
Zincbindpredict—Prediction of Zinc Binding Sites in Proteins



Abstract
:1. Introduction
2. Results and Discussion
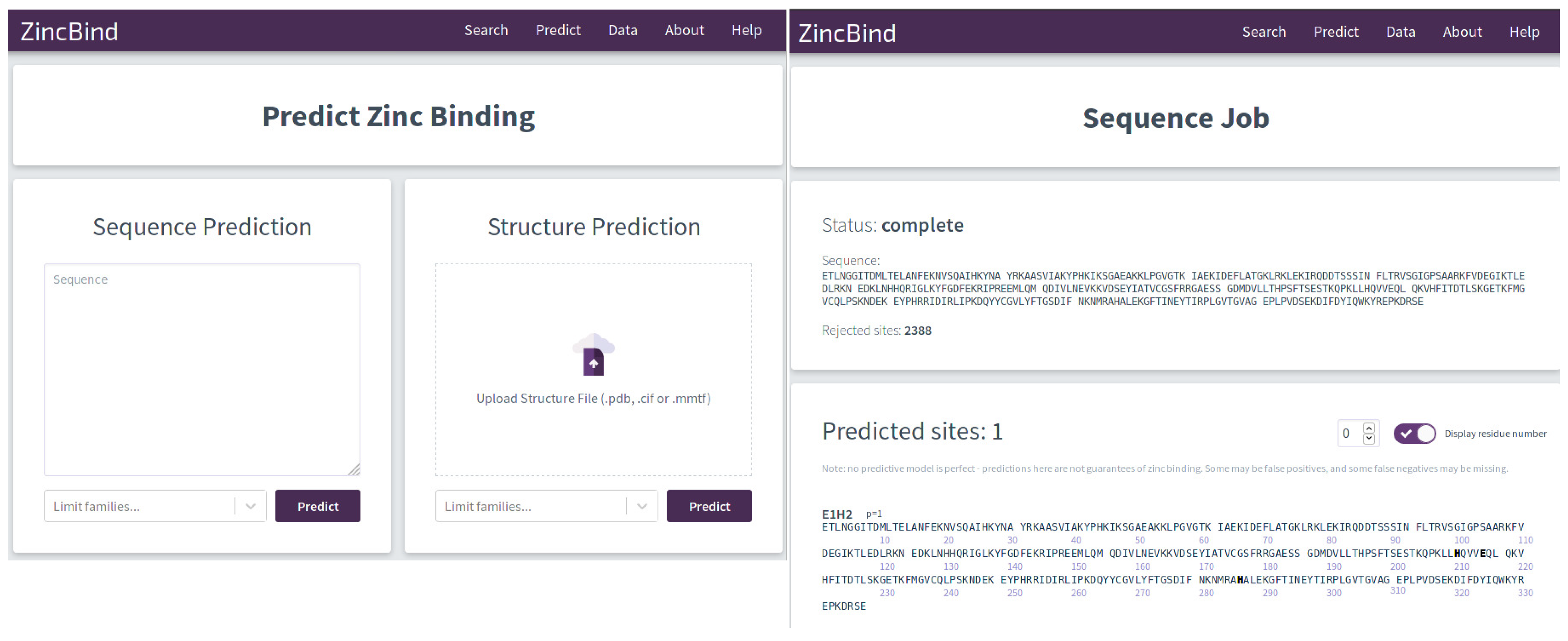
2.1. Deployment
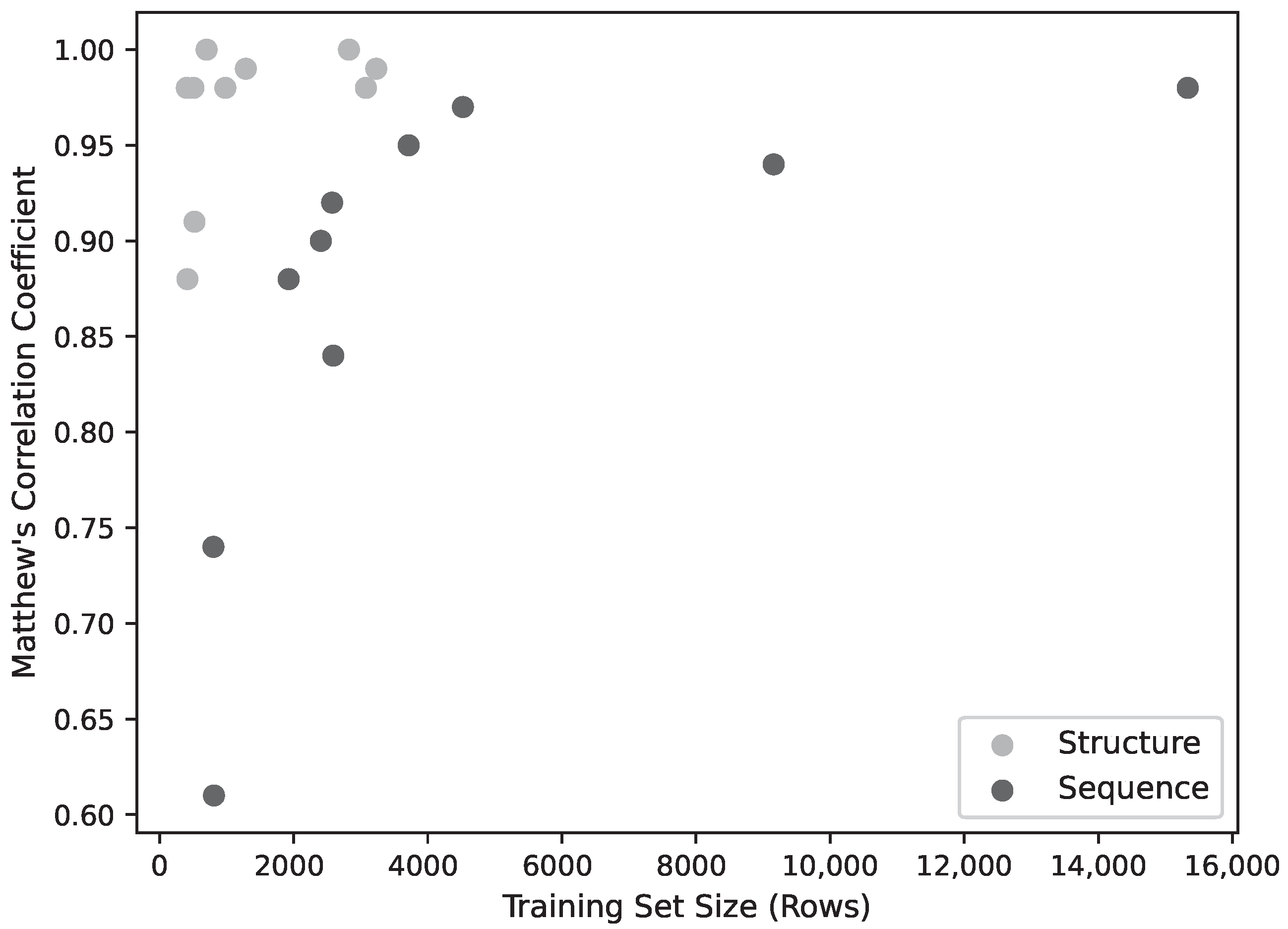
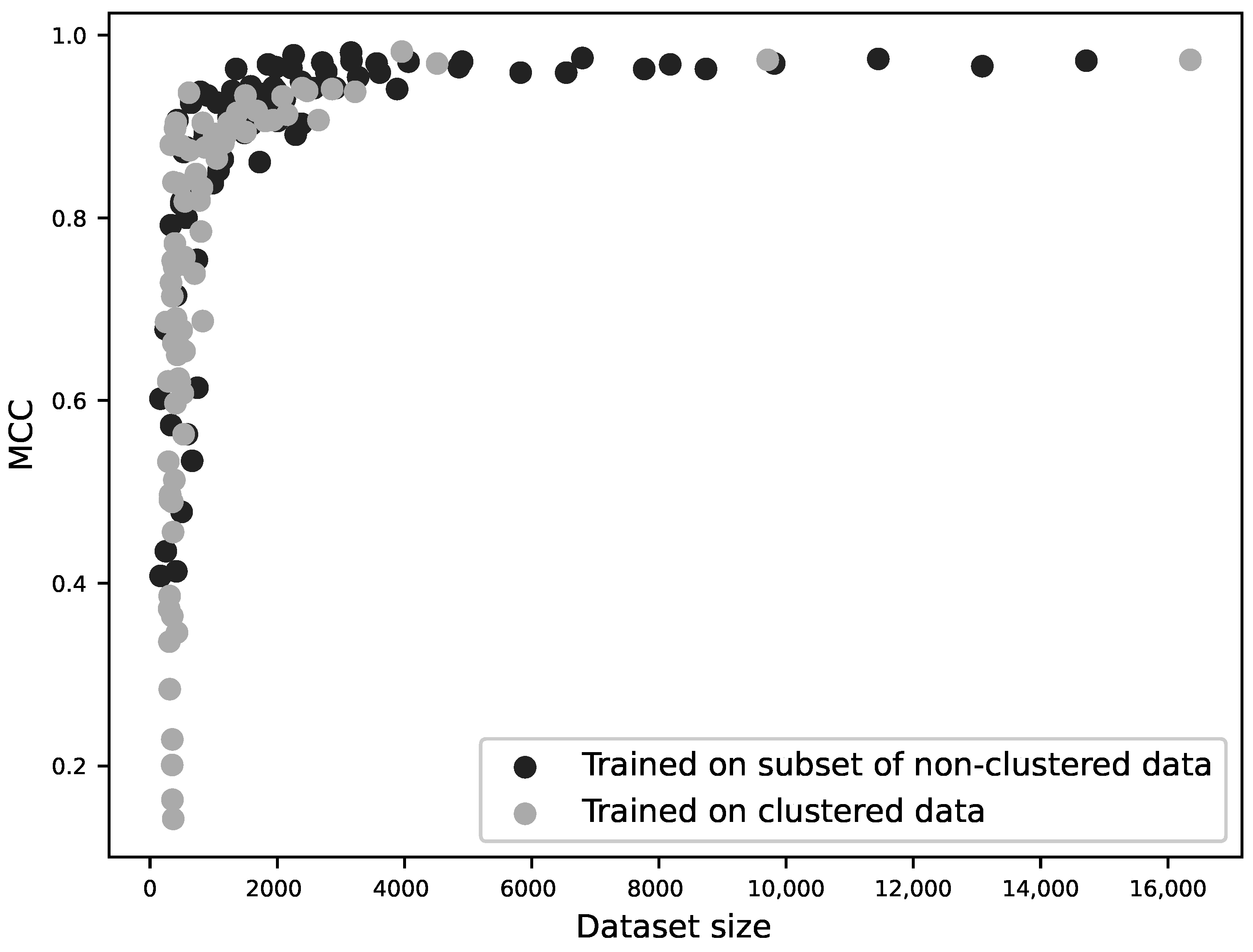
2.2. Training
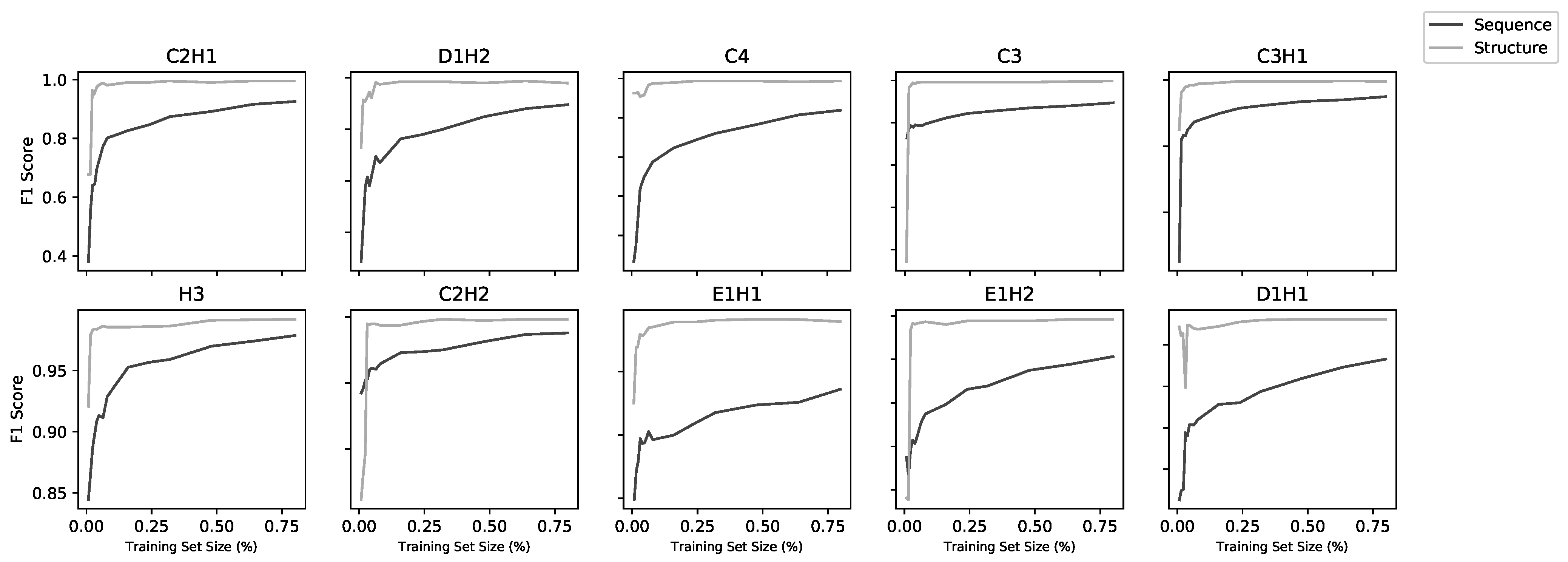
2.3. Models
3. Materials and Methods
3.1. Dataset Creation
3.2. Predictive Model Training
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| API | Application Programming Interface |
| SVM | Support Vector Machine |
| PDB | Protein Data Bank |
| MCC | Matthews Correlation Coefficient |
References
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Counting the zinc-proteins encoded in the human genome. J. Proteome Res. 2006, 5, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Vallee, B.L.; Auld, D.S. Zinc coordination, function, and structure of zinc enzymes and other proteins. Biochemistry 1990, 29, 5647–5659. [Google Scholar] [CrossRef]
- Miller, J.; McLachlan, A.; Klug, A. Repetitive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. EMBO J. 1985, 4, 1609–1614. [Google Scholar] [CrossRef]
- Barbosa, M.S.; Lowy, D.R.; Schiller, J.T. Papillomavirus polypeptides E6 and E7 are zinc-binding proteins. J. Virol. 1989, 63, 1404–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallee, B.L.; Auld, D.S. Short and long spacer sequences and other structural features of zinc binding sites in zinc enzymes. FEBS Lett. 1989, 257, 138–140. [Google Scholar] [CrossRef] [Green Version]
