
Doxorubicin Encapsulation in Carbon Nanotubes Having Haeckelite or Stone–Wales Defects as Drug Carriers: A Molecular Dynamics Approach



Abstract
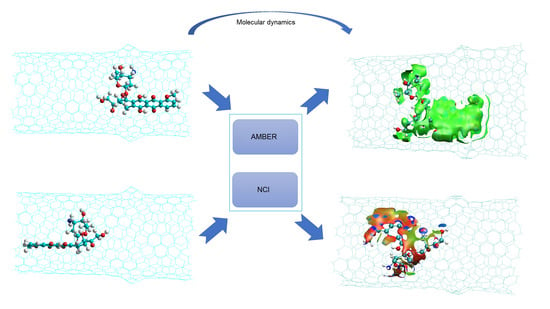
:
1. Introduction
2. Results
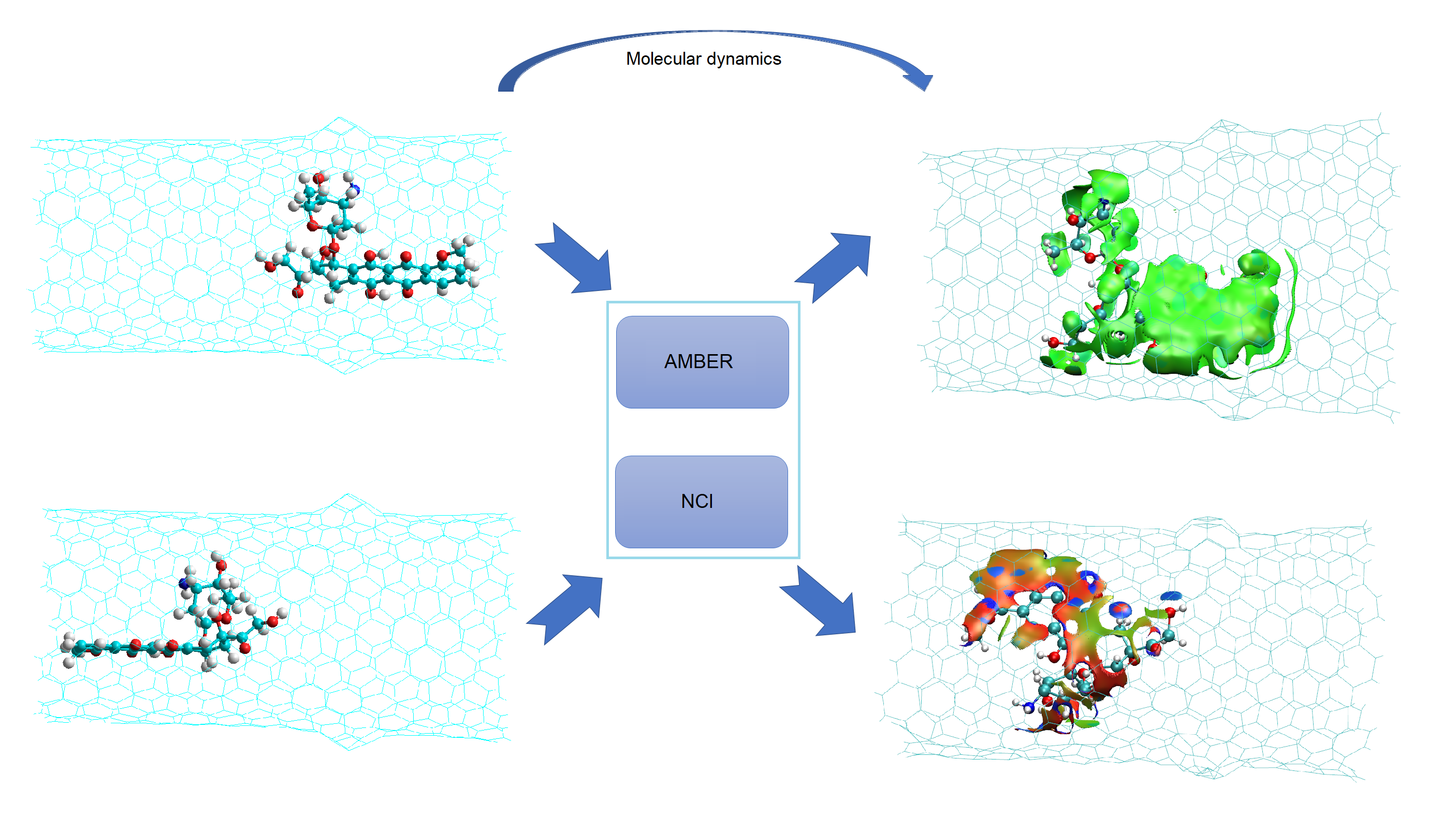
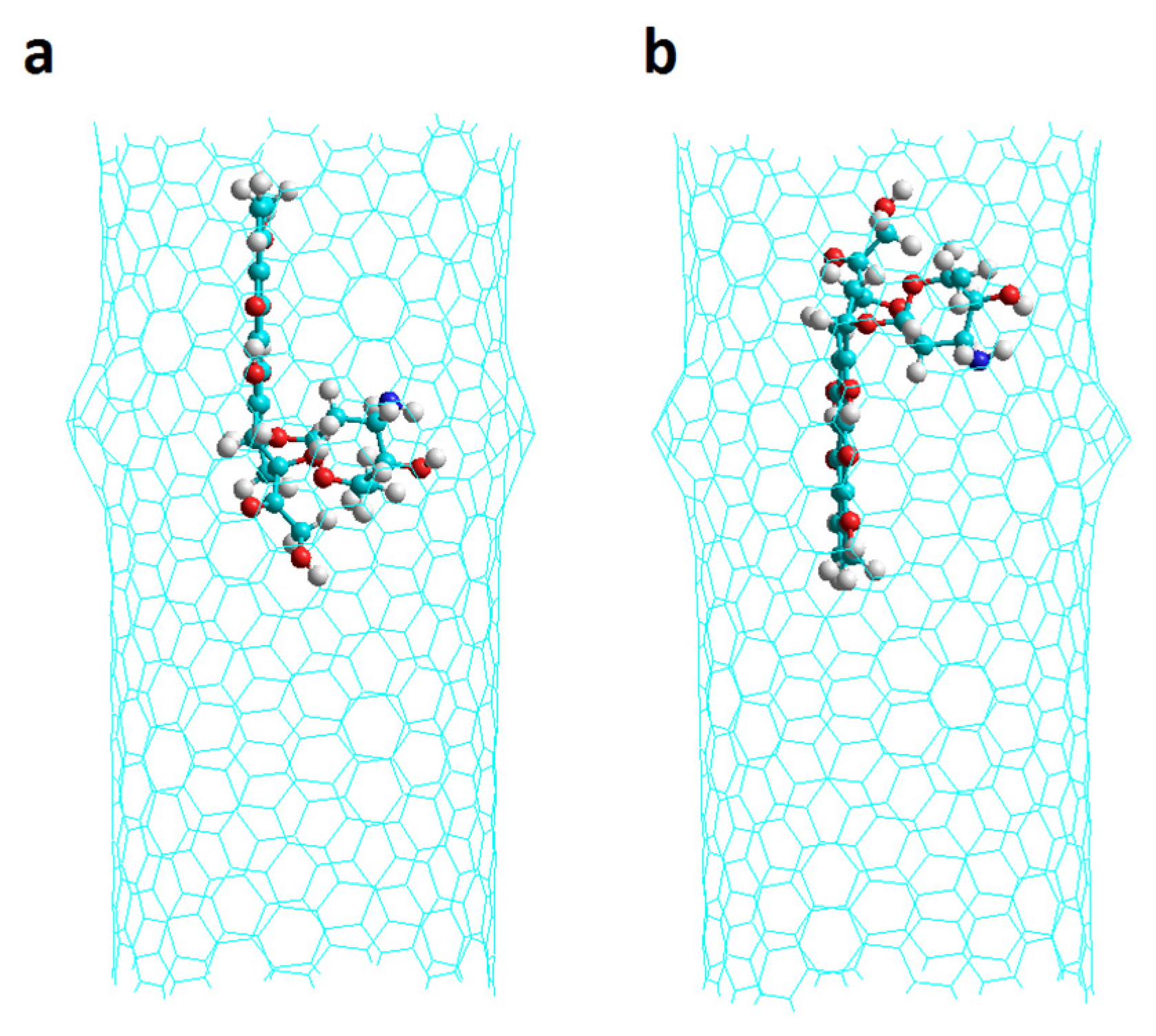
