
Computational Study on the Mechanism of the Photouncaging Reaction of Vemurafenib: Toward an Enhanced Photoprotection Approach for Photosensitive Drugs

Abstract
:1. Introduction
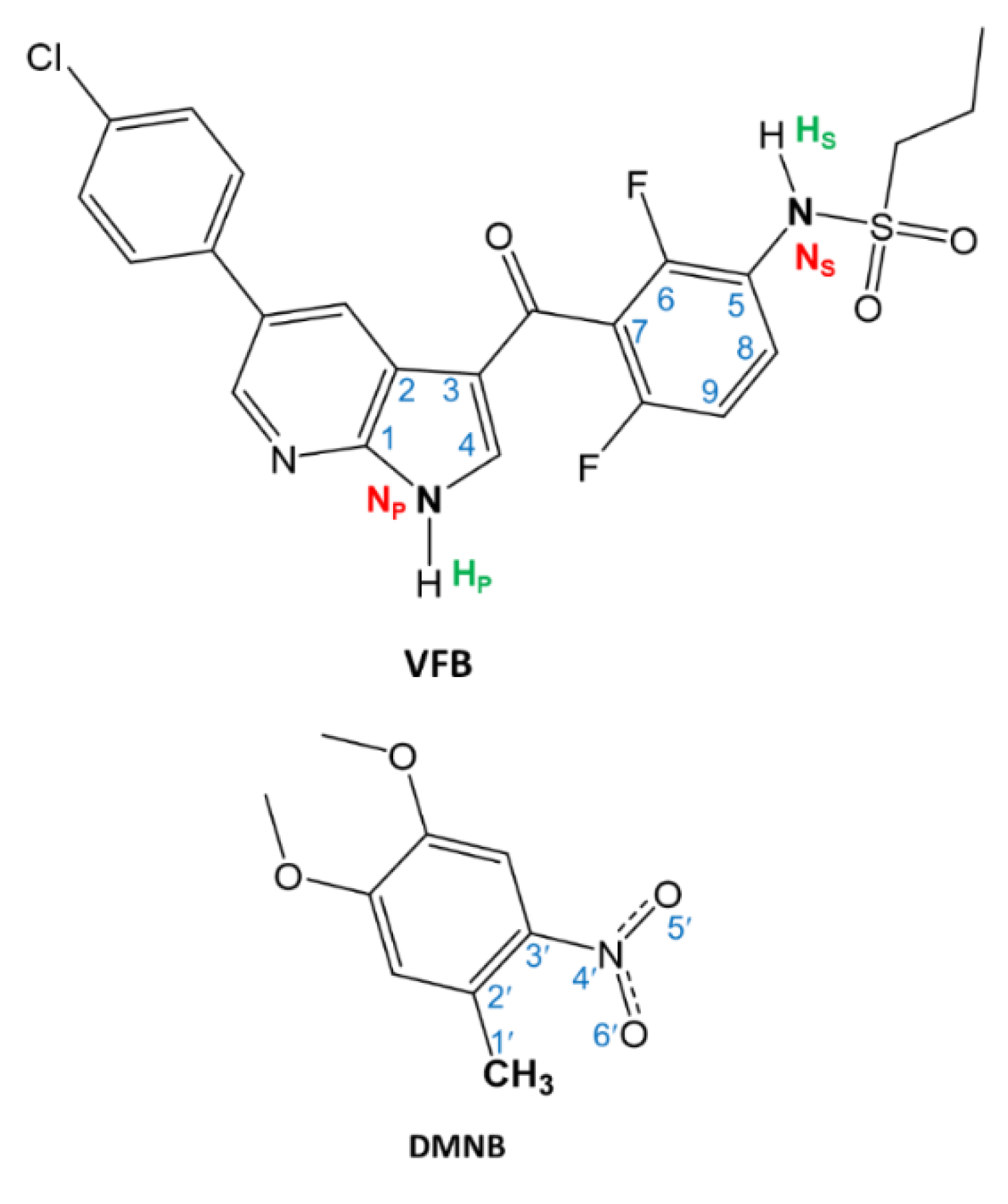
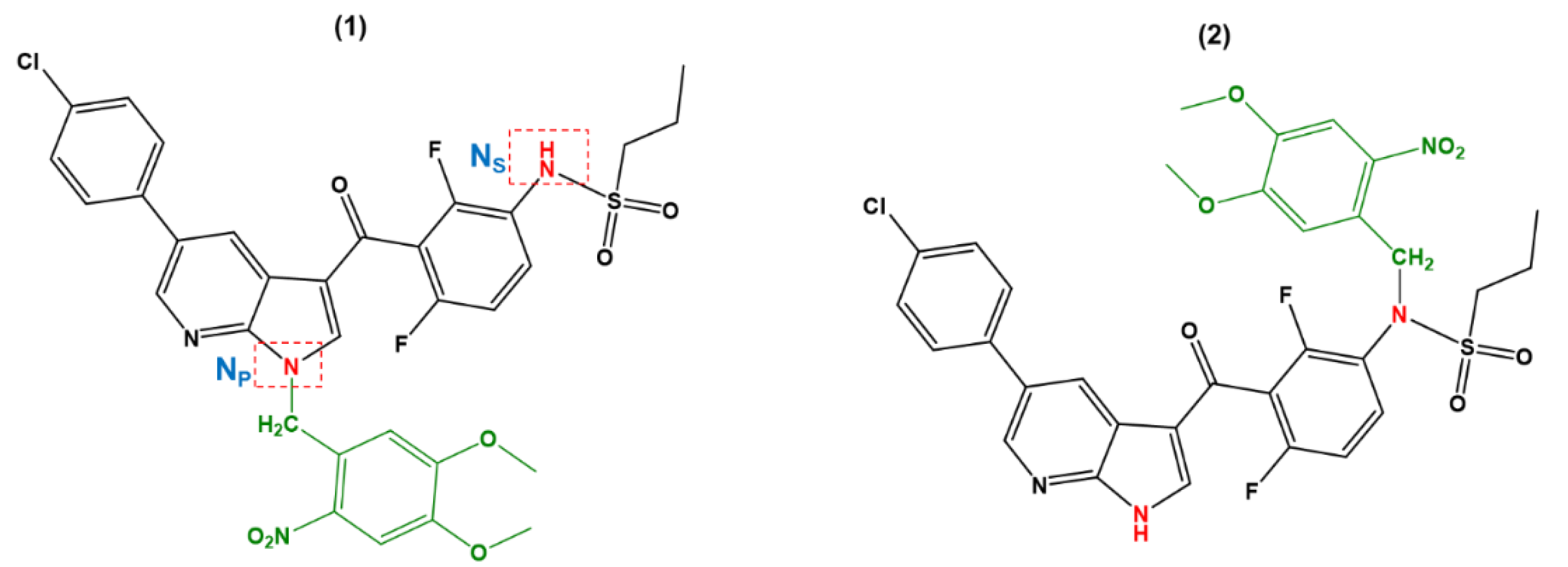
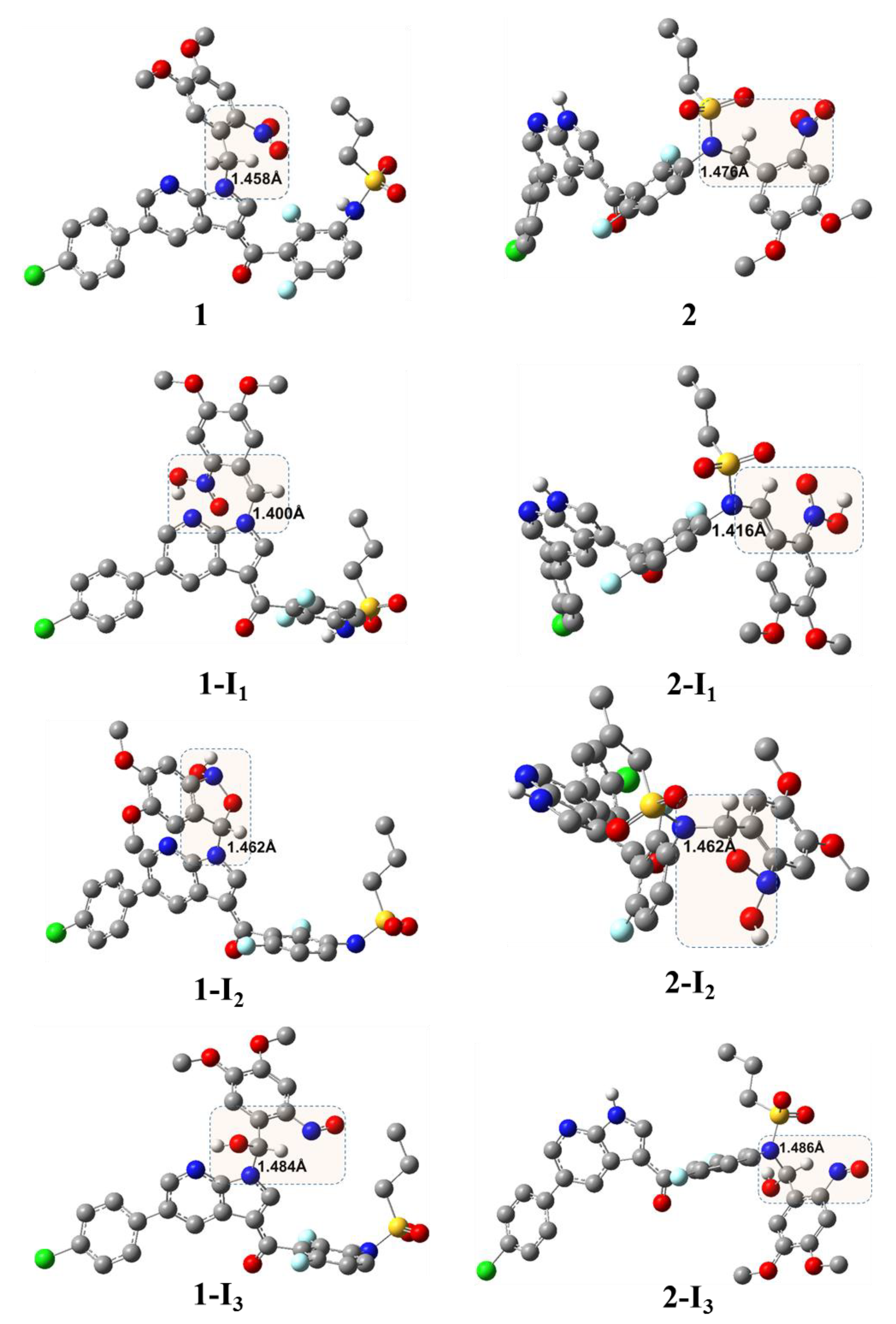
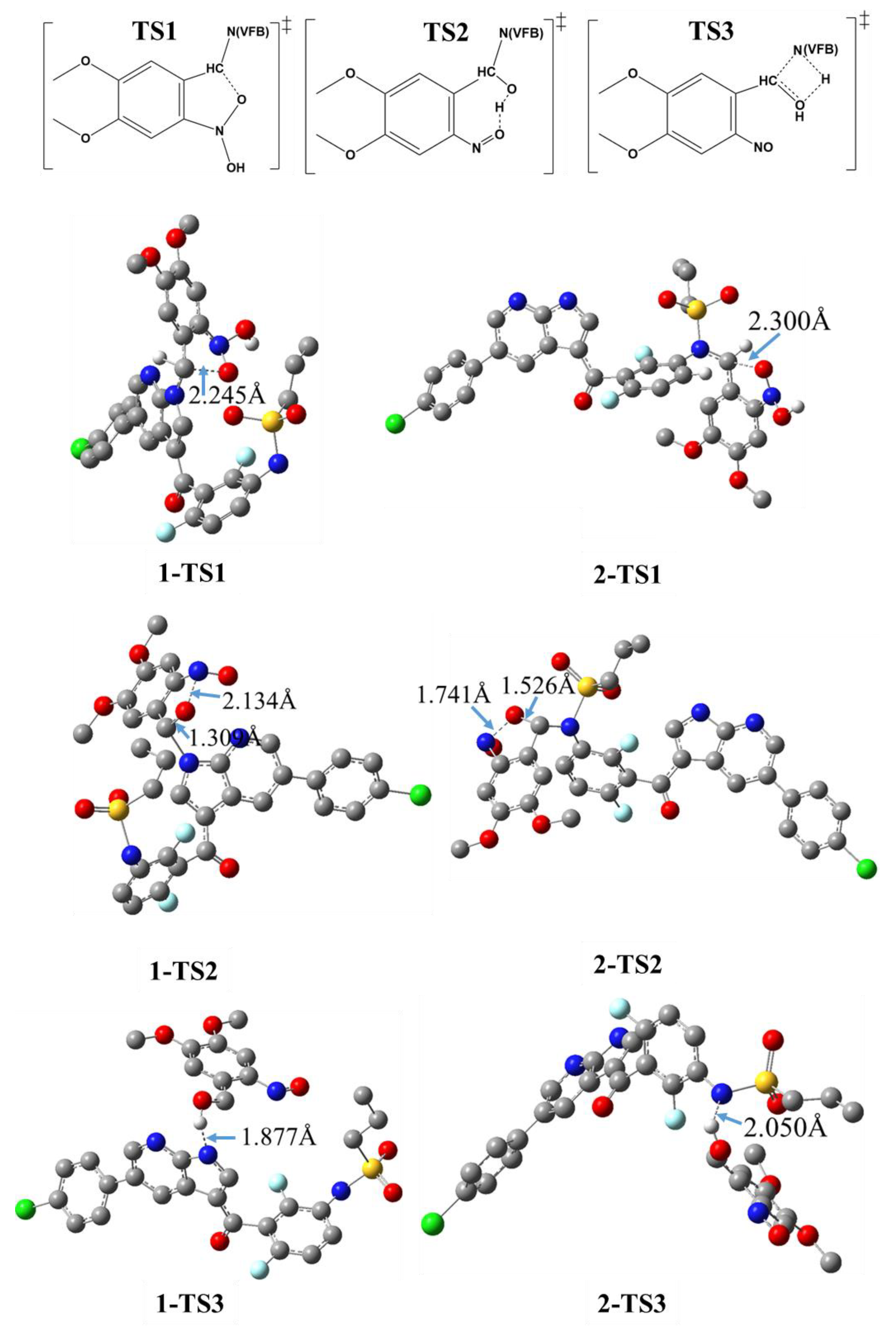
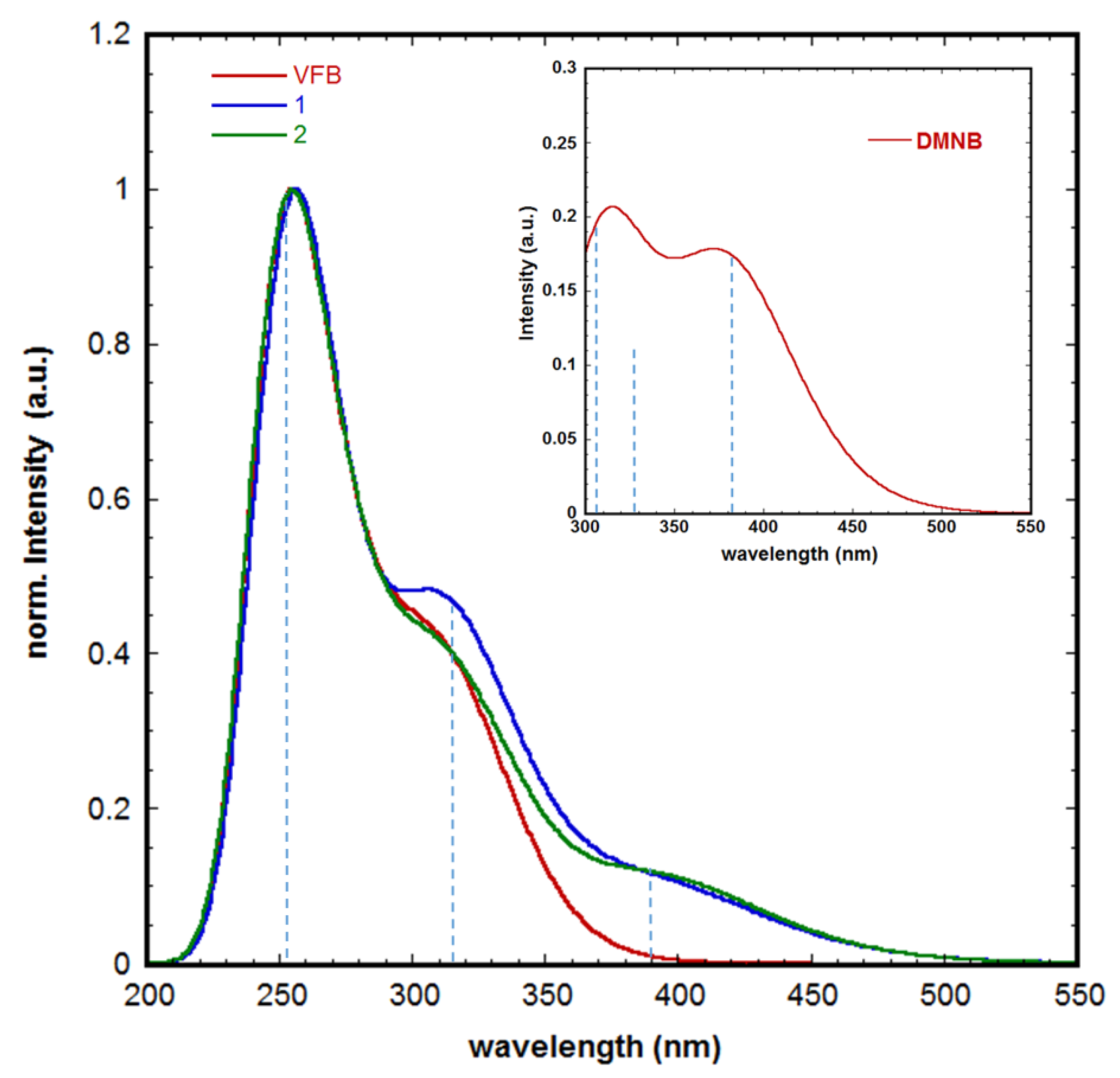
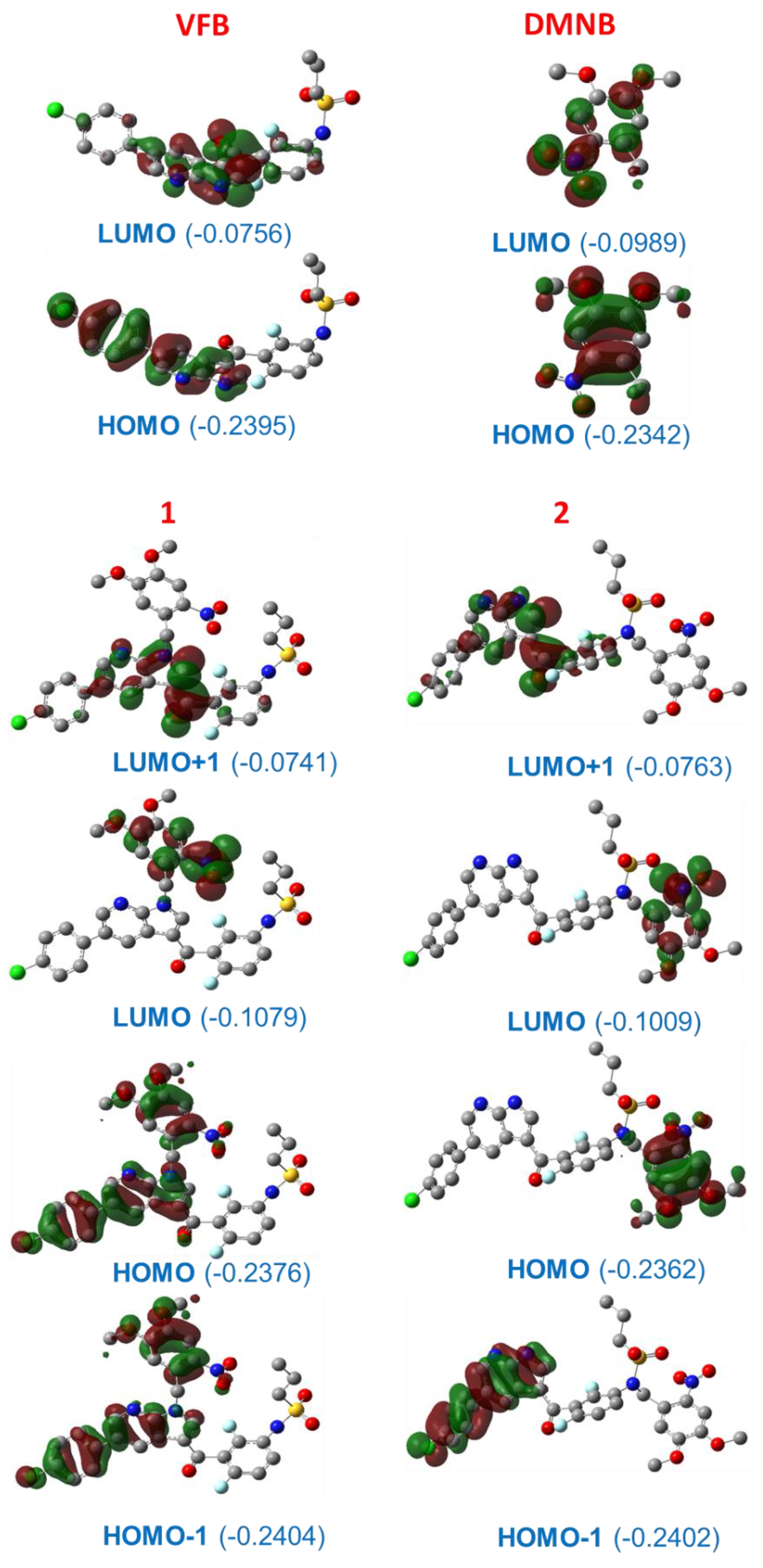
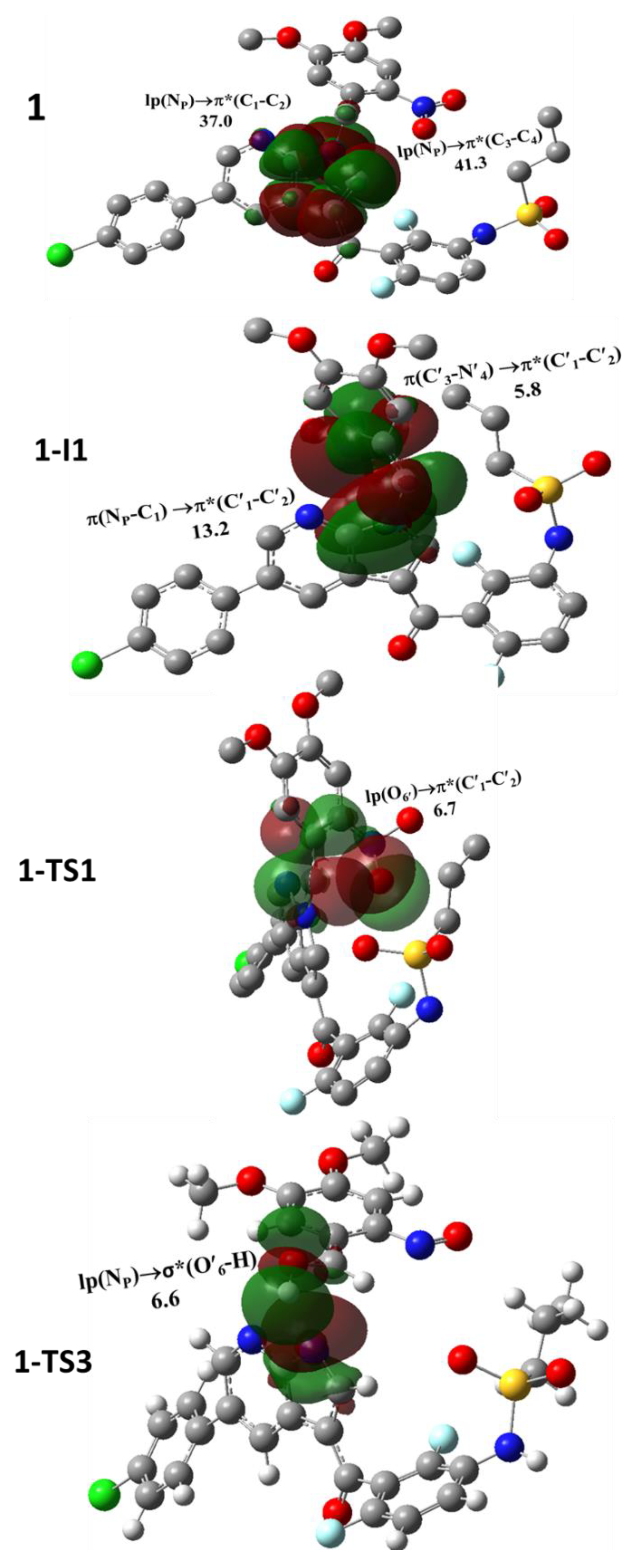
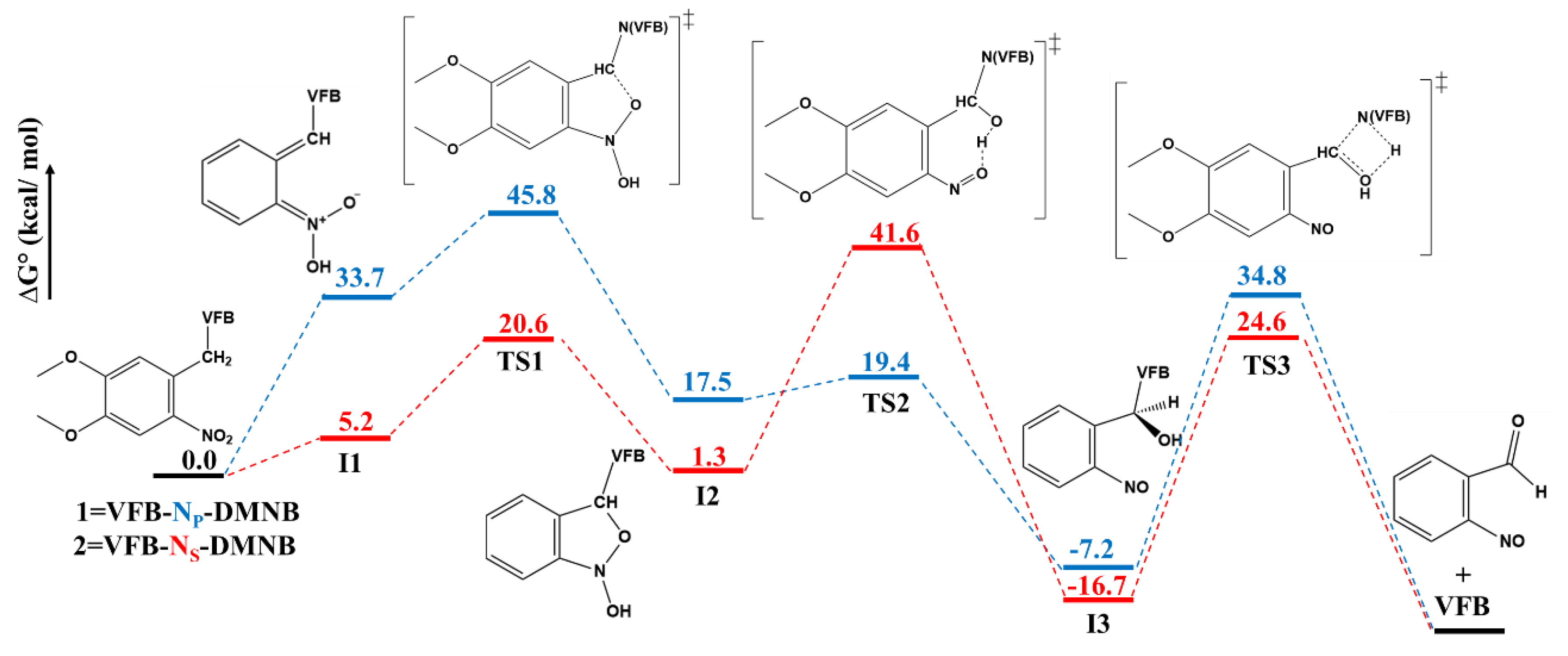
2. Results and Discussion
3. Computational Methods
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Caron, G.; Ermondi, G. Updating molecular properties during early drug discovery. Drug Discov. Today 2017, 22, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Caron, G.; Kihlberg, J.; Ermondi, G. Intramolecular hydrogen bonding: An opportunity for improved design in medicinal chemistry. Med. Res. Rev. 2019, 39, 1707–1729. [Google Scholar] [CrossRef] [PubMed]
- Dawoud Bani-Yaseen, A. Spectrofluorimetric study on the interaction between antimicrobial drug sulfamethazine and bovine serum albumin. J. Lumin. 2011, 131, 1042–1047. [Google Scholar] [CrossRef]
- Varma, M.V.; Lai, Y.; El-Kattan, A.F. Molecular properties associated with transporter-mediated drug disposition. Adv. Drug Deliv. Rev. 2017, 116, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Bani-Yaseen, A.D. Synchronous spectrofluorimetric study of the supramolecular host-guest interaction of β-cyclodextrin with propranolol: A comparative study. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 2015, 148, 93–98. [Google Scholar] [CrossRef]
