
Stemodane Diterpenes and Diterpenoids: Isolation, Structure Elucidation, Biogenesis, Biosynthesis, Biological Activity, Biotransformations, Metabolites and Derivatives Biological Activity, Rearrangements †

 , ,
, ,  and
and
Abstract
:1. Introduction
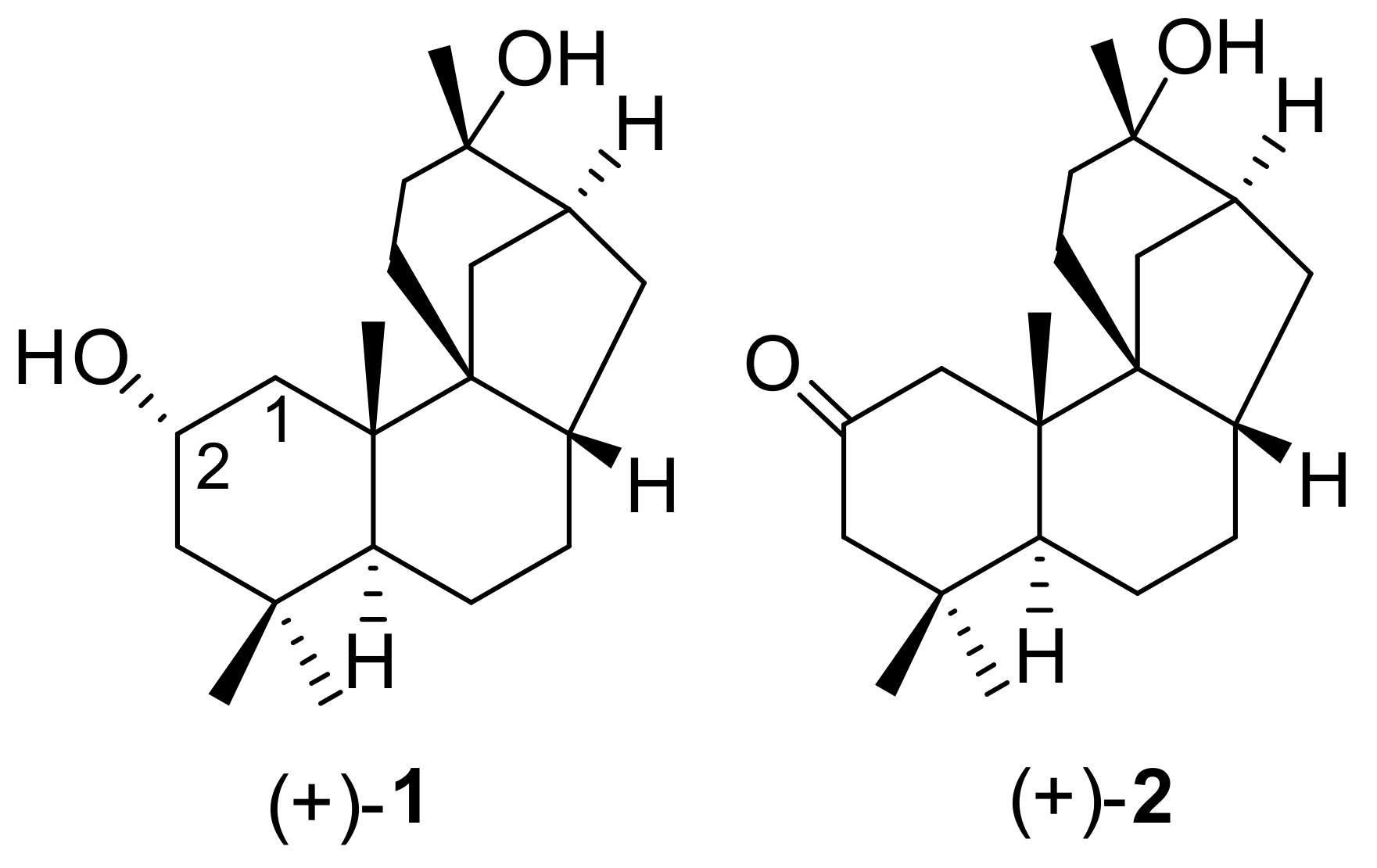
2. Isolation and Structure Elucidation
3. Biogenesis
4. Biosynthesis
5. Biological Activity
6. Biotransformations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Starting Material | Microorganism | Metabolites | Reference/Year |
|---|---|---|---|---|
| 1 | (+)-1 | Cunninghamella echinulata | (−)-32 (−)-33 34 | [65]/1991 |
| 2 | (+)-1 | Polyangium cellulosum | (−)-35 (−)-36 | [65]/1991 |
| 3 | (+)-1 | Rhizopus arrhizus ATCC 11145 | (−)-32 (−)-33 (−)-37 38 | [66]/1991 |
| 4 | (+)-1 | Streptomyces sp. NRRL 5691 | (−)-39 (−)-40 41 | [66]/1991 |
| 5 | (+)-1 | Cephalosporium aphidicola | (−)-32 (−)-33 (−)-35 (−)-37 | [67]/1994 |
| 6 | (+)-2 | Cephalosporium aphidicola | 42 43 | [67]/1994 |
| 7 | (+)-1 | Beauveria bassiana ATCC 7159 | (−)-37 | [68]/2001 |
| 8 | (+)-2 | Beauveria bassiana ATCC 7159 | (+)-42 | [68]/2001 |
| 9 | (+)-44 | Beauveria bassiana ATCC 7159 | (+)-45 | [68]/2001 |
| 10 | (+)-1 | Aspergillus niger ATCC 9142 | (+)-13 (−)-33 46 | [69]/2002 |
| 11 | (+)-2 | Aspergillus niger ATCC 9142 | (+)-42 (−)-47 | [69]/2002 |
| 12 | (+)-1 | Rhizopus oryzae ATCC 11145 | (+)-33 (−)-48 | [12]/2004 |
| 13 | (+)-2 | Rhizopus oryzae ATCC 11145 | (+)-49 (+)-50 | [12]/2004 |
| 14 | (+)-51 | Rhizopus oryzae ATCC 11145 | (+)-52 | [12]/2004 |
| 15 | (+)-53 | Rhizopus oryzae ATCC 11145 | (−)-54 | [12]/2004 |
| 16 | (+)-12 | Mucor plumbeus | 55 56 57 58 59 60 61 62 63 64 65 | [72]/2004 |
| 17 | (−)-11 | Mucor plumbeus | 66 67 68 | [72]/2004 |
| 18 | (+)-1 | Mucor plumbeus | (+)-13 (+)-34 69 (−)-70 | [73]/2005 |
| 19 | (+)-2 | Mucor plumbeus | (+)-49 (+)-71 | [73]/2005 |
| 20 | (+)-1 | Whetzelinia sclerotiorum ATCC. 18687 | 33 (−)-70 | [73]/2005 |
| 21 | (+)-2 | Whetzelinia sclerotiorum ATCC. 18687 | (+)-50 | [73]/2005 |
| 22 | (+)-1 | Cunninghamella echinulata var. elegans | (+)-13 32 33 | [74]/2006 |
| 23 | (+)-44 | Cunninghamella echinulata var. elegans | (−)-72 (+)-73 | [74]/2006 |
| 24 | (+)-2 | Cunninghamella echinulata var. elegans | (+)-50 (+)-74 | [74]/2006 |
| 25 | (+)-1 | Phanerochaete chrysosporium | (+)-13 33 (−)-70 | [74]/2006 |
| 26 | (+)-44 | Phanerochaete chrysosporium | untransformed | [74]/2006 |
| 27 | (+)-2 | Phanerochaete chrysosporium | 42 | [74]/2006 |
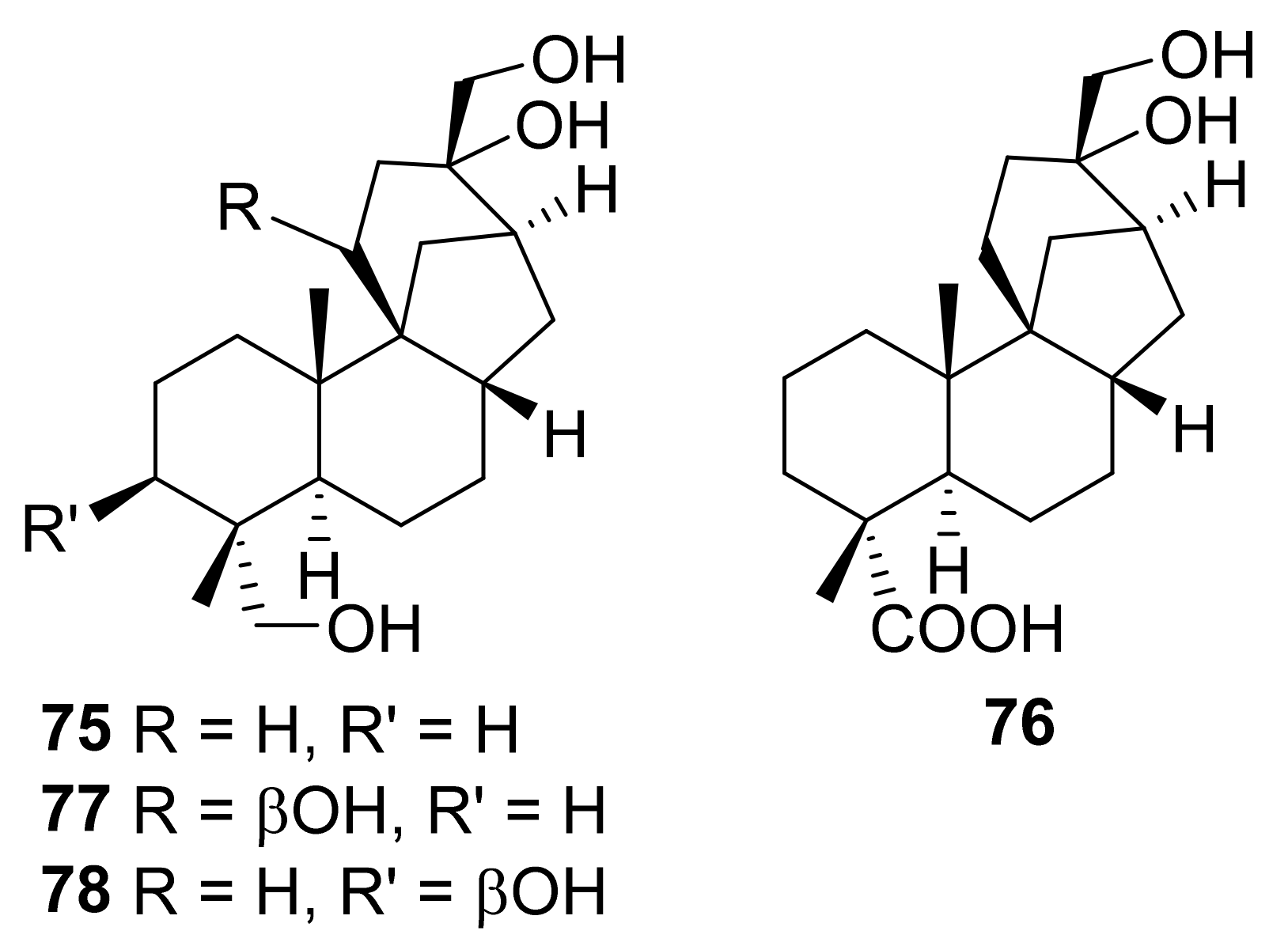
| 28 | 12 | Cephalosporium aphidicola | 56 65 75 76 77 78 | [75]/2012 |
| 29 | (+)-1 | Cyathus africanus | (+)-2 | [76]/2012 |
| 30 | (+)-2 | Cyathus africanus | (+)-1 | [76]/2012 |
| 31 | (+)-6 | Cyathus africanus | no metabolites | [76]/2012 |
| 32 | (+)-19 | Cyathus africanus | (+)-79 | [76]/2012 |
7. Stemodane Diterpenoid Metabolites and Derivatives Biological Activity
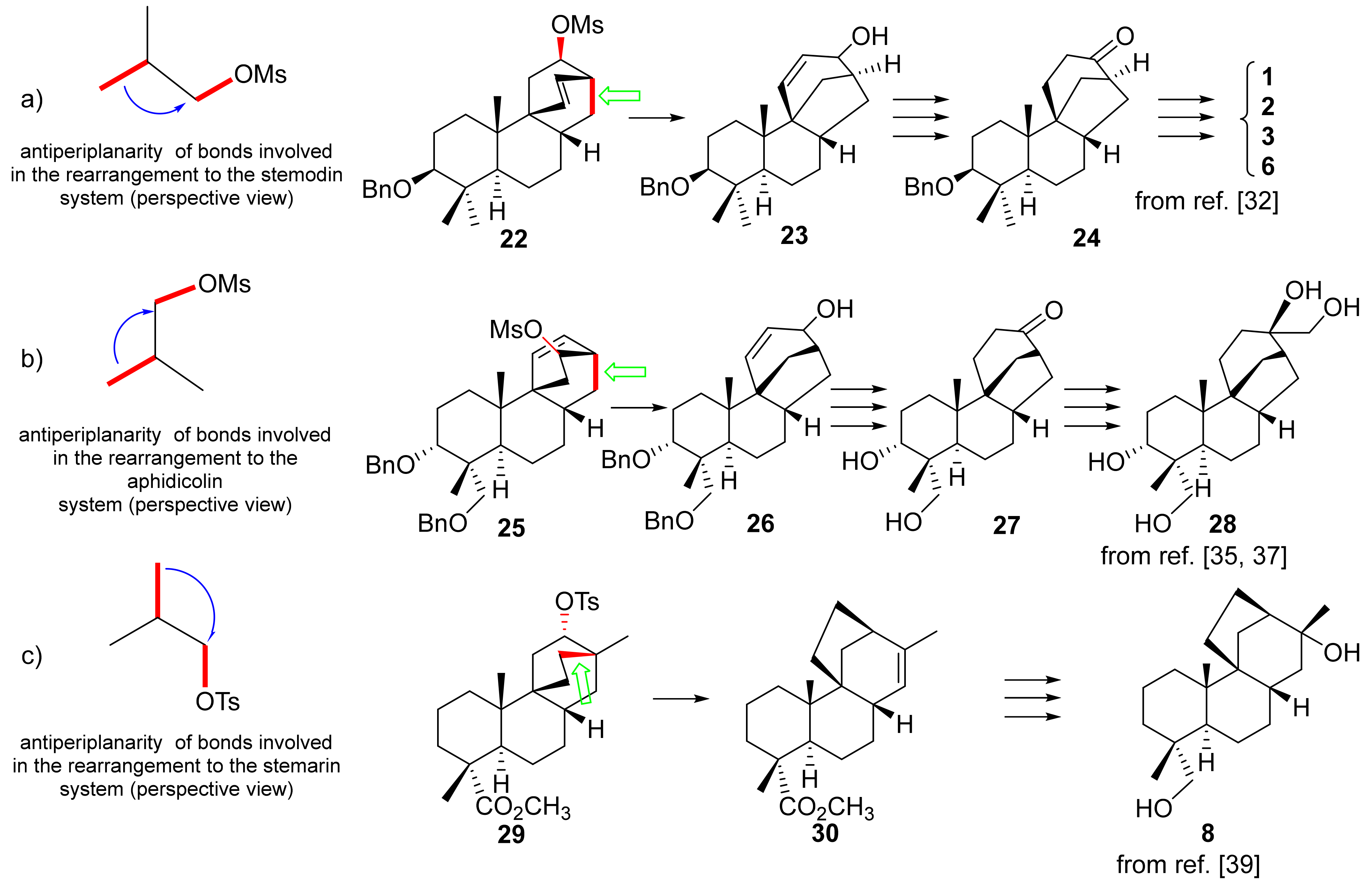
8. Rearrangements
9. Conclusions
Funding
Conflicts of Interest
References
- Manchand, P.S.; White, J.D.; Wright, H.; Clardy, J. Structures of Stemodin and Stemodinone. J. Am. Chem. Soc. 1973, 95, 2705–2706. [Google Scholar] [CrossRef]
- Hufford, C.D.; Guerrero, R.O.; Doorenbos, N.J. Two New Diterpenes from Stemodia maritima L. J. Pharm. Sci. 1976, 65, 778–780. [Google Scholar] [CrossRef] [PubMed]