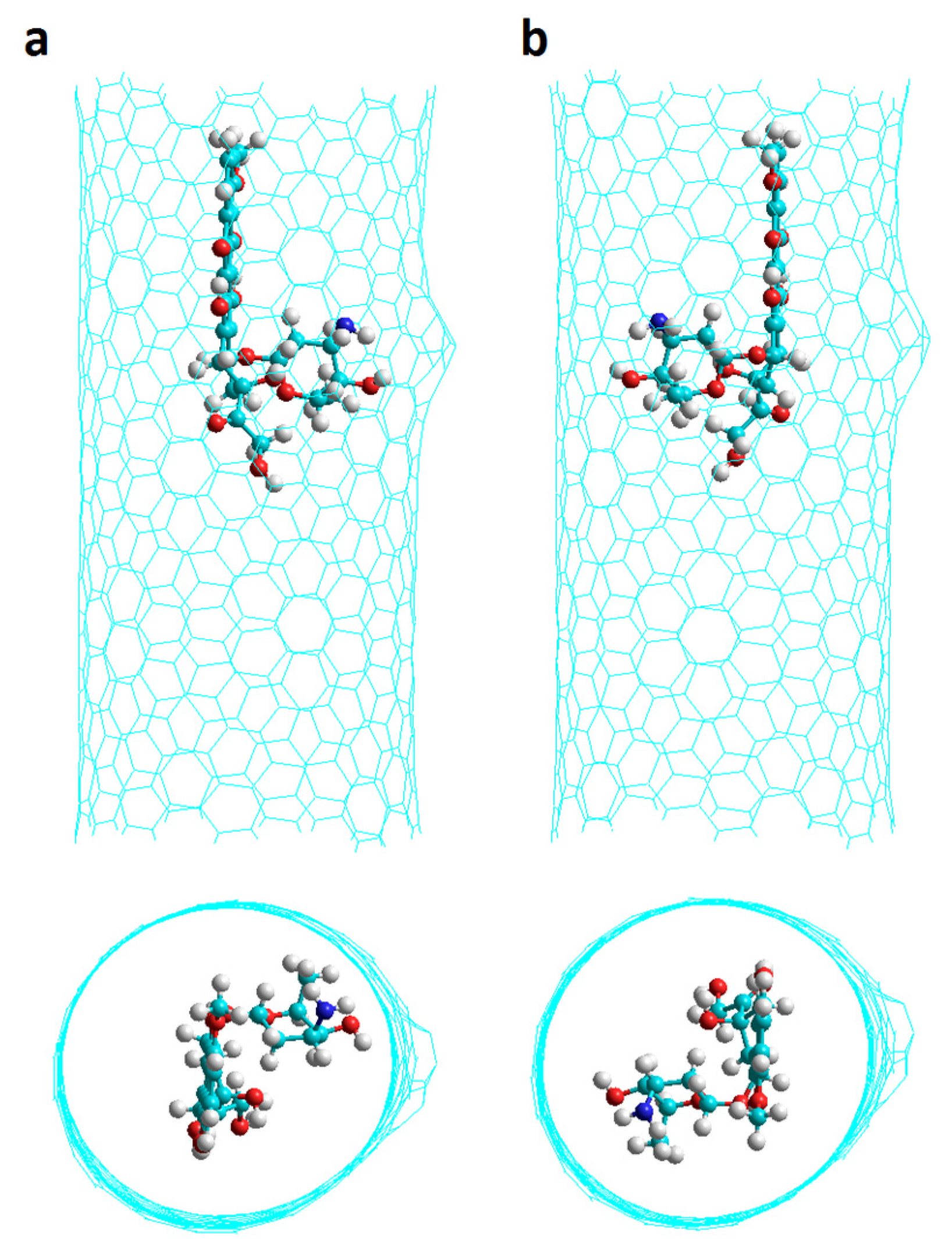
2.1. Chiral Nanotubes with Hk Defects
2.1.1. Chiral Nanotubes with HK1 Defects
2.1.2. Chiral Nanotubes with Hk2 Defects
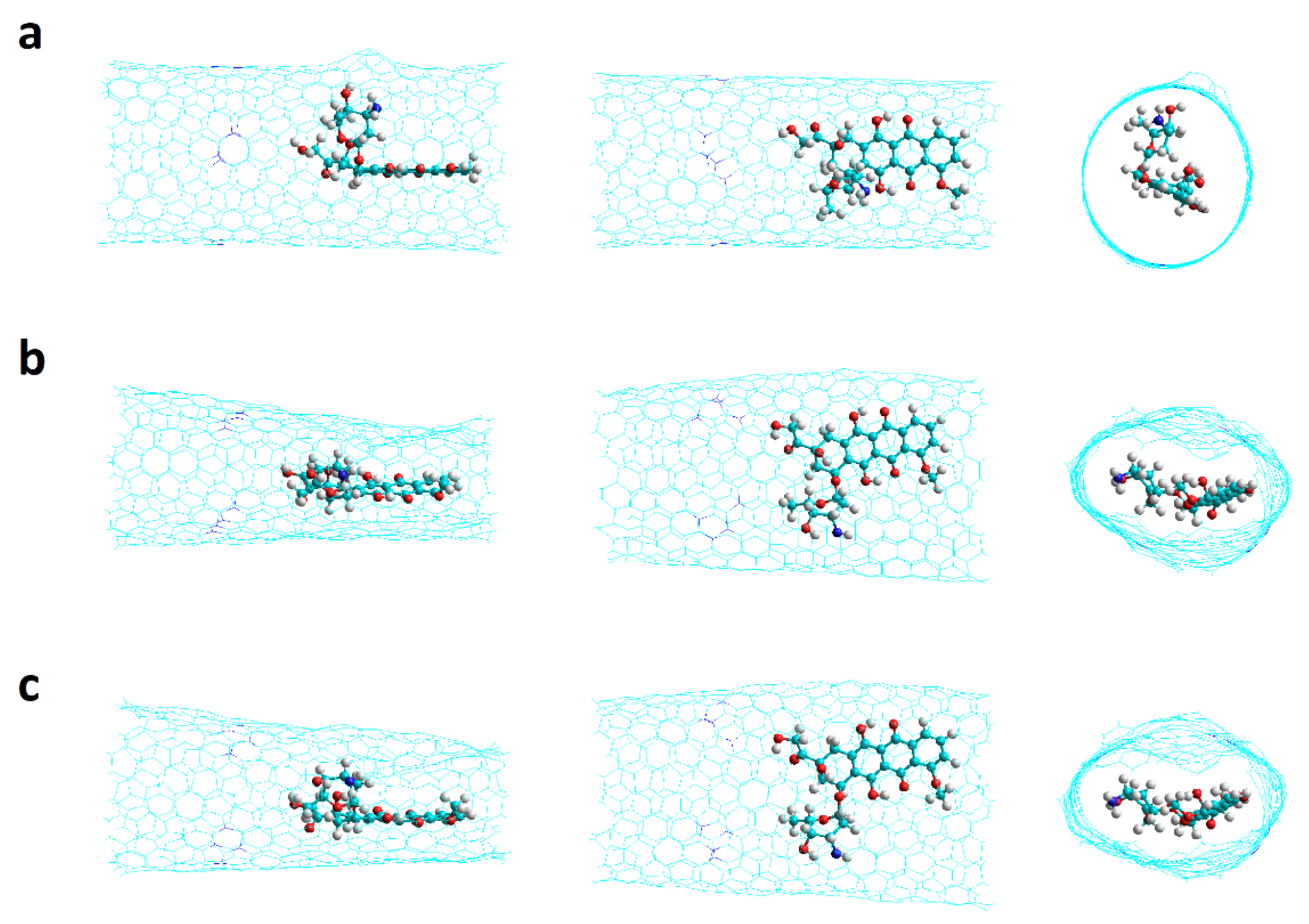
2.2. Nanotubes with Stone–Wales Defects
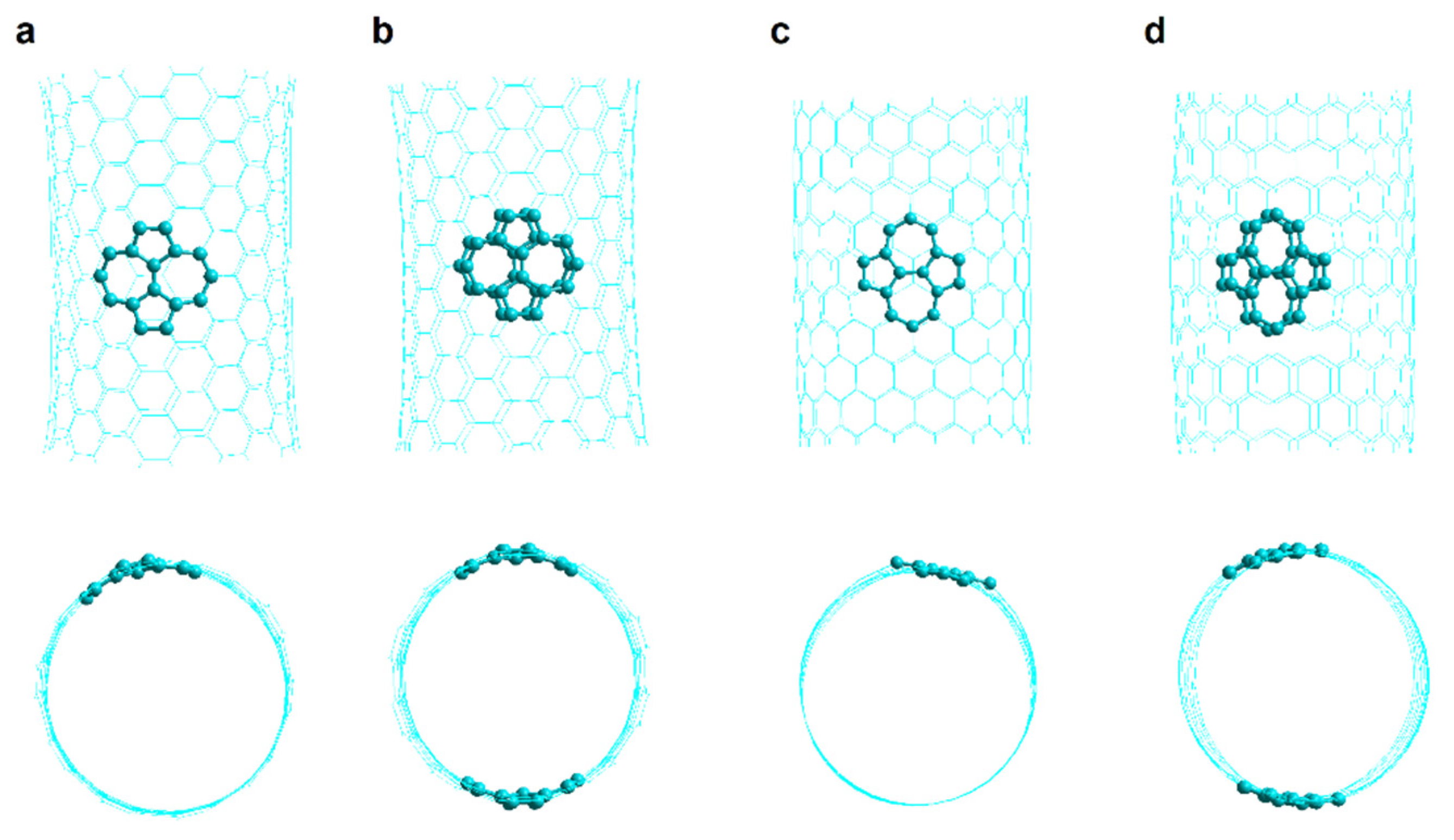
2.2.1. SW1 and SW2 Armchair Nanotubes