- Tarcsay, Á.; Keserű, G.M. Contributions of Molecular Properties to Drug Promiscuity. J. Med. Chem. 2013, 56, 1789–1795. [Google Scholar] [CrossRef]
- Bani-Yaseen, A.D. Computational molecular perspectives on the interaction of propranolol with β-cyclodextrin in solution: Towards the drug-receptor mechanism of interaction. J. Mol. Liq. 2017, 227, 280–290. [Google Scholar] [CrossRef]
- Bani-Yaseen, A.D.; Al-Balawi, M. The solvatochromic, spectral, and geometrical properties of nifenazone: A DFT/TD-DFT and experimental study. Phys. Chem. Chem. Phys. 2014, 16, 15519–15526. [Google Scholar] [CrossRef]
- González, M.; Ellahioui, Y.; Álvarez, R.; Gallego-Yerga, L.; Caballero, E.; Vicente-Blázquez, A.; Ramudo, L.; Marín Folgado, M.; Sanz, C.; Medarde, M.; et al. The Masked Polar Group Incorporation (MPGI) Strategy in Drug Design: Effects of Nitrogen Substitutions on Combretastatin and Isocombretastatin Tubulin Inhibitors. Molecules 2019, 24, 4319. [Google Scholar] [CrossRef] [Green Version]
- Bani-Yaseen, A.D.; Hammad, F.; Ghanem, B.S.; Mohammad, E.G. On the Photophysicochemical Properties of Selected Fluoroquinolones: Solvatochromic and Fluorescence Spectroscopy Study. J. Fluoresc. 2013, 23, 93–101. [Google Scholar] [CrossRef]
- Das, S.S.; Alkahtani, S.; Bharadwaj, P.; Ansari, M.T.; ALKahtani, M.D.F.; Pang, Z.; Hasnain, M.S.; Nayak, A.K.; Aminabhavi, T.M. Molecular insights and novel approaches for targeting tumor metastasis. Int. J. Pharm. 2020, 585, 119556. [Google Scholar] [CrossRef] [PubMed]