- Manchand, P.S.; Blount, J.F. X-Ray Structure and Absolute Stereochemistry of Stemarin, a Diterpene with a New Skeleton. J. Chem. Soc. Chem. Commun. 1975, 21, 894–895. [Google Scholar] [CrossRef]
- Kelly, R.B.; Harley, M.L.; Alward, S.J.; Manchand, P.S. Total synthesis of racemic 2-desoxystemodinone and stemodinol; the identity of natural “stemodinol” with stemarin. Can. J. Chem. 1982, 60, 675–677. [Google Scholar] [CrossRef]
- Kelly, R.B.; Harley, M.L.; Alward, S.J.; Rej, R.N.; Gowda, G.; Mukhopadhyay, A.; Manchand, P.S. Total synthesis of the stemodane-type diterpenoids, (±)-2-desoxystemodinone, (+)-2-desoxystemodinone, and (±)-stemodinol. Can. J. Chem. 1983, 61, 269–275. [Google Scholar] [CrossRef]
- Da Silva, F.R.L.; Rodrigues, F.E.A.; Gomes, A.R.S.; Arriaga, A.M.C.; Mafezoli, J.; Lemos, T.L.G.; de Almeida, M.C.S.; Santiago, G.M.P.; Braz-Filho, R.; da Costa, J.G.M.; et al. Phytochemical Study, Antioxidant and Antibacterial Activities of Stemodia maritima. Quím. Nova 2014, 37, 1474–1478. [Google Scholar]
- Hufford, C.D. 1H- and 13C-NMR assignments for the Stemodia Diterpenes, Stemodin, Stemodinone, and Maritimol. J. Nat. Prod. 1988, 51, 367–369. [Google Scholar] [CrossRef]
- Chamy, M.C.; Piovano, M.; Garbarino, J.A.; Gambaro, V. Stemodane diterpenoids from Stemodia Chilensis. Phytochemistry 1991, 30, 1719–1721. [Google Scholar] [CrossRef]
- Leonelli, F.; Mostarda, A.; De Angelis, L.; Lamba, D.; Demitri, N.; La Bella, A.; Ceccacci, F.; Migneco, L.M.; Marini Bettolo, R. Proof of the Structure of the Stemodia chilensis Tetracyclic Diterpenoid (+)-19-Acetoxystemodan-12-ol by Synthesis from (+)-Podocarpic Acid: X-ray Structure Determination of a Key Intermediate. J. Nat. Prod. 2016, 79, 1155–1159. [Google Scholar] [CrossRef]
- Hufford, C.D.; Oguntimein, B.O.; Muhammad, I. New Stemodane Diterpenes from Stemodia maritima. J. Nat. Prod. 1992, 55, 48–52. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Wang, X.-Y.; Lai, G.-F.; Lu, C.-H.; Luo, S.-D. Three New Sesquiterpenoids from the Aerial Parts of Homalomena occulta. Chem. Biodivers. 2007, 4, 925–931. [Google Scholar] [CrossRef]
- Martin, G.D.A.; Reynolds, W.F.; Reese, P.B. Investigation of the importance of the C-2 oxygen function in the transformation of stemodin analogues by Rhizopus oryzae ATCC 11145. Phytochemistry 2004, 65, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.E.A.; Lima, J.Q.; de Oliveira, M.d.C.F.; Vasconcelos, J.N.; Santiago, G.M.P.; Mafezoli, J.; Braz-Filho, R.; Arriaga, A.M.C. Diterpene and other Constituents from Stemodia maritima (Scrophulariaceae). J. Braz. Chem. Soc. 2010, 21, 1581–1586. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Niu, H.; Guan, C.; Sun, F.; Yang, L.; Fu, Y. A New Stemodane Diterpenoid from the Stems and Leaves of Trigonostemon heterophyllus. Chin. J. Org. Chem. 2018, 38, 3137–3140. [Google Scholar] [CrossRef]
- Wenkert, E. Structural and Biogenetic Relationships in the Diterpene Series. Chem. Ind. 1955, 1, 282–284. [Google Scholar]
- MacMillan, J.; Beale, M.H. Diterpene Biosynthesis. In Comprehensive Natural Products Chemistry: Isoprenoids Including Carotenoids and Steroids; Barton, D., Nakanishi, K., Eds.; Pergamon Press: Oxford, UK, 1999; Volume 2, pp. 238–240. [Google Scholar]