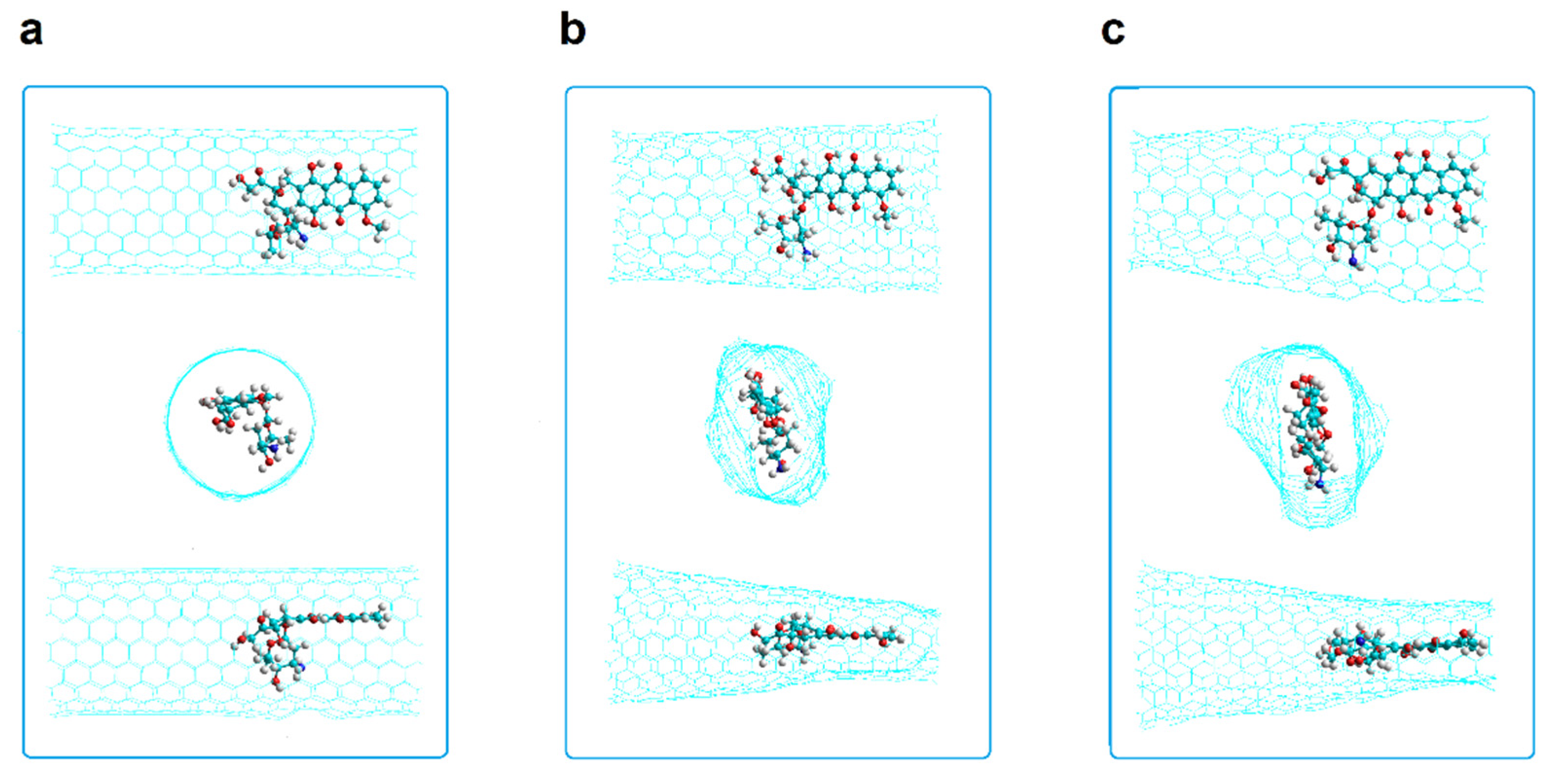
2.2.2. SW1 and SW2 Zigzag Nanotubes
3. Discussion
4. Simulation Methods
5. Conclusions
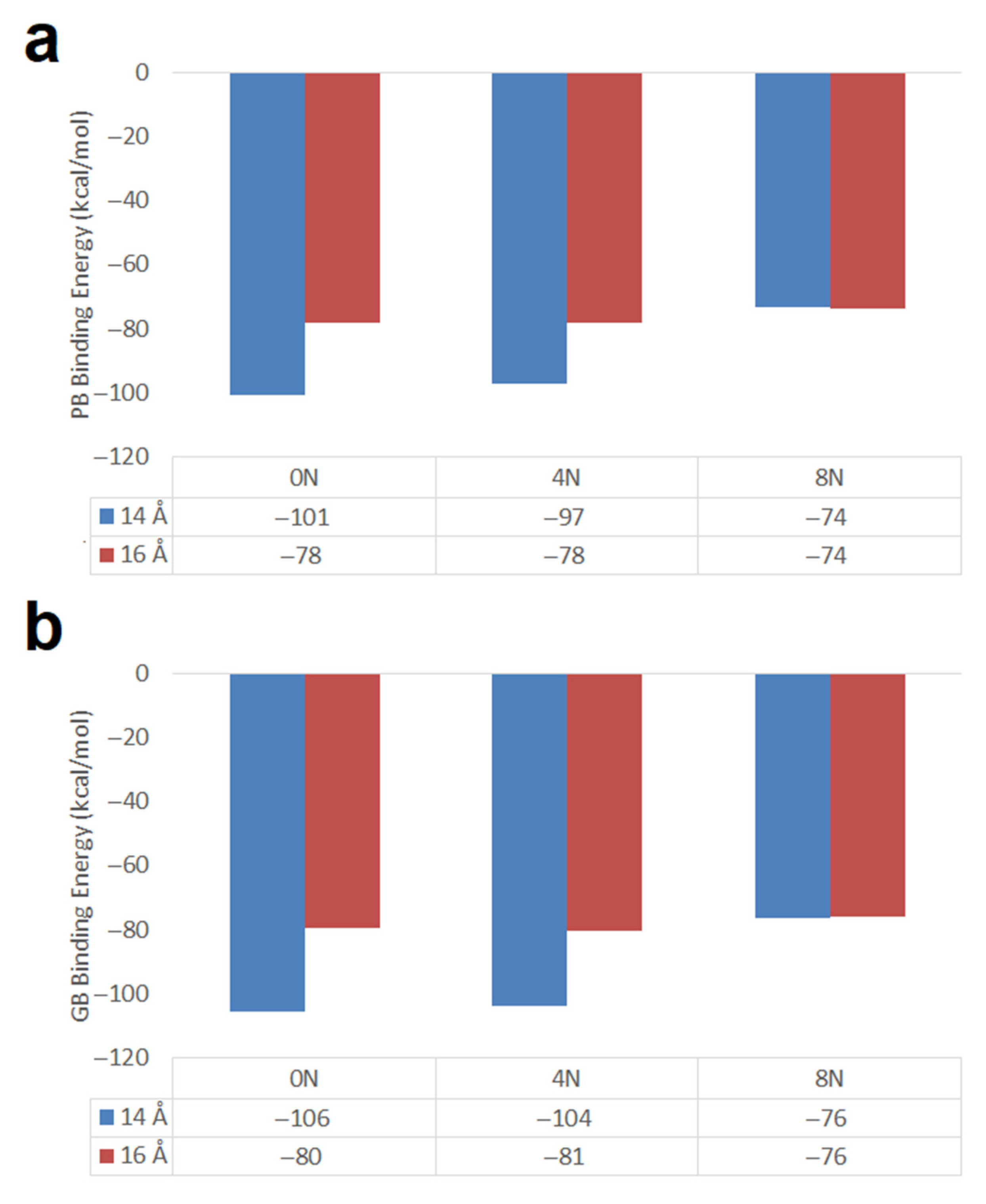
- When using RESP charges, DOX encapsulation inside Hk2 nanotubes of 14 Å diameter, predicts that the DOX-CNT attractive forces decrease in the following order of nanotube chirality: chiral > armchair > zigzag in agreement with that reported in a MD study under similar conditions, for defect-free nanotubes.
- Hk2 chiral nanotubes (short and long) favor DOX-CNT interactions which decrease in the presence of nitrogen as a dopant in the order 0N > 4N > 8N.