- Steyn, S.J.; Varma, M.V.S. Cytochrome-P450-Mediated Drug – Drug Interactions of Substrate Drugs: Assessing Clinical Risk Based on Molecular Properties and an Extended Clearance Classification System. Mol. Pharm. 2020, 17, 3024–3032. [Google Scholar] [CrossRef] [PubMed]
- Bani-Yaseen, A.D. Solvatochromic and Fluorescence Behavior of Sulfisoxazole. J. Fluoresc. 2011, 21, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Su, K.A.; Habel, L.A.; Achacoso, N.S.; Friedman, G.D.; Asgari, M.M. Photosensitizing antihypertensive drug use and risk of cutaneous squamous cell carcinoma. Br. J. Dermatol. 2018, 179, 1088–1094. [Google Scholar] [CrossRef]
- Monteiro, A.F.; Rato, M.; Martins, C. Drug-induced photosensitivity: Photoallergic and phototoxic reactions. Clin. Dermatol. 2016, 34, 571–581. [Google Scholar] [CrossRef]
- Peukert, S.; Nunez, J.; He, F.; Dai, M.; Yusuff, N.; DiPesa, A.; Miller-Moslin, K.; Karki, R.; Lagu, B.; Harwell, C.; et al. A method for estimating the risk of drug-induced phototoxicity and its application to smoothened inhibitors. Medchemcomm 2011, 2, 973. [Google Scholar] [CrossRef]
- Kim, W.B.; Shelley, A.J.; Novice, K.; Joo, J.; Lim, H.W.; Glassman, S.J. Drug-induced phototoxicity: A systematic review. J. Am. Acad. Dermatol. 2018, 79, 1069–1075. [Google Scholar] [CrossRef]
- Khandpur, S.; Porter, R.M.; Boulton, S.J.; Anstey, A. Drug-induced photosensitivity: New insights into pathomechanisms and clinical variation through basic and applied science. Br. J. Dermatol. 2017, 176, 902–909. [Google Scholar] [CrossRef]
