
Nickel(II) Complex of N4 Schiff Base Ligand as a Building Block for a Conducting Metallopolymer with Multiple Redox States


Abstract
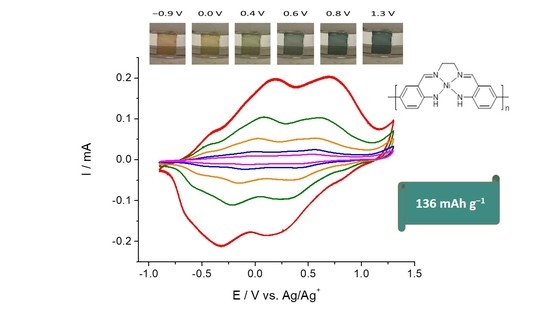
:
1. Introduction
2. Results
2.1. Electrochemical Studies of [NiAmben]
2.2. Combined Cyclic Voltammetry and Electrochemical Quartz Crystal Microbalance Characterization of [NiAmben] Polymerization and Redox Processes in Pol-[NiAmben]
2.3. In Situ Conductance Studies of Poly-[NiAmben]
2.4. In Situ UV-Vis Spectroelectrochemical Studies of Poly-[NiAmben]
3. Discussion
4. Materials and Methods
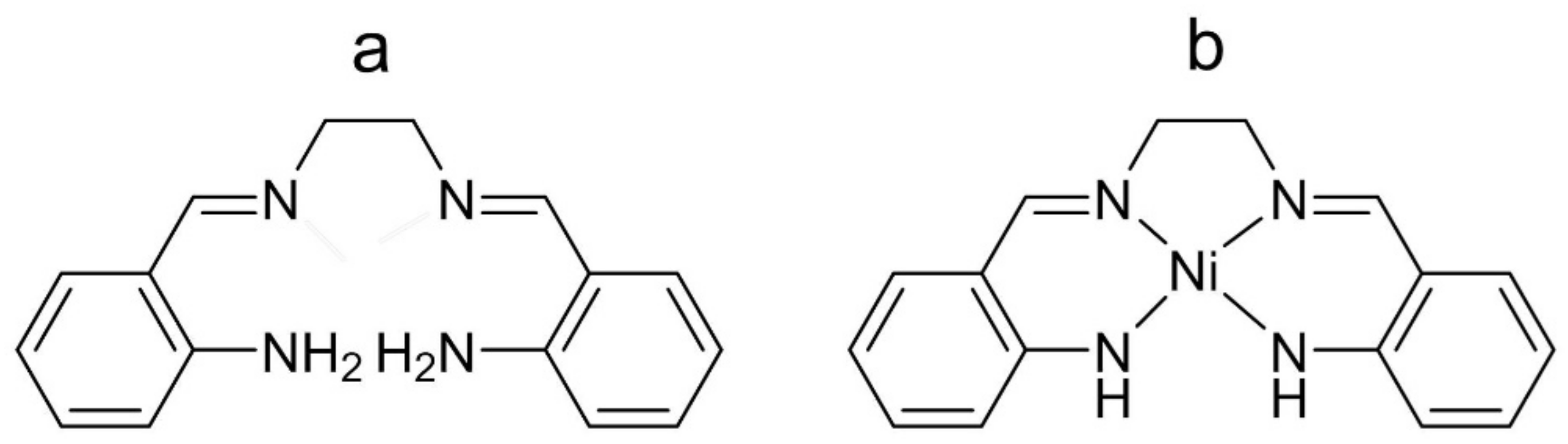
4.1. Chemicals and Synthesis
4.2. Cyclic Voltammetry and EQCM Studies
4.3. In Situ Conductance Measurements
4.4. UV-Vis Spectroscopy and In Situ UV-Vis Spectroelectrochemical Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Wang, J.; Dooner, M.; Clarke, J. Overview of current development in electrical energy storage technologies and the application potential in power system operation. Appl. Energy 2015, 137, 511–536. [Google Scholar] [CrossRef] [Green Version]
- Cuce, E.; Harjunowibowo, D.; Cuce, P.M. Renewable and sustainable energy saving strategies for greenhouse systems: A comprehensive review. Renew. Sustain. Energy Rev. 2016, 64, 34–59. [Google Scholar] [CrossRef]
- Mauger, A.; Julien, C. Surface modifications of electrode materials for lithium-ion batteries: Status and trends. Ionics 2014, 20, 751–787. [Google Scholar] [CrossRef]
- Mortimer, R.J. Electrochromic Materials. Annu. Rev. Mater. Res. 2011, 41, 241–268. [Google Scholar] [CrossRef]
- Xiang, J.; Ho, C.-L.; Wong, W.-Y. Metallopolymers for energy production, storage and conservation. Polym. Chem. 2015, 6, 6905–6930. [Google Scholar] [CrossRef]
- Wang, Y.; Astruc, D.; Abd-El-Aziz, A.S. Metallopolymers for advanced sustainable applications. Chem. Soc. Rev. 2019, 48, 558–636. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, L.; Wong, W.-Y. Energy materials based on metal Schiff base complexes. Coord. Chem. Rev. 2018, 355, 180–198. [Google Scholar] [CrossRef]
- Freire, C.; Nunes, M.; Pereira, C.; Fernandes, D.M.; Peixoto, A.F.; Rocha, M. Metallo(salen) complexes as versatile building blocks for the fabrication of molecular materials and devices with tuned properties. Coord. Chem. Rev. 2019, 394, 104–134. [Google Scholar] [CrossRef]
- Chepurnaya, I.A.; Karushev, M.P.; Alekseeva, E.V.; Lukyanov, D.A.; Levin, O.V. Redox-conducting polymers based on metal-salen complexes for energy storage applications. Pure Appl. Chem. 2020, 92, 1239–1258. [Google Scholar] [CrossRef]
- Vilas-Boas, M.; Freire, C.; De Castro, B.; Christensen, P.A.; Hillman, A.R. New Insights into the Structure and Properties of Electroactive Polymer Films Derived from [Ni(salen)]. Inorg. Chem. 1997, 36, 4919–4929. [Google Scholar] [CrossRef]