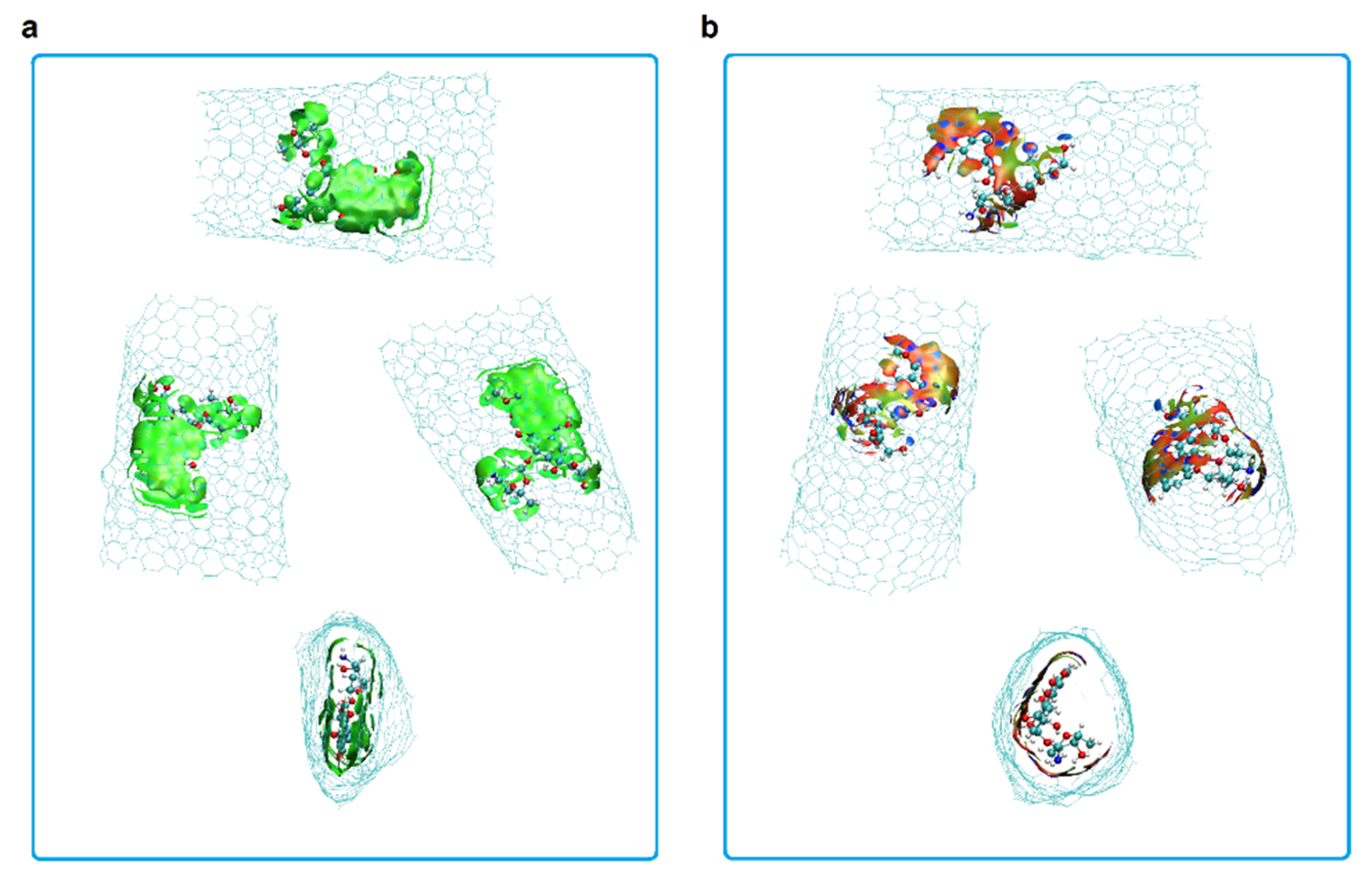
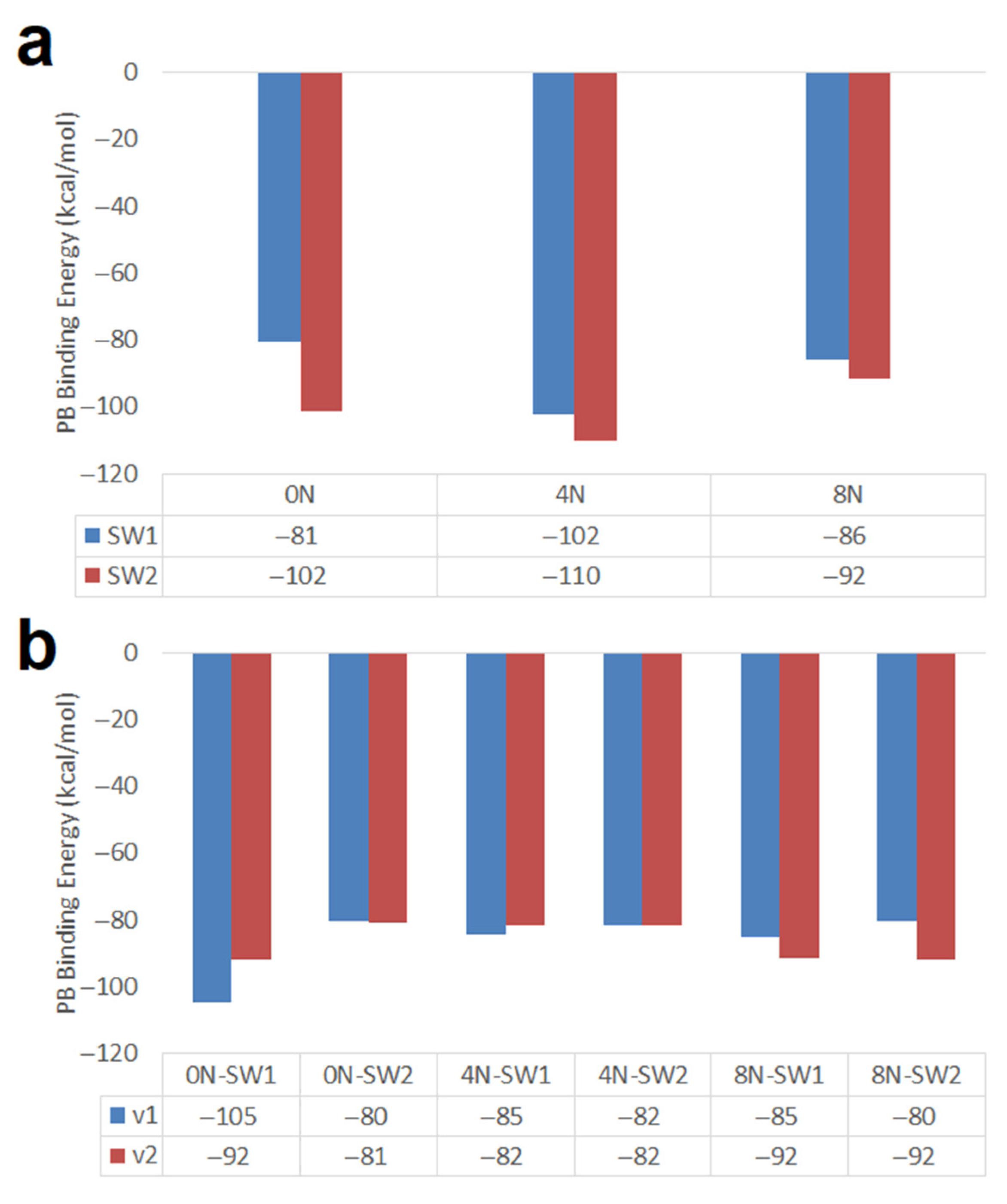
- Short armchair nanotubes promote more favorable DOX-CNT interactions if they are SW2; but long armchair nanotubes favor the DOX-CNT interactions if they are SW1. When the DOX is located close to the defect, a better interaction occurs than when it is located in the defect-free zone of the nanotube. The interaction is also improved if the DOX nitrogen atom is close to the defect.
