available online: https://arxiv.org/abs/2009.01910 (accessed on 3 February 2021).
- Wan, X.; Yin, Q.; Savrasov, S.Y. Calculation of magnetic exchange interactions in Mott-Hubbard systems. Phys. Rev. Lett. 2006, 97, 266403. [Google Scholar] [CrossRef] [Green Version]
- Boukhvalov, D.W.; Dobrovitski, V.V.; Katsnelson, M.I.; Lichtenstein, A.I.; Harmon, B.N.; Kogerler, P. Electronic structure and exchange interactions in V-15 magnetic molecules: LDA+U results. Phys. Rev. B 2004, 70, 054417. [Google Scholar] [CrossRef]
- Mostofi, A.A.; Yates, J.R.; Lee, Y.-S.; Souza, I.; Vanderbilt, D.; Marzari, N. wannier90: A tool for obtaining maximally-localised Wannier functions. Comput. Phys. Commun. 2008, 178, 685–699. [Google Scholar] [CrossRef] [Green Version]
- Pizzi, G.; Vitale, V.; Arita, R.; Bluegel, S.; Freimuth, F.; Geranton, G.; Gibertini, M.; Gresch, D.; Johnson, C.; Koretsune, T.; et al. Wannier90 as a community code: New features and applications. J. Phys. Condens. Matter 2020, 32, 165902. [Google Scholar] [CrossRef]
- Wang, X.D.; Wang, D.S.; Wu, R.Q.; Freeman, A.J. Validity of the force theorem for magnetocrystalline anisotropy. J. Magn. Magn. Mater. 1996, 159, 337–341. [Google Scholar] [CrossRef]
- Flygare, W.H. Magnetic-interactions in molecules and an analysis of molecular electronic charge distribution from magnetic parameters. Chem. Rev. 1974, 74, 653–687. [Google Scholar] [CrossRef]
- Brown, J.M.; Sears, T.J. Reduced form of the spin-rotation hamiltonian for asymmetric-top molecules, with applications to ho2 and nh2. J. Mol. Spectrosc. 1979, 75, 111–133. [Google Scholar] [CrossRef]
- Lahti, P.M.; Ichimura, A.S. Semiempirical study of electron exchange interaction in organic high-spin pi-systems—classifying structural effects in organic magnetic molecules. J. Org. Chem. 1991, 56, 3030–3042. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Lenz, B.; Kanungo, S.; Panda, S.K.; Biermann, S.; Schnelle, W.; Manna, K.; Kataria, R.; Uhlarz, M.; Skourski, Y.; et al. Pronounced 2/3 magnetization plateau in a frustrated S = 1 isolated spin-triangle compound: Interplay between Heisenberg and biquadratic exchange interactions. Phys. Rev. B 2019, 100, 094427. [Google Scholar] [CrossRef] [Green Version]
- Takano, K.; Sano, K. Determination of exchange parameters from magnetic susceptibility. J. Phys. Soc. Jpn. 1997, 66, 1846–1847. [Google Scholar] [CrossRef] [Green Version]
- Turek, I.; Kudrnovsky, J.; Drchal, V.; Bruno, P. Exchange interactions, spin waves, and transition temperatures in itinerant magnets. Philos. Mag. 2006, 86, 1713–1752. [Google Scholar] [CrossRef]
- Lancon, D.; Ewings, R.A.; Guidi, T.; Formisano, F.; Wildes, A.R. Magnetic exchange parameters and anisotropy of the quasi-two-dimensional antiferromagnet NiPS3. Phys. Rev. B 2018, 98, 134414. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chung, J.-H.; Gao, B.; Chen, T.; Stone, M.B.; Kolesnikov, A.I.; Huang, Q.; Dai, P. Topological Spin Excitations in Honeycomb Ferromagnet CrI3. Phys. Rev. X 2018, 8, 041028. [Google Scholar] [CrossRef] [Green Version]
- Oba, M.; Nakamura, K.; Akiyama, T.; Ito, T.; Weinert, M.; Freeman, A.J. Electric-Field-Induced Modification of the Magnon Energy, Exchange Interaction, and Curie Temperature of Transition-Metal Thin Films. Phys. Rev. Lett. 2015, 114, 107202. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Yoshioka, Y.; Yamaguchi, K. Effective exchange interactions and magnetic phase transition temperatures in Prussian blue analogs: A study by density functional theory. Chem. Phys. Lett. 1998, 297, 51–59. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Yu, H.; Lou, F.; Feng, J.; Whangbo, M.-H.; Xiang, H. Spin Hamiltonians in Magnets: Theories and Computations. Molecules 2021, 26, 803. https://doi.org/10.3390/molecules26040803
Li X, Yu H, Lou F, Feng J, Whangbo M-H, Xiang H. Spin Hamiltonians in Magnets: Theories and Computations. Molecules. 2021; 26(4):803. https://doi.org/10.3390/molecules26040803
Chicago/Turabian StyleLi, Xueyang, Hongyu Yu, Feng Lou, Junsheng Feng, Myung-Hwan Whangbo, and Hongjun Xiang. 2021. "Spin Hamiltonians in Magnets: Theories and Computations" Molecules 26, no. 4: 803. https://doi.org/10.3390/molecules26040803
APA StyleLi, X., Yu, H., Lou, F., Feng, J., Whangbo, M. -H., & Xiang, H. (2021). Spin Hamiltonians in Magnets: Theories and Computations. Molecules, 26(4), 803. https://doi.org/10.3390/molecules26040803