- Łępicka, K.; Pieta, P.; Shkurenko, A.; Borowicz, P.; Majewska, M.; Rosenkranz, M.; Avdoshenko, S.; Popov, A.A.; Kutner, W. Spectroelectrochemical Approaches to Mechanistic Aspects of Charge Transport in meso-Nickel(II) Schiff Base Electrochromic Polymer. J. Phys. Chem. C 2017, 121, 16710–16720. [Google Scholar] [CrossRef]
- Dmitrieva, E.; Rosenkranz, M.; Danilova, J.S.; Smirnova, E.A.; Karushev, M.P.; Chepurnaya, I.A.; Timonov, A.M. Radical formation in polymeric nickel complexes with N2O2 Schiff base ligands: An in situ ESR and UV–vis–NIR spectroelectrochemical study. Electrochim. Acta 2018, 283, 1742–1752. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Hesse, T.; Pfitzner, H.; Scholl, W.; Thielert, H. Innere Komplexsalze der Aldimin- und Azoreihe. J. Prakt. Chem. 1937, 149, 217–296. [Google Scholar] [CrossRef]
- Green, M.; Tasker, P.A. NN′-ethylenebis-(o-aminobenzylideneiminato)nickel(II) and its derivatives. J. Chem. Soc. A 1970, 2531–2539. [Google Scholar] [CrossRef]
- Green, M.; Tasker, P. New macrocyclic complexes of cobalt(II), nickel(II) and copper(II). Inorg. Chim. Acta 1971, 5, 65–69. [Google Scholar] [CrossRef]
- Higson, B.; McKenzie, E. The redox properties of some planar Schiff compounds of cobalt, nickel and copper. Inorg. Nucl. Chem. Lett. 1970, 6, 209–213. [Google Scholar] [CrossRef]
- Higson, B.M.; McKenzie, E.D. The structure, redox properties, and reactions of some planar [M II N4] chelate compounds of cobalt, nickel, and copper, and their oxidised products, including paramagnetic cobalt(III) species. J. Chem. Soc. Dalton Trans. 1972, 269. [Google Scholar] [CrossRef]
- Brewer, G.; Jasinski, J.; Mahany, W.; May, L.; Prytkov, S. Spin crossover complexes of iron(III) with an N4 tetradentate Schiff base ligand. Inorg. Chim. Acta 1995, 232, 183–187. [Google Scholar] [CrossRef]
- Busse, S.; Elias, H.; Fischer, J.; Poggemann, M.; Wannowius, K.J.; Boca, R. Kinetics and Mechanism of Metal Substitution and the Irving−Williams Series: Anion-Catalyzed Substitution of Nickel for Copper in Cu(amben) [=(N,N′-Ethylenebis(2-aminobenzaldiminato))copper(II)]. Inorg. Chem. 1998, 37, 3999–4005. [Google Scholar] [CrossRef]
- Kochem, A.; Gellon, G.; Leconte, N.; Baptiste, B.; Philouze, C.; Jarjayes, O.; Orio, M.; Thomas, F. Stable Anilinyl Radicals Coordinated to Nickel: X-ray Crystal Structure and Characterization. Chem. A Eur. J. 2013, 19, 16707–16721. [Google Scholar] [CrossRef] [PubMed]
- Shagisultanova, G.A.; Ardasheva, L.P. Electrochemical Polymerization of Ni(II) and Pd(II) Complexes with 1,2-Bis(o-aminobenzylidene)ethylenediamine. Russ. J. Coord. Chem. 2004, 30, 94–99. [Google Scholar] [CrossRef]
- Freire, C.; De Castro, B. Spectroscopic characterisation of electrogenerated nickel(III) species. Complexes with N2O2 Schiff-base ligands derived from salicylaldehyde. J. Chem. Soc. Dalton Trans. 1998, 1491–1498. [Google Scholar] [CrossRef]