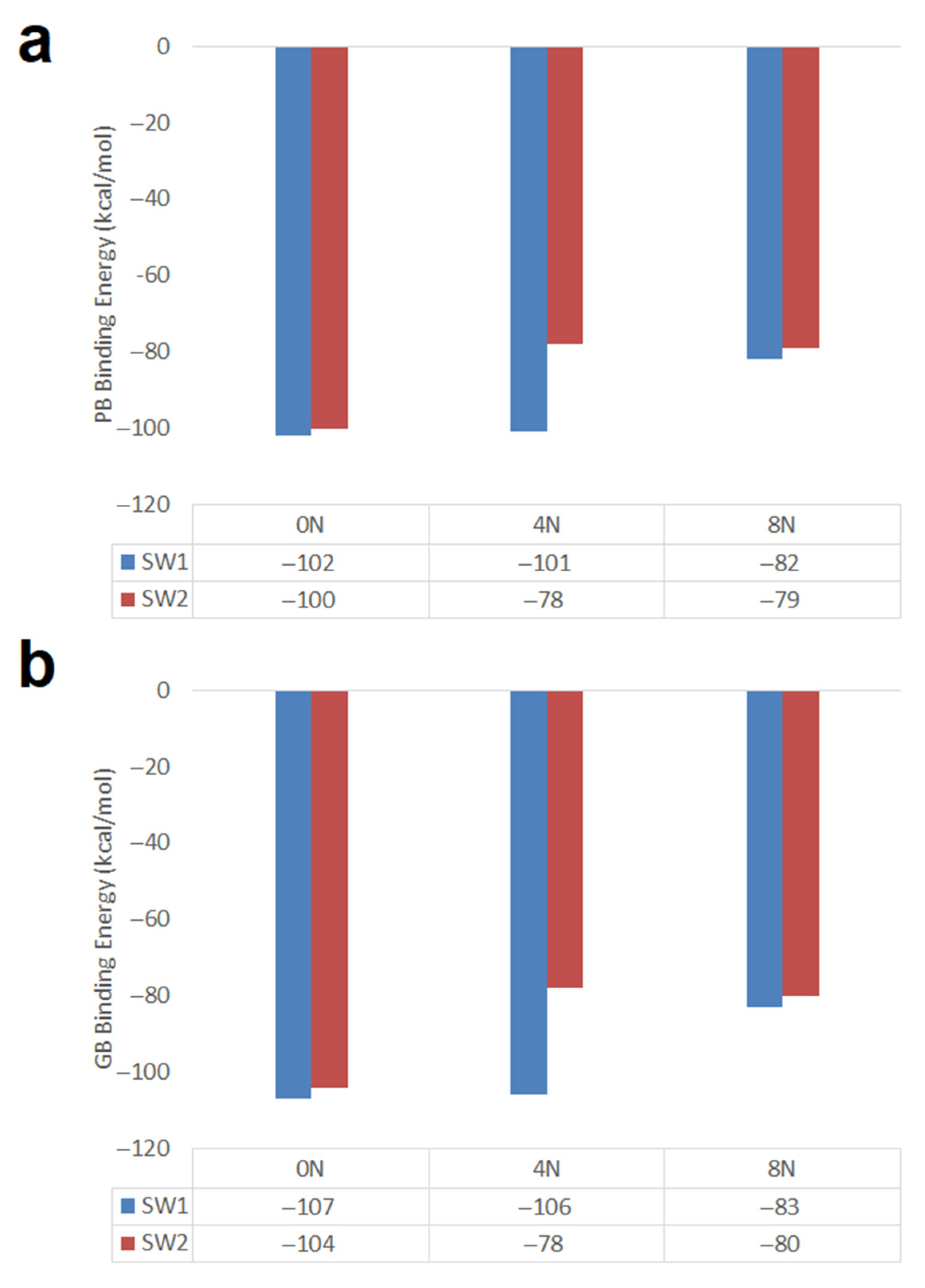
- Short SW1 zigzag nanotubes favor DOX-CNT interactions which decrease in the presence of nitrogen as a dopant in the order 0N > 4N > 8N, like the Hk2 chiral nanotubes. SW1 zigzag nanotubes exhibit more exothermic DOX-CNT binding energies than SW2 zigzag nanotubes. In both cases, the undoped SW defective zigzag nanotubes are predicted to exhibit stronger DOX-CNT interactions than corresponding nitrogen-doped nanotubes.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Etheridge, M.L.; Campbell, S.A.; Erdman, A.G.; Haynes, C.L.; Wolf, S.M.; McCullough, J. The big picture on nanomedicine: The state of investigational and approved nanomedicine products. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marketing Bureau. Zydus Cadila Receives US FDA Approval for Generic Doxil Liposome Injection. Available online: http://www.pharmabiz.com/NewsDetails.aspx?aid=131053&sid=2 (accessed on 12 September 2020).
- Patel, A.G.; Kaufmann, S.H. How does doxorubicin work? eLife 2012, 1, e00387. [Google Scholar] [CrossRef]
- Denard, B.; Lee, C.; Ye, J. Doxorubicin blocks proliferation of cancer cells through proteolytic activation of CREB3L1. eLife 2012, 1, e00090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.L.; Oliveira, P.J.; Cunha-Oliveira, T. Epigenetics in Doxorubicin Cardiotoxicity; Elsevier BV: Amsterdam, The Netherlands, 2019; pp. 837–846. [Google Scholar]
- Rodríguez-Galván, A.; Amelines-Sarria, O.; Rivera, M.; Carreón-Castro, M.D.P.; Basiuk, V.A. Adsorption and Self-Assembly of Anticancer Antibiotic Doxorubicin on Single-Walled Carbon Nanotubes. Nano 2016, 11, 1650038. [Google Scholar] [CrossRef]
- Lalan, M.; Jani, D. Toxicological Aspects of Carbon Nanotubes, Fullerenes and Graphenes. Curr. Pharm. Des. 2021, 27, 556–564. [Google Scholar] [CrossRef]
- Contreras , M.L.; Rozas, R. Carbon Nanotubes: Toxicological Properties, Use as Drug Delivery Material and Computational Contribution to Predict their Properties Including Structures with Topological Defects. Adv. Clin. Toxicol. 2020, 5, 1–7. [Google Scholar] [CrossRef]
- Mamidi, N. Cytotoxicity Evaluation of Carbon Nanotubes for Biomedical and Tissue Engineering Applications. Perspect. Carbon Nanotub. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Ali-Boucetta, H.; Kostarelos, K. Pharmacology of carbon nanotubes: Toxicokinetics, excretion and tissue accumulation. Adv. Drug Deliv. Rev. 2013, 65, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Nayak, T.; Leow, P.; Ee, P.-L.; Arockiadoss, T.; Ramaprabhu, S.; Pastorin, G. Crucial Parameters Responsible for Carbon Nanotubes Toxicity. Curr. Nanosci. 2010, 6, 141–154. [Google Scholar] [CrossRef]
- Venkataraman, A.; Amadi, E.V.; Chen, Y.; Papadopoulos, C. Carbon Nanotube Assembly and Integration for Applications. Nanoscale Res. Lett. 2019, 14, 1–47. [Google Scholar] [CrossRef]
- Pei, B.; Wang, W.; Dunne, N.; Li, X. Applications of Carbon Nanotubes in Bone Tissue Regeneration and Engineering: Superiority, Concerns, Current Advancements, and Prospects. Nanomaterials 2019, 9, 1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saliev, T. The Advances in Biomedical Applications of Carbon Nanotubes. C J. Carbon Res. 2019, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Wang, R.; Hu, Y.; Sun, R.; Song, T.; Shi, X.; Yin, S. Stacking of doxorubicin on folic acid-targeted multiwalled carbon nanotubes for in vivo chemotherapy of tumors. Drug Deliv. 2018, 25, 1607–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, C.M.Q.; Cao, X.T.; Kim, D.W.; Ban, U.H.; Lee, S.H.; Lim, K.T. Preparation of poly(styrene-alt-maleic anhydride) grafted multi-walled carbon nanotubes for pH-responsive release of doxorubicin. Mol. Cryst. Liq. Cryst. 2017, 654, 181–189. [Google Scholar] [CrossRef]
- Kumar, D.S.; Hasumura, T.; Nagaoka, Y.; Yoshida, Y.; Maekawa, T.; Jeyamohan, P. Accelerated killing of cancer cells using a multifunctional single-walled carbon nanotube-based system for targeted drug delivery in combination with photothermal therapy. Int. J. Nanomed. 2013, 8, 2653–2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Sun, X.; Nakayama-Ratchford, N.; Dai, H. Supramolecular Chemistry on Water-Soluble Carbon Nanotubes for Drug Loading and Delivery. ACS Nano 2007, 1, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Z. Interaction mechanism of doxorubicin and SWCNT: Protonation and diameter effects on drug loading and releasing. RSC Adv. 2016, 6, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Das, D. Carbon Nanotube and Graphene Nanoribbon Interconnects; CRC Press: Boca Raton, FL, USA, 2014; pp. 1–196. [Google Scholar]
- Charlier, J.-C. Defects in Carbon Nanotubes. Accounts Chem. Res. 2002, 35, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Crespi, V.H.; Cohen, M.L.; Rubio, A. In Situ Band Gap Engineering of Carbon Nanotubes. Phys. Rev. Lett. 1997, 79, 2093–2096. [Google Scholar] [CrossRef] [Green Version]
- Saito, R.; Fujita, M.; Dresselhaus, G.; Dresselhaus, M.S. Electronic structure of chiral graphene tubules. Appl. Phys. Lett. 1992, 60, 2204–2206. [Google Scholar] [CrossRef]
- Ebbesen, T.W.; Takada, T. Topological and SP3 defect structures in nanotubes. Carbon 1995, 33, 973–978. [Google Scholar] [CrossRef]
- Sternberg, M.; Gruen, D.M.; Kedziora, G.; Horner, D.A.; Zapol, P.; Curtiss, L.A.; Redfern, P.C. Carbon Ad-Dimer Defects in Carbon Nanotubes. Phys. Rev. Lett. 2006, 96, 075506. [Google Scholar] [CrossRef] [Green Version]
- Terrones, H.; Hernández, E.; Grobert, N.; Charlier, J.-C.; Ajayan, P.M. New Metallic Allotropes of Planar and Tubular Carbon. Phys. Rev. Lett. 2000, 84, 1716–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, A.J.; Wales, D.J. Theoretical studies of icosahedral C60 and some related species. Chem. Phys. Lett. 1986, 128, 501–503. [Google Scholar] [CrossRef]
- Orlikowski, D.; Bernholc, J.; Roland, C.; Nardelli, M.B. Ad-dimers on Strained Carbon Nanotubes: A New Route for Quantum Dot Formation? Phys. Rev. Lett. 1999, 83, 4132–4135. [Google Scholar] [CrossRef]