- Oikawa, H.; Toshima, H.; Ohashi, S.; König, W.A.; Kenmoku, H.; Sassa, T. Diversity of diterpene hydrocarbons in fungus Phoma betae. Tetrahedron Lett. 2001, 42, 2329–2332. [Google Scholar] [CrossRef]
- Valenta, Z.; Wiesner, K. Biogenetic Interrelationships of Diterpene Alkaloids. Chem. Ind. 1956, 17, 354. [Google Scholar]
- Wiesner, K.; Valenta, Z. Recent Progress in the Chemistry of the Aconite-Garrya Alkaloids. In Progress in the Chemistry of Organic Natural Products; Zechmeister, L., Ed.; Springer: Wien, Austria, 1958; Volume XVI, pp. 26–89. [Google Scholar]
- Cookson, R.C.; Trevett, M.E. Aconitum and Delphinium Alkaloids. Part II. Interrelation of the Functional Groups of Delpheline. J. Chem. Soc. 1956, 62, 3121–3129. [Google Scholar] [CrossRef]
- Wiesner, K.; Tsai, T.Y.R.; Huber, K.; Bolton, S.E.; Vlahov, R. Total Synthesis of Talatisamine, a Delphinine Type Alkaloid. J. Am. Chem. Soc. 1974, 96, 4990–4992. [Google Scholar] [CrossRef]
- Wiesner, K. The Total Synthesis of Racemic Talatisamine. Pure Appl. Chem. 1975, 41, 93–112. [Google Scholar] [CrossRef]
- Tsai, T.Y.R.; Tsai, C.S.J.; Sy, W.W.; Shanbhag, M.N.; Liu, W.C.; Lee, S.F.; Wiesner, K. A Stereospecific Total Synthesis of Chasmanine. Heterocycles 1977, 7, 217–226. [Google Scholar]
- Wiesner, K. Centenary Lecture. Systematic Development of Strategy in the Synthesis of Polycyclic Polysubstituted Natural Products: The Aconite Alkaloids. Chem. Soc. Rev. 1977, 6, 413–430. [Google Scholar] [CrossRef]
- Atwal, K.S.; Marini Bettolo, R.; Sanchez, I.H.; Tsai, T.Y.R.; Wiesner, K. On the construction of the C/D ring systems of chasmanine and napelline by diene addition. Can. J. Chem. 1978, 56, 1102–1113. [Google Scholar] [CrossRef]
- Tsai, T.Y.R.; Nambiar, K.P.; Krikorian, D.; Botta, M.; Marini Bettolo, R.; Wiesner, K. A new synthesis of chasmanine and 13-desoxydelphonine: A preferred route to the aromatic intermediate. Can. J. Chem. 1979, 57, 2124–2134. [Google Scholar] [CrossRef]
- Wiesner, K. Total Synthesis of Delphinine-Type Alkaloids by Simple, Fourth Generation Methods. Pure Appl. Chem. 1979, 51, 689–703. [Google Scholar] [CrossRef]
- Sethi, S.P.; Atwal, K.S.; Marini Bettolo, R.; Tsai, T.Y.R.; Wiesner, K. A stereospecific synthesis of napelline. Can. J. Chem. 1980, 58, 1889–1891. [Google Scholar] [CrossRef]
- van Tamelen, E.E.; Carlson, J.G.; Russell, R.K.; Zawacky, S.R. Total Synthesis of (±)-Maritimol. J. Am. Chem. Soc. 1981, 103, 4615–4616. [Google Scholar] [CrossRef]
- Bravetti, D.; Marini Bettolo, R.; Lupi, A. On the Construction of the C/D-Ring Systems of Aphidicolin and Stemodin; a Regio and Stereospecific Synthesis of 17-Noraphidicolan-16-one and 17-Norstemodan-16-one. Helv. Chim. Acta 1982, 65, 371–376. [Google Scholar] [CrossRef]
- La Bella, A.; Leonelli, F.; Migneco, L.M.; Marini Bettolo, R. (+)-Podocarpic Acid as Chiral Template in the Synthesis of Aphidicolane, Stemodane and Stemarane Diterpenoids. Molecules 2016, 21, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marini Bettolo, R.; Tagliatesta, P.; Lupi, A.; Bravetti, D. A Stereoselective Total Synthesis of (±)-Maritimol, (±)-2-Deoxystemodinone, (±)-Stemodinone, and (±)-Stemodin. Helv. Chim. Acta 1983, 66, 760–770. [Google Scholar] [CrossRef]
- Lupi, A.; Patamia, M.; Grgurina, I.; Marini Bettolo, R.; Di Leo, O.; Gioia, P.; Antonaroli, S. Biogenetic-Type Total Synthesis of (±)-2-Deoxystemodinone. Helv. Chim. Acta 1984, 67, 2261–2263. [Google Scholar] [CrossRef]