- Wu, G.; Zhao, T.; Kang, D.; Zhang, J.; Song, Y.; Namasivayam, V.; Kongsted, J.; Pannecouque, C.; De Clercq, E.; Poongavanam, V.; et al. Overview of Recent Strategic Advances in Medicinal Chemistry. J. Med. Chem. 2019, 62, 9375–9414. [Google Scholar] [CrossRef]
- Horbert, R.; Pinchuk, B.; Davies, P.; Alessi, D.; Peifer, C. Photoactivatable Prodrugs of Antimelanoma Agent Vemurafenib. ACS Chem. Biol. 2015, 10, 2099–2107. [Google Scholar] [CrossRef]
- Tamura, R.; Balabanova, A.; Frakes, S.A.; Bargmann, A.; Grimm, J.; Koch, T.H.; Yin, H. Photoactivatable Prodrug of Doxazolidine Targeting Exosomes. J. Med. Chem. 2019, 62, 1959–1970. [Google Scholar] [CrossRef] [PubMed]
- Zindler, M.; Pinchuk, B.; Renn, C.; Horbert, R.; Döbber, A.; Peifer, C. Design, Synthesis, and Characterization of a Photoactivatable Caged Prodrug of Imatinib. ChemMedChem 2015, 10, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Döbber, A.; Phoa, A.F.; Abbassi, R.H.; Stringer, B.W.; Day, B.W.; Johns, T.G.; Abadleh, M.; Peifer, C.; Munoz, L. Development and Biological Evaluation of a Photoactivatable Small Molecule Microtubule-Targeting Agent. ACS Med. Chem. Lett. 2017, 8, 395–400. [Google Scholar] [CrossRef] [Green Version]
- Basa, P.N.; Antala, S.; Dempski, R.E.; Burdette, S.C. A Zinc(II) Photocage Based on a Decarboxylation Metal Ion Release Mechanism for Investigating Homeostasis and Biological Signaling. Angew. Chem. 2015, 127, 13219–13223. [Google Scholar] [CrossRef] [Green Version]