- Fuentes, M.L.C.; Soto, R.R. Carbon Nanotubes: Molecular and Electronic Properties of Regular and Defective Structures. Density Funct. Calc. 2018, 9, 197–218. [Google Scholar] [CrossRef] [Green Version]
- Pakdel, M.; Raissi, H.; Shahabi, M. Predicting doxorubicin drug delivery by single-walled carbon nanotube through cell membrane in the absence and presence of nicotine molecules: A molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2019, 38, 1488–1498. [Google Scholar] [CrossRef] [PubMed]
- Contreras, M.L.; Torres, C.; Villarroel, I.; Rozas, R. Molecular dynamics assessment of doxorubicin–carbon nanotubes molecular interactions for the design of drug delivery systems. Struct. Chem. 2019, 30, 369–384. [Google Scholar] [CrossRef]
- Torres, C.; Villarroel, I.; Rozas, R.; Contreras, L. Carbon Nanotubes Having Haeckelite Defects as Potential Drug Carriers. Molecular Dynamics Simulation. Molecules 2019, 24, 4281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Peng, G.; Li, J.; Liang, L.; Kong, Z.; Wang, H.; Jia, L.; Wang, X.; Zhang, W.; Shen, J.-W. Molecular dynamics study on the configuration and arrangement of doxorubicin in carbon nanotubes. J. Mol. Liq. 2018, 262, 295–301. [Google Scholar] [CrossRef]
- Izadyar, A.S.; Farhadian, N.; Chenarani, N. Molecular dynamics simulation of doxorubicin adsorption on a bundle of functionalized CNT. J. Biomol. Struct. Dyn. 2015, 34, 1797–1805. [Google Scholar] [CrossRef]
- Sornmee, P.; Rungrotmongkol, T.; Saengsawang, O.; Arsawang, U.; Remsungnen, T.; Hannongbua, S. Understanding the Molecular Properties of Doxorubicin Filling Inside and Wrapping Outside Single-Walled Carbon Nanotubes. J. Comput. Theor. Nanosci. 2011, 8, 1385–1391. [Google Scholar] [CrossRef]
- Wolski, P.; Nieszporek, K.; Panczyk, T. Carbon nanotubes and short cytosine-rich telomeric DNA oligomers as platforms for controlled release of doxorubicin-a molecular dynamics study. Int. J. Mol. Sci. 2020, 21, 3619. [Google Scholar] [CrossRef]
- Maleki, R.; Afrouzi, H.H.; Hosseini, M.; Toghraie, D.; Piranfar, A.; Rostami, S. pH-sensitive loading/releasing of doxorubicin using single-walled carbon nanotube and multi-walled carbon nanotube: A molecular dynamics study. Comput. Methods Programs Biomed. 2020, 186, 105210. [Google Scholar] [CrossRef] [PubMed]
- Pennetta, C.; Floresta, G.; Graziano, A.C.E.; Cardile, V.; Rubino, L.; Galimberti, M.; Rescifina, A.; Barbera, V. Functionalization of Single and Multi-Walled Carbon Nanotubes with Polypropylene Glycol Decorated Pyrrole for the Development of Doxorubicin Nano-Conveyors for Cancer Drug Delivery. Nanomaterials 2020, 10, 1073. [Google Scholar] [CrossRef]
- Kordzadeh, A.; Amjad-Iranagh, S.; Zarif, M.; Modarress, H. Adsorption and encapsulation of the drug doxorubicin on covalent functionalized carbon nanotubes: A scrutinized study by using molecular dynamics simulation and quantum mechanics calculation. J. Mol. Graph. Model. 2019, 88, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Karnati, K.R.; Wang, Y. Understanding the co-loading and releasing of doxorubicin and paclitaxel using chitosan functionalized single-walled carbon nanotubes by molecular dynamics simulations. Phys. Chem. Chem. Phys. 2018, 20, 9389–9400. [Google Scholar] [CrossRef]
- Wolski, P.; Nieszporek, K.; Panczyk, T. Pegylated and folic acid functionalized carbon nanotubes as pH controlled carriers of doxorubicin. Molecular dynamics analysis of the stability and drug release mechanism. Phys. Chem. Chem. Phys. 2017, 19, 9300–9312. [Google Scholar] [CrossRef] [PubMed]
- Rungnim, C.; Rungrotmongkol, T.; Poo-Arporn, R.P. pH-controlled doxorubicin anticancer loading and release from carbon nanotube noncovalently modified by chitosan: MD simulations. J. Mol. Graph. Model. 2016, 70, 70–76. [Google Scholar] [CrossRef]
- Janas, D. From Bio to Nano: A Review of Sustainable Methods of Synthesis of Carbon Nanotubes. Sustain. J. Rec. 2020, 12, 4115. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Veclani, D.; Tolazzi, M.; Melchior, A. Molecular Interpretation of Pharmaceuticals’ Adsorption on Carbon Nanomaterials: Theory Meets Experiments. Processes 2020, 8, 642. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Khalak, Y.; Tresadern, G.; de Groot, B.L.; Gapsys, V. Non-equilibrium approach for binding free energies in cyclodextrins in SAMPL7: Force fields and software. J. Comput. Mol. Des. 2021, 35, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Westermaier, Y.; Ruiz-Carmona, S.; Theret, I.; Perron-Sierra, F.; Poissonnet, G.; Dacquet, C.; Boutin, J.A.; Ducrot, P.; Barril, X. Binding mode prediction and MD/MMPBSA-based free energy ranking for agonists of REV-ERBα/NCoR. J. Comput. Mol. Des. 2017, 31, 755–775. [Google Scholar] [CrossRef] [PubMed]
- Chéron, N.; Shakhnovich, E.I. Effect of sampling on BACE-1 ligands binding free energy predictions via MM-PBSA calculations. J. Comput. Chem. 2017, 38, 1941–1951. [Google Scholar] [CrossRef]
- Eken, Y.; Almeida, N.M.S.; Wang, C.; Wilson, A.K. SAMPL7: Host–guest binding prediction by molecular dynamics and quantum mechanics. J. Comput. Mol. Des. 2021, 35, 63–77. [Google Scholar] [CrossRef]
- Ruangpornvisuti, V. Molecular modeling of dissociative and non-dissociative chemisorption of nitrosamine on close-ended and open-ended pristine and Stone-Wales defective (5,5) armchair single-walled carbon nanotubes. J. Mol. Model. 2009, 16, 1127–1138. [Google Scholar] [CrossRef]
- Peng, C.; Wang, J.; Yu, Y.; Wang, G.; Chen, Z.; Xu, Z.; Cai, T.; Shao, Q.; Shi, J.; Zhu, W. Improving the accuracy of predicting protein–ligand binding-free energy with semiempirical quantum chemistry charge. Futur. Med. Chem. 2019, 11, 303–321. [Google Scholar] [CrossRef] [PubMed]
- Contreras, M.L.; Ávila, D.; Alvarez, J.; Rozas, R. Computational algorithms for fast-building 3D carbon nanotube models with defects. J. Mol. Graph. Model. 2012, 38, 389–395. [Google Scholar] [CrossRef] [PubMed]
- HyperChem Release, Version 7.5; Hypercube Inc.: Gainesville, FL, USA, 2004.