- Bishop, W.R.; Kirschmeier, P.; George, S.J.; Cramer, S.P.; Hendrickson, W.A. Identification and characterization of zinc binding sites in protein kinase C. Science 1991, 254, 1776–1779. [Google Scholar]
- Furukawa, Y.; Lim, C.; Tosha, T.; Yoshida, K.; Hagai, T.; Akiyama, S.; Watanabe, S.; Nakagome, K.; Shiro, Y. Identification of a novel zinc-binding protein, C1orf123, as an interactor with a heavy metal-associated domain. PLoS ONE 2018, 13, e0204355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigrist, C.J.A.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucleic Acids Res. 2012, 41, D344–D347. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Han, L.; Zhang, H.; Zheng, C.; Xie, B.; Cao, Z.W.; Chen, Y.Z. Prediction of the functional class of metal-binding proteins from sequence derived physicochemical properties by support vector machine approach. BMC Bioinform. 2006, 7, S13. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Kumar, M. Prediction of zinc binding sites in proteins using sequence derived information. J. Biomol. Struct. Dyn. 2018, 36, 4413–4423. [Google Scholar] [CrossRef]
- Li, H.; Pi, D.; Chen, C.; Li, H. A Novel Prediction Method for Zinc-Binding Sites in Proteins by an Ensemble of SVM and Sample-Weighted Probabilistic Neural Network. IEEE Access 2019, 7, 186147–186157. [Google Scholar] [CrossRef]
- Zheng, C.; Wang, M.; Takemoto, K.; Akutsu, T.; Zhang, Z.; Song, J. An Integrative Computational Framework Based on a Two-Step Random Forest Algorithm Improves Prediction of Zinc-Binding Sites in Proteins. PLoS ONE 2012, 7, e49716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S. Prediction of Metal Ion Binding Sites in Proteins from Amino Acid Sequences by Using Simplified Amino Acid Alphabets and Random Forest Model. Genom. Inform. 