- Van Tamelen, E.E.; Zawacky, S.R.; Russell, R.K.; Carlson, J.G. Biogenetic-Type Total Synthesis of (±)-Aphidicolin. J. Am. Chem. Soc. 1983, 105, 142–143. [Google Scholar] [CrossRef]
- Marini Bettolo, R.; Tagliatesta, P.; Lupi, A.; Bravetti, D. A Total Synthesis of Aphidicolin: Stereospecific Synthesis of (±)-3α,18-Dihydroxy-17-noraphidicolan-16-one. Helv. Chim. Acta 1983, 66, 1922–1928. [Google Scholar] [CrossRef]
- Kelly, R.B.; Lal, G.S.; Gowda, G.; Rej, R.N. A synthesis of (+)-aphidicol-15-ene. Can. J. Chem. 1984, 62, 1930–1932. [Google Scholar] [CrossRef]
- Lupi, A.; Patamia, M.; Marini Bettolo, R. A Total Syntesis of (±)-Aphidicolin: Regio- and Stereoselective Conversion of 3α,18-Di-O-benzyl-17-nor-14-aphidicolen-16-one into (±)Aphidicolin. Helv. Chim. Acta 1988, 71, 872–875. [Google Scholar] [CrossRef]
- Marini Bettolo, R. The Total Synthesis of the Antiviral Agent Aphidicolin and Related Diterpenes. Korean J. Med. Chem. 1996, 6, 329–332. [Google Scholar]
- Kelly, R.B.; Harley, M.L.; Alward, S.J. A total synthesis of (±)-stemarin, a diterpenoid with a unique bicyclic C/D ring system. Can. J. Chem. 1980, 58, 755–756. [Google Scholar] [CrossRef]
- Berettoni, M.; Marini Bettolo, R.; Montanari, V.; Prencipe, T.; Romeo, S. Studies for a Diastereoselective Synthesis of the Tetracyclic Diterpenic Diol Stemarin: A Model Study for a New Preparation of the Key Intermediate and the Synthesis of (+)-18-Deoxystemarin. Helv. Chim. Acta 1991, 74, 1671–1678. [Google Scholar] [CrossRef]
- Leonelli, F.; Caschera, B.; Silvestri, L.; Prastaro, A.; Corso, G.; Ceccacci, F.; La Bella, A.; Migneco, L.M.; Marini Bettolo, R. Synthesis of (+)-13-Stemarene and (+)-18-Deoxystemarin: Expeditious Preparation of the Key 6-exo-Hydroxybicyclo[2.2.2]octan-2-one Ethylene Dithioacetal. Helv. Chim. Acta 2008, 91, 598–607. [Google Scholar] [CrossRef]
- Leonelli, F.; Latini, V.; Trombetta, A.; Bartoli, G.; Ceccacci, F.; La Bella, A.; Sferrazza, A.; Lamba, D.; Migneco, L.M.; Marini Bettolo, R. Regio- and Diastereoselective Synthesis and X-ray Structure Determination of (+)-2-Deoxyoryzalexin S from (+)-Podocarpic Acid. Structural Nonidentity with Its Nominal Natural Isolated Enantiomer. J. Nat. Prod. 2012, 75, 1944–1950. [Google Scholar] [CrossRef] [PubMed]
- Leonelli, F.; Blesi, F.; Dirito, P.; Trombetta, A.; Ceccacci, F.; La Bella, A.; Migneco, L.M.; Marini Bettolo, R. Diastereoselective Total Synthesis of (+)-13-Stemarene by Fourth Generation Methods: A Formal Total Synthesis of (+)-18-Deoxystemarin. J. Org. Chem. 2011, 76, 6871–6876. [Google Scholar] [CrossRef]
- Leonelli, F.; Valletta, A.; Migneco, L.M.; Marini Bettolo, R. Stemarane Diterpenes and Diterpenoids. Int. J. Mol. Sci. 2019, 20, 2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walborsky, H.M.; Baum, M.E.; Youssef, A.A. Acetolysis of Bicyclo[2.2.2]octyl-2 p-Bromobenzenesulfonate and the Absolute Configurations of Bicyclo[2.2.2]octanol-2 and cis- and trans-Bicyclo[3.2.1]octanol-2. J. Am. Chem. Soc. 1961, 83, 988–993. [Google Scholar] [CrossRef]
- Goering, H.L.; Greiner, R.W.; Sloan, M.F. Ionic Reactions in Bicyclic Systems. I. The Preparation and Assignment of Configuration of the Isomeric Bicyclo[3.2.1]oct-3-en-2-ols and Bicyclo[3.2.1]octan-2-ols. J. Am. Chem. Soc. 1961, 83, 1391–1397. [Google Scholar] [CrossRef]