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer , N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufrie, A.; Simmerling, C.; Wang, B.; Woods, R. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | Type 1 | PB | GB | dp-NT | dN-NT |
|---|---|---|---|---|---|
| 1 | Ch(13,08)0N-HK1-DoxDIn.v1o | −78 | −77 | 3.7 | 3.7 |
| 2 | Ch(13,08)0N-HK1-DoxDIn.v1p | −102 | −104 | 3.6 | 3.5 |
| 3 | Ch(13,08)0N-HK1-DoxDIn.v2o | −79 | −80 | 3.6 | 3.9 |
| 4 | Ch(13,08)0N-HK1-DoxDIn.v2p | −85 | −87 | 3.8 | 3.6 |
| 5 | Ch(13,08)0N-HK1-DoxRIn.v1o | −78 | −77 | 3.4 | 3.1 |
| 6 | Ch(13,08)0N-HK1-DoxRIn.v1p | −80 | −80 | 3.4 | 3.6 |
| 7 | Ch(13,08)0N-HK1-DoxRIn.v2o | −79 | −78 | 3.8 | 4.9 |
| 8 | Ch(13,08)0N-HK1-DoxRIn.v2p | −80 | −80 | 3.5 | 3.2 |
| 9 | Ch(13,08)4N-HK1-DoxDIn.v1o | −77 | −76 | 3.5 | 3.4 |
| 10 | Ch(13,08)4N-HK1-DoxDIn.v1p | −99 | −102 | 3.4 | 3.3 |
| 11 | Ch(13,08)4N-HK1-DoxDIn.v2o | −78 | −80 | 3.8 | 3.9 |
| 12 | Ch(13,08)4N-HK1-DoxDIn.v2p | −79 | −80 | 3.4 | 3.0 |
| 13 | Ch(13,08)4N-HK1-DoxRIn.v1o | −77 | −77 | 3.6 | 4.0 |
| 14 | Ch(13,08)4N-HK1-DoxRIn.v1p | −79 | −81 | 3.6 | 3.5 |
| 15 | Ch(13,08)4N-HK1-DoxRIn.v2o | −78 | −79 | 4.2 | 4.6 |
| 16 | Ch(13,08)4N-HK1-DoxRIn.v2p | −79 | −80 | 3.5 | 3.2 |
| 17 | Ch(13,08)8N-HK1-DoxDIn.v1o | −78 | −79 | 3.6 | 3.5 |
| 18 | Ch(13,08)8N-HK1-DoxDIn.v1p | −102 | −104 | 3.4 | 3.2 |
| 19 | Ch(13,08)8N-HK1-DoxDIn.v2o | −78 | −79 | 3.8 | 3.6 |
| 20 | Ch(13,08)8N-HK1-DoxDIn.v2p | −78 | −79 | 3.4 | 3.4 |
| 21 | Ch(13,08)8N-HK1-DoxRIn.v1o | −79 | −80 | 3.4 | 3.4 |
| 22 | Ch(13,08)8N-HK1-DoxRIn.v1p | −81 | −81 | 3.6 | 3.4 |
| 23 | Ch(13,08)8N-HK1-DoxRIn.v2o | −73 | −76 | 3.5 | 3.2 |
| 24 | Ch(13,08)8N-HK1-DoxRIn.v2p | −81 | −82 | 3.5 | 3.1 |
| Run | Type 1 | PB | GB | dp-NT | dN-NT |
|---|---|---|---|---|---|
| 1 | Ch(13,08)0N-HK2-DoxDin.v1 | −109 | −112 | 3.4 | 3.4 |
| 2 | Ch(13,08)0N-HK2-DoxDin.v2 | −82 | −83 | 3.7 | 3.4 |
| 3 | Ch(13,08)0N-HK2-DoxRin.v1 | −80 | −79 | 3.3 | 3.4 |
| 4 | Ch(13,08)0N-HK2-DoxRin.v2 | −79 | −79 | 3.2 | 4.8 |
| 5 | Ch(13,08)4N-HK2-DoxDin.v1 | −104 | −108 | 3.5 | 4.3 |
| 6 | Ch(13,08)4N-HK2-DoxDin.v2 | −82 | −83 | 3.8 | 3.5 |
| 7 | Ch(13,08)4N-HK2-DoxRin.v1 | −79 | −78 | 4.1 | 3.4 |
| 8 | Ch(13,08)4N-HK2-DoxRin.v2 | −80 | −80 | 3.3 | 3.5 |
| 9 | Ch(13,08)8N-HK2-DoxDin.v1 | −80 | −78 | 3.4 | 3.3 |
| 10 | Ch(13,08)8N-HK2-DoxDin.v2 | −87 | −89 | 3.6 | 3.2 |
| 11 | Ch(13,08)8N-HK2-DoxRin.v1 | −80 | −80 | 3.6 | 3.3 |
| 12 | Ch(13,08)8N-HK2-DoxRin.v2 | −80 | −79 | 3.8 | 3.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Contreras, L.; Villarroel, I.; Torres, C.; Rozas, R. Doxorubicin Encapsulation in Carbon Nanotubes Having Haeckelite or Stone–Wales Defects as Drug Carriers: A Molecular Dynamics Approach. Molecules 2021, 26, 1586. https://doi.org/10.3390/molecules26061586
Contreras L, Villarroel I, Torres C, Rozas R. Doxorubicin Encapsulation in Carbon Nanotubes Having Haeckelite or Stone–Wales Defects as Drug Carriers: A Molecular Dynamics Approach. Molecules. 2021; 26(6):1586. https://doi.org/10.3390/molecules26061586
Chicago/Turabian StyleContreras, Leonor, Ignacio Villarroel, Camila Torres, and Roberto Rozas. 2021. "Doxorubicin Encapsulation in Carbon Nanotubes Having Haeckelite or Stone–Wales Defects as Drug Carriers: A Molecular Dynamics Approach" Molecules 26, no. 6: 1586. https://doi.org/10.3390/molecules26061586
APA StyleContreras, L., Villarroel, I., Torres, C., & Rozas, R. (2021). Doxorubicin Encapsulation in Carbon Nanotubes Having Haeckelite or Stone–Wales Defects as Drug Carriers: A Molecular Dynamics Approach. Molecules, 26(6), 1586. https://doi.org/10.3390/molecules26061586