2017, 15, 162–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi, M.; Wu, D.; Wang, Z.; Shen, Y. DeepAffinity: Interpretable deep learning of compound–protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 2019, 35, 3329–3338. [Google Scholar] [CrossRef] [Green Version]
- Haberal, I.; Ogul, H. Prediction of Protein Metal Binding Sites Using Deep Neural Networks. Mol. Inform. 2019, 38, 1800169. [Google Scholar] [CrossRef]
- Yamashita, M.M.; Wesson, L.; Eisenman, G.; Eisenberg, D. Where metal ions bind in proteins. Proc. Natl. Acad. Sci. USA 1990, 87, 5648–5652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, D.S.; Martin, A.C.R.; Cheetham, J.C.; Rees, A.R. The prediction and characterization of metal binding sites in proteins. Protein Eng. Des. Sel. 1993, 6, 29–35. [Google Scholar] [CrossRef]
- Wallace, A.C.; Borkakoti, N.; Thornton, J.M. TESS: A geometric hashing algorithm for deriving 3D coordinate templates for searching structural databases. Application to enzyme active sites. Protein Sci. 1997, 6, 2308–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Xu, M.; Liang, Z.; Ding, B.; Niu, L.; Liu, H.; Teng, M. Structure-based de novo prediction of zinc-binding sites in proteins of unknown function. Bioinformatics 2011, 27, 1262–1268. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wang, Y.; Zhou, C.; Xue, Y.; Zhao, W.; Liu, H. Computationally characterizing and comprehensive analysis of zinc-binding sites in proteins. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 171–180. [Google Scholar] [CrossRef]