- Santos, I.; Vilas-Boas, M.; Piedade, M.; Freire, C.; Duarte, M.; de Castro, B. Electrochemical and X-ray studies of nickel(II) Schiff base complexes derived from salicylaldehyde. Polyhedron 2000, 19, 655–664. [Google Scholar] [CrossRef]
- Kapturkiewicz, A.; Behr, B. Voltammetric studies of Co(salen) and Ni(salen) in nonaqueous solvents at Pt electrode. Inorg. Chim. Acta 1983, 69, 247–251. [Google Scholar] [CrossRef]
- Bradner, F.; Shapiro, J. Improvement in the quality of polypyrrole films prepared electrochemically on a mercury anode. Synth. Met. 1988, 26, 69–77. [Google Scholar] [CrossRef]
- Ferreira, C.; Aeiyach, S.; Delamar, M.; Lacaze, P. Electropolymerization of pyrrole on iron electrodes. J. Electroanal. Chem. Interfacial Electrochem. 1990, 284, 351–369. [Google Scholar] [CrossRef]
- Zotti, G.; Zecchin, S.; Schiavon, G.; Groenendaal, L. “Bert” Conductive and Magnetic Properties of 3,4-Dimethoxy- and 3,4-Ethylenedioxy-Capped Polypyrrole and Polythiophene. Chem. Mater. 2000, 12, 2996–3005. [Google Scholar] [CrossRef]
- Muench, S.; Wild, A.; Friebe, C.; Häupler, B.; Janoschka, T.; Schubert, U.S. Polymer-Based Organic Batteries. Chem. Rev. 2016, 116, 9438–9484. [Google Scholar] [CrossRef]
- Hager, M.D.; Esser, B.; Feng, X.; Schuhmann, W.; Theato, P.; Schubert, U.S. Polymer-Based Batteries—Flexible and Thin Energy Storage Systems. Adv. Mater. 2020, 32, 2000587. [Google Scholar] [CrossRef]
- Vilas-Boas, M.; Henderson, M.J.; Freire, C.; Hillman, A.R.; Vieil, E. A Combined Electrochemical Quartz-Crystal Microbalance Probe Beam Deflection (EQCM-PBD) Study of Solvent and Ion Transfers at a Poly[Ni(saltMe)]-Modified Electrode During Redox Switching. Chem. A Eur. J. 2000, 6, 1160–1167. [Google Scholar] [CrossRef]
- Vilas-Boas, M.; Santos, I.C.; Henderson, M.J.; Freire, C.; Hillman, A.R.; Vieil, E. Electrochemical Behavior of a New Precursor for the Design of Poly[Ni(salen)]-Based Modified Electrodes. Langmuir 2003, 19, 7460–7468. [Google Scholar] [CrossRef] [Green Version]
- Salinas, G.; Frontana-Uribe, B.A. Analysis of Conjugated Polymers Conductivity by in situ Electrochemical-Conductance Method. ChemElectroChem 2019, 6, 4105–4117. [Google Scholar] [CrossRef]
- Schiavon, G.; Sitran, S.; Zotti, G. A simple two-band electrode for in situ conductivity measurements of polyconjugated conducting polymers. Synth. Met. 1989, 32, 209–217. [Google Scholar] [CrossRef]
- Link, S.; Richter, T.; Yurchenko, O.; Heinze, J.; Ludwigs, S. Electrochemical Behavior of Electropolymerized and Chemically Synthesized Hyperbranched Polythiophenes. J. Phys. Chem. B 2010, 114, 10703–10708. [Google Scholar] [CrossRef]