- Goering, H.L.; Sloan, M.F. Ionic Reactions in Bicyclic Systems. II. Carbonium Ion Reactions in Bicyclo[2.2.2]octane and Bicyclo[3.2.1]octane Derivatives. J. Am. Chem. Soc. 1961, 83, 1397–1401. [Google Scholar] [CrossRef]
- Goering, H.L.; Sloan, M.F. Ionic Reactions in Bicyclic Systems. III. Solvolysis of Bicycloöctanyl and Bicycloöctenyl p-Toluenesulfonates. J. Am. Chem. Soc. 1961, 83, 1992–1999. [Google Scholar] [CrossRef]
- Kraus, W.; Chassin, C.; Chassin, R.; Schmutte, P. Bicyclische Verbindungen, XVIII Solvolytische und reduktive Umlagerung von Bicyclo[2.2.2]octyltosylaten. Liebigs Ann. Chem. 1970, 738, 97–112. [Google Scholar] [CrossRef]
- Hong, Y.J.; Tantillo, D.J. A Maze of Dyotropic Rearrangements and Triple Shifts: Carbocation Rearrangements Connecting Stemarene, Stemodene, Betaerdene, Aphidicolene, and Scopadulanol. J. Org. Chem. 2018, 83, 3780–3793. [Google Scholar] [CrossRef] [PubMed]
- Brundret, K.M.; Dalziel, W.; Hesp, B.; Jarvis, J.A.J.; Neidle, S. X-Ray Crystallographic Determination of the Structure of the Antibiotic Aphidicolin: A Tetracyclic Diterpenoid Containing a New Ring System. J. Chem. Soc. Chem. Commun. 1972, 18, 1027–1028. [Google Scholar] [CrossRef]
- Dalziel, W.; Hesp, B.; Stevenson, K.M.; Jarvis, J.A.J. The Structure and Absolute Configuration of the Antibiotic Aphidicolin: A Tetracyclic Diterpenoid Containing a New Ring System. J. Chem. Soc. Perkin I 1973, 1, 2841–2851. [Google Scholar] [CrossRef]
- Adams, M.R.; Bu’Lock, J.D. Biosynthesis of the Diterpene Antibiotic Aphidicolin, by Radioisotope and 13C Nuclear Magnetic Resonance Methods. J. Chem. Soc. Chem. Commun. 1975, 10, 389–391. [Google Scholar] [CrossRef]
- Ackland, M.J.; Hanson, J.R.; Yeoh, B.L.; Ratcliffe, A.H. Studies in Terpenoid Biosynthesis. Part 32. The Incorporation of Aphidicol-16-ene and Aphidicolan-16-ol into the Diterpenoid Aphidicolin by the Fungus Cephalosporium aphidicola. J. Chem. Soc. Perkin Trans. I 1985, 1, 2705–2707. [Google Scholar] [CrossRef]
- Bohlmann, J.; Meyer-Gauen, G.; Croteau, R. Plant terpenoid synthases: Molecular biology and phylogenetic analysis. Proc. Natl. Acad. Sci. USA 1998, 95, 4126–4133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christianson, D.W. Structural Biology and Chemistry of the Terpenoid Cyclases. Chem. Rev. 2006, 106, 3412–3442. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wilderman, P.R.; Morrone, D.; Xu, J.; Roy, A.; Margis-Pinheiro, M.; Upadhyaya, N.M.; Coates, R.M.; Peters, R.J. Functional characterization of the rice kaurene synthase-like gene family. Phytochemistry 2007, 68, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Morrone, D.; Jin, Y.; Xu, M.; Choi, S.-Y.; Coates, R.M.; Peters, R.J. An unexpected diterpene cyclase from rice: Functional identification of a stemodene synthase. Arch. Biochem. Biophys. 2006, 448, 133–140. [Google Scholar] [CrossRef]
- Austin, D.F. Florida Ethnobotany; CRC Press: Boca Raton, FL, USA, 2004; p. 162. [Google Scholar]
- Garbarino, J.A.; Chamy, M.C.; Piovano, M. Chemistry of the Calceolaria Genus. Structural and Biological Aspects. Molecules 2000, 5, 302–303. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, D.M.; Riet-Correa, F.; Medeiros, R.M.T.; de Oliveira, O.F. Plantas tóxica para ruminantes e eqüídeos no Seridó Ocidental e Oriental do Rio Grande do Norte. Pesq. Vet. Bras. 2006, 26, 223–236. [Google Scholar] [CrossRef] [Green Version]