- Ireland, S.M.; Martin, A.C.R. ZincBind—The database of zinc binding sites. Database 2019, 2019, baz006. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Pi, D.; Liang, Y.; Chen, C.; Liu, Y. Integrative computing method for the prediction of zinc-binding sites in proteins. In Proceedings of the 2017 International Joint Conference on Neural Networks (IJCNN), Anchorage, AK, USA, 14–19 May 2017. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Y.; Zhai, Y.F.; Song, J.; Zhang, Z. ZincExplorer: An accurate hybrid method to improve the prediction of zinc-binding sites from protein sequences. Mol. Biosyst. 2013, 9, 2213. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Ireland, S.M.; Martin, A.C.R. Atomium—A Python structure parser. Bioinformatics 2020, 36, 2750–2754. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation Sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21, 6. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Madeira, F.; mi Park, Y.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Dataset Size | Recall | Precision | F1 | MCC |
|---|---|---|---|---|---|
| C2H2 | 702 | 1.00 | 1.00 | 1.00 | 1.00 |
| C4 | 2825 | 1.00 | 1.00 | 1.00 | 1.00 |
| C3H1 | 3232 | 1.00 | 0.99 | 1.00 | 0.99 |
| E1H2 | 1287 | 1.00 | 0.99 | 1.00 | 0.99 |
| C2H1 | 506 | 1.00 | 0.98 | 0.99 | 0.98 |
| H3 | 3078 | 1.00 | 0.98 | 0.99 | 0.98 |