- Klán, P.; Šolomek, T.; Bochet, C.G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy. Chem. Rev. 2013, 113, 119–191. [Google Scholar] [CrossRef] [PubMed]
- Pelliccioli, A.P.; Wirz, J. Photoremovable protecting groups: Reaction mechanisms and applications. Photochem. Photobiol. Sci. 2002, 1, 441–458. [Google Scholar] [CrossRef]
- Šolomek, T.; Wirz, J.; Klán, P. Searching for Improved Photoreleasing Abilities of Organic Molecules. Acc. Chem. Res. 2015, 48, 3064–3072. [Google Scholar] [CrossRef]
- Brugière, C.; Stefan, A.; Morice, C.; Cornet, E.; Moreau, A.; Allouche, S.; Verneuil, L. Vemurafenib skin phototoxicity is indirectly linked to ultraviolet A minimal erythema dose decrease. Br. J. Dermatol. 2014, 171, 1529–1532. [Google Scholar] [CrossRef]
- Woods, J.A.; Ferguson, J.S.; Kalra, S.; Degabriele, A.; Gardner, J.; Logan, P.; Ferguson, J. The phototoxicity of vemurafenib: An investigation of clinical monochromator phototesting and in vitro phototoxicity testing. J. Photochem. Photobiol. B Biol. 2015, 151, 233–238. [Google Scholar] [CrossRef]
- Sinha, R.; Larkin, J.; Gore, M.; Fearfield, L. Cutaneous toxicities associated with vemurafenib therapy in 107 patients with BRAF V600E mutation-positive metastatic melanoma, including recognition and management of rare presentations. Br. J. Dermatol. 2015, 173, 1024–1031. [Google Scholar] [CrossRef]