- Chidsey, C.E.D.; Murray, R.W. Redox capacity and direct current electron conductivity in electroactive materials. J. Phys. Chem. 1986, 90, 1479–1484. [Google Scholar] [CrossRef]
- Ofer, D.; Crooks, R.M.; Wrighton, M.S. Potential dependence of the conductivity of highly oxidized polythiophenes, polypyrroles, and polyaniline: Finite windows of high conductivity. J. Am. Chem. Soc. 1990, 112, 7869–7879. [Google Scholar] [CrossRef]
- Zotti, G.; Schiavon, G. Spin and spinless conductivity in polypyrrole. Evidence for mixed-valence conduction. Chem. Mater. 1991, 3, 62–65. [Google Scholar] [CrossRef]
- Yurchenko, O.; Heinze, J.; Ludwigs, S. Electrochemically Induced Formation of Independent Conductivity Regimes in Polymeric Tetraphenylbenzidine Systems. ChemPhysChem 2010, 11, 1637–1640. [Google Scholar] [CrossRef]
- Malacrida, C.; Lu, Y.; Dirnberger, K.; Gámez-Valenzuela, S.; Delgado, M.C.R.; Ludwigs, S. Towards highly conducting bicarbazole redox polymer films with plateau-like conductivities. J. Mater. Chem. C 2020, 8, 15393–15405. [Google Scholar] [CrossRef]
- Heinze, J.; Frontana-Uribe, B.A.; Ludwigs, S. Electrochemistry of Conducting Polymers—Persistent Models and New Concepts†. Chem. Rev. 2010, 110, 4724–4771. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, C.P.; Zuo, Q. Oxidative electropolymerization of iron and ruthenium complexes containing aniline-substituted 2,2′-bipyridine ligands. Inorg. Chem. 1992, 31, 1607–1613. [Google Scholar] [CrossRef]
- Qiu, D.; Zhao, Q.; Bao, X.; Liu, K.; Wang, H.; Guo, Y.; Zhang, L.; Zeng, J.; Wang, H. Electropolymerization and characterization of an alternatively conjugated donor–acceptor metallopolymer: Poly-[Ru(4′-(4-(Diphenylamino)phenyl)-2,2′:6′,2″-Terpyridine)2]2+. Inorg. Chem. Commun. 2011, 14, 296–299. [Google Scholar] [CrossRef]
- Witker, D.; Reynolds, J.R. Soluble Variable Color Carbazole-Containing Electrochromic Polymers. Macromolecules 2005, 38, 7636–7644. [Google Scholar] [CrossRef]
- Clarke, R.M.; Herasymchuk, K.; Storr, T. Electronic structure elucidation in oxidized metal–salen complexes. Coord. Chem. Rev. 2017, 352, 67–82. [Google Scholar] [CrossRef]
- Peverari, C.R.; David-Parra, D.N.; Barsan, M.M.; Teixeira, M.F. Mechanistic study of the formation of multiblock π-conjugated metallopolymer. Polyhedron 2016, 117, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Comisso, N.; Daolio, S.; Mengoli, G.; Salmaso, R.; Zecchin, S.; Zotti, G. Chemical and electrochemical synthesis and characterization of polydiphenylamine and poly-N-methylaniline. J. Electroanal. Chem. Interfacial Electrochem. 1988, 255, 97–110. [Google Scholar] [CrossRef]
- Guay, J.; Dao, L.H. Formation of poly(4-phenylaniline) by electropolymerization of 4-aminobiphenyl or diphenylamine. J. Electroanal. Chem. Interfacial Electrochem. 1989, 274, 135–142. [Google Scholar] [CrossRef]