| D1H2 | 982 | 1.00 | 0.98 | 0.99 | 0.98 |
| C3 | 407 | 1.00 | 0.98 | 0.99 | 0.98 |
| D1H1 | 522 | 1.00 | 0.91 | 0.95 | 0.91 |
| E1H1 | 416 | 0.93 | 0.95 | 0.94 | 0.88 |
| Mean | 0.99 | 0.98 | 0.99 | 0.97 |
| Family | Dataset Size | Recall | Precision | F1 | MCC |
|---|---|---|---|---|---|
| C4 | 15,332 | 1.00 | 0.98 | 0.99 | 0.98 |
| H3 | 4524 | 0.98 | 0.99 | 0.98 | 0.97 |
| C2H2 | 3715 | 0.97 | 0.99 | 0.98 | 0.95 |
| C3H1 | 9158 | 0.98 | 0.96 | 0.97 | 0.94 |
| E1H2 | 2574 | 0.95 | 0.97 | 0.96 | 0.92 |
| D1H2 | 2406 | 0.94 | 0.95 | 0.94 | 0.90 |
| C2H1 | 1926 | 0.93 | 0.95 | 0.94 | 0.88 |
| C3 | 2591 | 0.95 | 0.89 | 0.92 | 0.84 |
| D1H1 | 804 | 0.80 | 0.93 | 0.86 | 0.74 |
| E1H1 | 812 | 0.81 | 0.83 | 0.82 | 0.61 |
| Mean | 0.93 | 0.94 | 0.94 | 0.87 |
| Family | Dataset Size | Recall | Precision | F1 | MCC |
|---|---|---|---|---|---|
| C2H2 | 3960 | 0.99 | 0.95 | 0.97 | 0.94 |
| C3H1 | 9710 | 0.29 | 0.87 | 0.44 | 0.33 |
| C2H1 | 2154 | 0.24 | 0.88 | 0.37 | 0.30 |
| D1H1 | 818 | 0.05 | 0.80 | 0.09 | 0.11 |
| C3 | 2868 | 0.13 | 0.61 | 0.21 | 0.07 |
| E1H1 | 828 | 0.06 | 0.62 | 0.11 | 0.06 |
| D1H2 | 2470 | 0.03 | 0.53 | 0.06 | 0.01 |
| H3 | 5058 | 0.01 | 0.19 | 0.02 | −0.10 |
| E1H2 | 2648 | 0.02 | 0.33 | 0.04 | −0.06 |
| Mean | 0.18 | 0.58 | 0.23 | 0.17 |
| Species | Percentage of Genome |
|---|---|
| Predicted Zinc Binding | |
| Campylobacter jejuni | 6.4% |
| Clostridioides difficile | 5.8% |
| Enterococcus faecalis | 7.5% |
| Listeria monocytogenes | 7.9% |
| Mycobacterium tuberculosis | 11.3% |
| Salmonella enterica | 11.1% |