- Boudon, S.M.; Plappert-Helbig, U.; Odermatt, A.; Bauer, D. Characterization of Vemurafenib Phototoxicity in a Mouse Model. Toxicol. Sci. 2014, 137, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Gelot, P.; Dutartre, H.; Khammari, A.; Boisrobert, A.; Schmitt, C.; Deybach, J.-C.; Nguyen, J.-M.; Seité, S.; Dréno, B. Vemurafenib: An unusual UVA-induced photosensitivity. Exp. Dermatol. 2013, 22, 297–298. [Google Scholar] [CrossRef] [PubMed]
- Boussemart, L.; Boivin, C.; Claveau, J.; Tao, Y.G.; Tomasic, G.; Routier, E.; Mateus, C.; Deutsch, E.; Robert, C. Vemurafenib and Radiosensitization. JAMA Dermatol. 2013, 149, 855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Il’ichev, Y.V.; Schwörer, M.A.; Wirz, J. Photochemical Reaction Mechanisms of 2-Nitrobenzyl Compounds: Methyl Ethers and Caged ATP. J. Am. Chem. Soc. 2004, 126, 4581–4595. [Google Scholar] [CrossRef]
- Rhee, H.; Lee, J.-S.; Lee, J.; Joo, C.; Han, H.; Cho, M. Photolytic Control and Infrared Probing of Amide I Mode in the Dipeptide Backbone-Caged with the 4,5-Dimethoxy-2-nitrobenzyl Group. J. Phys. Chem. B 2008, 112, 2128–2135. [Google Scholar] [CrossRef]
- Sebej, P.; Solomek, T.; Hroudna, L.; Brancova, P.; Klan, P. Photochemistry of 2-Nitrobenzylidene Acetals. J. Org. Chem. 2009, 74, 8647–8658. [Google Scholar] [CrossRef]
- Morlière, P.; Boscá, F.; Silva, A.M.S.; Teixeira, A.; Galmiche, A.; Mazière, J.-C.; Nourry, V.; Ferreira, J.; Santus, R.; Filipe, P. A molecular insight into the phototoxic reactions observed with vemurafenib, a first-line drug against metastatic melanoma. Photochem. Photobiol. Sci. 2015, 14, 2119–2127. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. THEOCHEM 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Shkoor, M.; Mehanna, H.; Shabana, A.; Farhat, T.; Bani-Yaseen, A.D. Experimental and DFT/TD-DFT computational investigations of the solvent effect on the spectral properties of nitro substituted pyridino [3,4-c]coumarins. J. Mol. Liq. 2020, 313, 113509. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Transition (Donor→Acceptor) | E(2) kcal/mol | Species | Transition (Donor→Acceptor) | E(2) kcal/mol |
|---|---|---|---|---|---|
| VFB | lp(NP)→π*(C1-C2) | 42.0 | 2 | lp(NS)→σ*(S-O) | 10.2 |
| lp(NP)→π*(C3-C4) | 35.9 | lp(NS)→σ*(C5-C6) | 7.4 | ||
| lp(NS)→σ*(S-O) | 8.2 | lp(NS)→σ*(C5-C8) | 7.8 | ||
| lp(NS)→σ*(C5-C6) | 7.8 | lp(NS)→σ*(C′1-C′2) | 8.0 | ||
| 1 | lp(NP)→π*(C1-C2) | 41.3 | 2-I1 | lp(NS)→σ*(S-O) | 8.2 |
| lp(NP)→π*(C3-C4) | 37.0 | lp(NS)→σ*(C5-C6) | 6.3 | ||
| lp(NP)→π*(C′1-C′2) | 6.7 | lp(NS)→σ*(C5-C8) | 7.1 | ||
| 1-I1 | π(NP-C1)→π*(C′1-C′2) | 13.2 | lp(NS)→σ*(C′1-C′2) | 33.8 | |
| π(C′3-N′4)→π*(C′1-C′2) | 5.8 | 2-I2 | lp(NS)→σ*(C′1-O′6) | 17.6 | |
| 1-I2 | lp(NP)→σ*(C′1-O′6) | 16.1 | lp(O′6)→σ*(NS-C′1) | 8.4 | |
| 1-I3 | lp(NP)→σ*(C′1-O′6) | 5.6 | 2-I3 | lp(O′6)→σ*(NS-C′1) | 13.9 |
| lp(O′6)→σ*(NP-C′1) | 8.5 | 2-TS1 | lp(NS)→σ*(C′1-C′2) | 26.1 | |
| lp(O′6)→s*(H) | 24.7 | lp(O′6)→π*(C′1-C′2) | 9.4 | ||
| 1-TS1 | lp(NP)→π*(C′1-C′2) | 46.9 | 2-TS2 | lp(NS)→σ*(C′1-O′6) | 22.2 |
| lp(O′6)→π*(C′1-C′2) | 6.7 | 2-TS3 | lp(NP)→σ*(O′6-H) | 7.1 | |
| 1-TS2 | lp(NP)→σ*(C′1-O′6) | 16.4 | |||
| 1-TS3 | lp(NP)→σ*(O′6-H) | 6.6 |
| Species | 1 | 2 | ||||
|---|---|---|---|---|---|---|