- Arriaga, A.M.C.; Rodrigues, F.E.A.; Lemos, T.L.G.; de Oliveira, M.D.C.F.; Lima, J.Q.; Santiago, G.M.P.; Braz-Filho, R.; Mafezoli, J. Composition and Larvicidal Activity of Essential Oil from Stemodia maritima L. Nat. Prod. Commun. 2007, 2, 1237–1239. [Google Scholar] [CrossRef]
- Teixeira, A.H.; de Oliveira Freire, J.M.; de Sousa, L.H.T.; Parente, A.T.; de Sousa, N.A.; Arriaga, A.M.C.; Lopes da Silva, F.R.; Melo, I.M.; Castro da Silva, I.I.; Pereira, K.M.A.; et al. Stemodia maritima L. Extract Decreases Inflammation, Oxidative Stress, and Alveolar Bone Loss in an Experimental Periodontitis Rat Model. Front. Physiol. 2017, 8, 988. [Google Scholar] [CrossRef] [Green Version]
- de Sousa, R.S.; da Silva, J.D.A.G.; de Oliveira Borba, E.F.; de Sousa, M.G.O.; da Silva, S.D.J.L.; da Silva, T.G. Análise Fitoquímica e Efeito Gastroprotetor do Extrato Hexânico de Stemodia Maritima, L.J. Biol. Pharm. Agric. Manag. 2021, 17, 456–474. [Google Scholar]
- Badria, F.A.; Hufford, C.D. Microbial Transformations of Stemodin, a Stemodia Diterpene. Phytochemistry 1991, 30, 2265–2268. [Google Scholar] [CrossRef]
- Hufford, C.D.; Badria, F.A.; Abou-Karam, M.; Shier, W.T.; Rogers, R.D. Preparation, Characterization, and Antiviral Activity of Microbial Metabolites of Stemodin. J. Nat. Prod. 1991, 54, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.R.; Reese, P.B.; Takahashi, J.A.; Wilson, M.R. Biotransformation of Some Stemodane Diterpenoids by Cephalosporium aphidicola. Phytochemistry 1994, 36, 1391–1393. [Google Scholar] [CrossRef]
- Buchanan, G.O.; Reese, P.B. Biotransformation of diterpenes and diterpene derivatives by Beauveria bassiana ATCC 7159. Phytochemistry 2001, 56, 141–151. [Google Scholar] [CrossRef]
- Chen, A.R.M.; Reese, P.B. Biotransformation of terpenes from Stemodia maritima by Aspergillus niger ATCC 9142. Phytochemistry 2002, 59, 57–62. [Google Scholar] [CrossRef]
- Martin, G.D.A.; Reynolds, W.F.; Reese, P.B. Investigation of the importance of the C-2 and C-13 oxygen functions in the transformation of stemodin analogues by Rhizopus oryzae ATCC 11145. Phytochemistry 2004, 65, 2211–2217. [Google Scholar] [CrossRef] [PubMed]
- Garbarino, J.A.; Fraga, B.M.; Hernandez, M.G.; Chamy, M.C.; Piovano, M. Chemistry and biotranformations in the Scrophulariaceae family. Pure Appl. Chem. 2001, 73, 579–582. [Google Scholar] [CrossRef] [Green Version]
- Fraga, B.M.; Guillermo, R.; Hernández, M.G.; Chamy, M.C.; Garbarino, J.A. Biotransformation of two stemodane diterpenes by Mucor plumbeus. Tetrahedron 2004, 60, 7921–7932. [Google Scholar] [CrossRef]
- Chen, A.R.M.; Ruddock, P.L.D.; Lamm, A.S.; Reynolds, W.F.; Reese, P.B. Stemodane and stemarane diterpenoid hydroxylation by Mucor plumbeus and Whetzelinia sclerotiorum. Phytochemistry 2005, 66, 1898–1902. [Google Scholar] [CrossRef]
- Lamm, A.S.; Reynolds, W.F.; Reese, P.B. Bioconversion of Stemodia maritima diterpenes and derivatives by Cunninghamella echinulata var. elegans and Phanerochaete chrysosporium. Phytochemistry 2006, 67, 1088–1093. [Google Scholar] [CrossRef]