| Shigella flexneri | 10.1% |
| Streptococcus pneumoniae | 7.6% |
| Model Type | Feature |
|---|---|
| Sequence | |
| Inter-residue distance (one per gap) | |
| Average hydrophobicity around residues (window 1) | |
| Average hydrophobicity around residues (window 3) | |
| Average hydrophobicity around residues (window 5) | |
| Average number of charges around residues (window 1) | |
| Average number of charges around residues (window 3) | |
| Average number of charges around residues (window 5) | |
| Structure | |
| Mean Inter-C distance | |
| Maximum Inter-C distance | |
| Minimum Inter-C distance | |
| Inter-C distance standard deviation | |
| Mean Inter-C distance | |
| Maximum Inter-C distance | |
| Minimum Inter-C distance | |
| Inter-C distance standard deviation | |
| Hydrophobic contrast (radius 4 Å) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ireland, S.M.; Martin, A.C.R. Zincbindpredict—Prediction of Zinc Binding Sites in Proteins. Molecules 2021, 26, 966. https://doi.org/10.3390/molecules26040966
Ireland SM, Martin ACR. Zincbindpredict—Prediction of Zinc Binding Sites in Proteins. Molecules. 2021; 26(4):966. https://doi.org/10.3390/molecules26040966
Chicago/Turabian StyleIreland, Sam M., and Andrew C. R. Martin. 2021. "Zincbindpredict—Prediction of Zinc Binding Sites in Proteins" Molecules 26, no. 4: 966. https://doi.org/10.3390/molecules26040966
APA StyleIreland, S. M., & Martin, A. C. R. (2021). Zincbindpredict—Prediction of Zinc Binding Sites in Proteins. Molecules, 26(4), 966. https://doi.org/10.3390/molecules26040966