| Np | Ns | C′1 | Np | Ns | C′1 | |
| VFB | −0.534 | −0.869 | ||||
| prodrug | −0.359 | −0.849 | −0.282 | −0.534 | −0.667 | −0.281 |
| I1 | −0.349 | −0.836 | 0.019 | −0.533 | −0.598 | −0.005 |
| TS1 | −0.336 | −0.832 | 0.123 | −0.533 | −0.600 | 0.057 |
| I2 | −0.382 | −0.836 | 0.231 | −0.534 | −0.663 | 0.217 |
| TS2 | −0.338 | −0.842 | 0.247 | −0.533 | −0.658 | 0.232 |
| I3 | −0.384 | −0.867 | 0.256 | −0.533 | −0.687 | 0.242 |
| TS3 | −0.591 | −0.839 | 0.381 | −0.546 | −0.832 | 0.361 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bani-Yaseen, A.D. Computational Study on the Mechanism of the Photouncaging Reaction of Vemurafenib: Toward an Enhanced Photoprotection Approach for Photosensitive Drugs. Molecules 2021, 26, 1846. https://doi.org/10.3390/molecules26071846
Bani-Yaseen AD. Computational Study on the Mechanism of the Photouncaging Reaction of Vemurafenib: Toward an Enhanced Photoprotection Approach for Photosensitive Drugs. Molecules. 2021; 26(7):1846. https://doi.org/10.3390/molecules26071846
Chicago/Turabian StyleBani-Yaseen, Abdulilah Dawoud. 2021. "Computational Study on the Mechanism of the Photouncaging Reaction of Vemurafenib: Toward an Enhanced Photoprotection Approach for Photosensitive Drugs" Molecules 26, no. 7: 1846. https://doi.org/10.3390/molecules26071846
APA StyleBani-Yaseen, A. D. (2021). Computational Study on the Mechanism of the Photouncaging Reaction of Vemurafenib: Toward an Enhanced Photoprotection Approach for Photosensitive Drugs. Molecules, 26(7), 1846. https://doi.org/10.3390/molecules26071846