- Fraga, B.M.; Guillermo, R.; Hernández, M.G.; Chamy, M.C.; Garbarino, J.A. The Incubation of 13, 17-Dihydroxystemodane with Cephalosporium aphidicola. Molecules 2012, 17, 1744–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCook, K.P.; Chen, A.R.M.; Reynolds, W.F.; Reese, P.B. The potential of Cyathus africanus for transformation of terpenes substrates. Phytochemistry 2012, 82, 61–66. [Google Scholar] [CrossRef]
- Azerad, R. Regio- and stereoselective microbial hydroxylation of terpenoids compounds. In Stereoselective Biocatalysis; Patel, R.N., Ed.; Dekker: New York, NY, USA, 2000; pp. 153–180. [Google Scholar]
- Asakawa, Y.; Noma, Y. Biotransformations of Di- and Triterpenoids, Steroids, and Miscellaneous Synthetic Substrates. Compr. Nat. Prod. II Chem. Biol. 2010, 3, 893–965. [Google Scholar]
- De Oliveira Silva, E.; Jacometti Cardoso Furtado, N.A.; Aleu, J.; Gonzáles Collado, I. Terpenoid biotransformations by Mucor species. Phytochem. Rev. 2013, 12, 857–876. [Google Scholar] [CrossRef]
- Rico-Martínez, M.; Medina, F.G.; Marrero, J.G.; Osegueda-Robles, S. Biotransformation of diterpenes. RCS Adv. 2014, 4, 10627–10647. [Google Scholar] [CrossRef]
- Bhatti, H.N.; Khera, R.A. Biotranformations of diterpenoids and triterpenoids: A review. J. Asian Nat. Prod. Res. 2014, 16, 70–104. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, Y.; Sekita, M.; Hashimoto, T. Biotranformation of Bicyclic Sesqui- and Diterpene 1,2-dials and Their Derivatives by the Fungus, Aspergillus niger. Nat. Prod. Commun. 2018, 13, 923–932. [Google Scholar]
- Russell, F.A.; Mulabagal, V.; Thompson, D.R.; Singh-Wilmot, M.A.; Reynolds, W.F.; Nair, M.G.; Langer, V.; Reese, P.B. Stemodin-derived analogues with lipid peroxidation, cyclooxygenase enzymes and human tumour cell proliferation inhibitory activities. Phytochemistry 2011, 72, 2361–2368. [Google Scholar] [CrossRef]
- Martin, G.D.A.; Reynolds, W.F.; Reese, P.B. Stemodane skeletal rearrangement: Chemistry and microbial transformation. Phytochemistry 2005, 66, 901–909. [Google Scholar] [CrossRef]



























| Entry | Plant | Collection Location | Collection Period | Plant Part Extracted | Extraction Method | Stemodane Isolated Compounds | Reference/Year |
|---|---|---|---|---|---|---|---|
| 1 | S. maritima | Palisadoes peninsula (Jamaica) | n.r. | leaves | n.r. | (+)-1, (+)-2 | [1]/1973 |
| 2 | S. maritima | Palisadoes peninsula (Jamaica) | n.r. | leaves | n.r. | (+)-6 | [3]/1975 |
| 3 | S. maritima | Curaçao | n.r. | above ground portion | available | (+)-1, (+)-2, (+)-3, (+)-8 | [2]/1976 |
| 4 | S. chilensis | Littoral zone Concón, Valparaiso Region (Chile) | December | aerial parts | available | (+)-6, (+)-9, (+)-10, (−)-11, (+)-12 | [8]/1991 |
| 5 | S. maritima | Curaçao | n.r. | whole plant | available | (+)-13, (+)-14, (+)-15, (+)-16 | [10]/1992 |
| 6 | S. maritima | n.r. | n.r. | n.r. | available | 1, 14, 15, (−)-17 | [12]/2004 |
| 7 | H. occulta | Xishuangbanna (Yunnan Province, P. R. China) | Jul. | aerial parts | available | (+)-13 | [11]/2007 |
| 8 | S. maritima | Flexeiras-CE (Brasil) | Flowering stage | fresh stems | available | (+)-1 | [13]/2010 |
| 9 | S. maritima | Flexeiras-CE (Brasil) | Flowering stage | leaves and roots | available | (+)-1, (+)-4, (+)-15 | [6]/2014 |
| 10 | T. heterophyllus | n.a.E. | n.a.E. | stems and leaves | n.a.E. | (+)-18 | [14]/2018 |
| Compound | Shigella flexneri | Shigella dysenteriae | Shigella sonnei | Mycobacterium tuberculosis | α-Hemolytic streptococcus | Streptococcus pneumoniae |