- Guay, J.; Paynter, R.; Dao, L.H. Synthesis and characterization of poly(diarylamines): A new class of electrochromic conducting polymers. Macromolecules 1990, 23, 3598–3605. [Google Scholar] [CrossRef]
- Wen, T.-C.; Chen, J.-B.; Gopalan, A. Soluble and methane sulfonic acid doped poly(diphenylamine)—Synthesis and characterization. Mater. Lett. 2002, 57, 280–290. [Google Scholar] [CrossRef]
- Visy, C.; Lakatos, M.; Szücs, Á.; Novák, M. Separation of faradaic and capacitive current regions in the redox transformation of poly(3-methylthiophene) with the exclusion of overoxidation processes. Electrochim. Acta 1997, 42, 651–657. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Breith, E.; Lübbe, E.; Tsumaki, T. Tricyclische orthokondensierte Nebenvalenzringe. Justus Liebig’s Ann. Chem. 1933, 503, 84–130. [Google Scholar] [CrossRef]
- Sauerbrey, G. Verwendung von Schwingquarzen zur Wägung Dünner Schichten und zur Mikrowägung. Z. Phys. 1959, 155, 206–222. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrolyte Solvent | Potential Range, V | Qredox/Qpoly |
|---|---|---|
| EC/DEC | −0.9 to 0.5 | 0.03 |
| −0.9 to 1.3 | 0.18 | |
| DCE | −0.9 to 0.65 | 0.22 |
| −0.9 to 1.3 | 0.55 |
| Scan Rate, vs (V s−1) | Electrons Exchanged Per Repeat Unit (Oxidation), nox | Electrons Exchanged Per Repeat Unit (Reduction), nred |
|---|---|---|
| 0.005 | 2.35 | 1.63 |
| 0.010 | 2.09 | 1.62 |
| 0.025 | 1.85 | 1.62 |
| 0.050 | 1.78 | 1.59 |
| 0.100 | 1.68 | 1.57 |
| 0.200 | 1.60 | 1.53 |
| Potential Range, V | Effective Molar Mass of Electrolyte Species, Mox (g mol−1) | Electrons Exchanged Per Repeat Unit, nox |
|---|---|---|
| −0.57 to −0.16 | 3 | 0.25 |
| −0.16 to 0.05 | 18 | 0.25 |
| 0.05 to 0.45 | 42 | 0.5 |
| 0.45 to 1.00 | 59 | 0.63 |
| 1.00 to 0.93 (reverse scan) | 17 | 0.46 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karushev, M.; Smirnova, E.; Chepurnaya, I. Nickel(II) Complex of N4 Schiff Base Ligand as a Building Block for a Conducting Metallopolymer with Multiple Redox States. Molecules 2021, 26, 2646. https://doi.org/10.3390/molecules26092646
Karushev M, Smirnova E, Chepurnaya I. Nickel(II) Complex of N4 Schiff Base Ligand as a Building Block for a Conducting Metallopolymer with Multiple Redox States. Molecules. 2021; 26(9):2646. https://doi.org/10.3390/molecules26092646
Chicago/Turabian StyleKarushev, Mikhail, Evgenia Smirnova, and Irina Chepurnaya. 2021. "Nickel(II) Complex of N4 Schiff Base Ligand as a Building Block for a Conducting Metallopolymer with Multiple Redox States" Molecules 26, no. 9: 2646. https://doi.org/10.3390/molecules26092646
APA StyleKarushev, M., Smirnova, E., & Chepurnaya, I. (2021). Nickel(II) Complex of N4 Schiff Base Ligand as a Building Block for a Conducting Metallopolymer with Multiple Redox States. Molecules, 26(9), 2646. https://doi.org/10.3390/molecules26092646