|---|---|---|---|---|---|---|
| (+)-13 | n.a. (a) | n.a. | 300 | 400 | 350 | 250 |
| Rifampicin (b) | 1 | 1 | 1 | 1 | 1 | 1 |
| Bacteria | (+)-1 | (+)-15 |
|---|---|---|
| Staphylococcus aureus (ATCC 12692) | 512 | ≥1024 |
| Staphylococcus aureus (ATCC 6538) | ND | 512 |
| Staphylococcus aureus (ATCC 358) | 512 | ND |
| Staphylococcus aureus (ATCC 12624) | ND | ≥1024 |
| Pseudomonas aeruginosa (ATCC 15442) | 512 | 512 |
| Bacillus cereus (ATCC 33018) | 512 | ≥1024 |
| Escherichia coli (ATCC 27) | 512 | ≥1024 |
| Escherichia coli (ATCC 25922) | ≥1024 | 512 |
| Aeromonas carveae (ATCC 15468) | ND | ≥1024 |
| Klebsiella pneumoniae (ATCC 10031) | ND | 256 |
| Shigella flexneri (ATC 12022) | ND | 512 |
| Vibrio cholerae (ATCC 15748) | ND | ≥1024 |
| Listeria monocytogenes (ATCC 7644) | ND | 256 |
| IC50/μmol·L−1 | |||||
|---|---|---|---|---|---|
| Compound | HL-60 | SMMC-7721 | A-549 | MCF-7 | SW480 |
| (+)-18 | 0.98 ±0.07 | 1.87 ± 0.10 | 2.05 ± 0.09 | 0.86 ± 0.06 | 1.69 ± 0.08 |
| cisplatin | 1.56 ± 0.06 | 12.57 ± 0.10 | 16.65 ± 0.11 | 21.58 ± 0.12 | 27.26 ± 0.21 |
| Entry | Compound | % Reduction in Number in Plaques | Minimum Antiviral Concentration (mg/mL) | Cytotoxicity (CD50) (mg/mL) |
|---|---|---|---|---|
| 1 | (+)-aphidicolin (28) | 100 | 0.005 | 0.20 |
| 2 | (+)-stemodin (1) | 67 | 0.02 | 0.04 |
| 3 | (−)-7α-hydroxystemodin (32) | 33 | 0.1 | 0.20 |
| 4 | (−)-7β-hydroxystemodin (33) | none | - | 0.07 |
| 5 | 14α-hydroxystemodin (34) | 27 | 0.1 | 0.15 |
| 6 | (−)-19-hydroxystemodin (35) | none | - | >0.399 |
| 7 | (−)-17,19-dihydroxystemodin (36) | none | - | >0.399 |
| 8 | (−)-18-hydroxystemodin (37) | 67 | 0.1 | 0.20 |
| 9 | 16,18-dihydroxystemodin (38) | none | - | 0.07 |
| 10 | (−)-8β-hydroxystemodin (39) | 93 | 0.1 | 0.30 |
| 11 | (−)-8β,18-dihydroxystemodin (40) | none | - | 0.66 |
| 12 | 7β,8β -dihydroxystemodin (41) | none | - | 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leonelli, F.; Migneco, L.M.; Valletta, A.; Marini Bettolo, R. Stemodane Diterpenes and Diterpenoids: Isolation, Structure Elucidation, Biogenesis, Biosynthesis, Biological Activity, Biotransformations, Metabolites and Derivatives Biological Activity, Rearrangements. Molecules 2021, 26, 2761. https://doi.org/10.3390/molecules26092761
Leonelli F, Migneco LM, Valletta A, Marini Bettolo R. Stemodane Diterpenes and Diterpenoids: Isolation, Structure Elucidation, Biogenesis, Biosynthesis, Biological Activity, Biotransformations, Metabolites and Derivatives Biological Activity, Rearrangements. Molecules. 2021; 26(9):2761. https://doi.org/10.3390/molecules26092761
Chicago/Turabian StyleLeonelli, Francesca, Luisa Maria Migneco, Alessio Valletta, and Rinaldo Marini Bettolo. 2021. "Stemodane Diterpenes and Diterpenoids: Isolation, Structure Elucidation, Biogenesis, Biosynthesis, Biological Activity, Biotransformations, Metabolites and Derivatives Biological Activity, Rearrangements" Molecules 26, no. 9: 2761. https://doi.org/10.3390/molecules26092761
APA StyleLeonelli, F., Migneco, L. M., Valletta, A., & Marini Bettolo, R. (2021). Stemodane Diterpenes and Diterpenoids: Isolation, Structure Elucidation, Biogenesis, Biosynthesis, Biological Activity, Biotransformations, Metabolites and Derivatives Biological Activity, Rearrangements. Molecules, 26(9), 2761. https://doi.org/10.3390/molecules26092761