
Synthesis, Pharmacological Evaluation, and Computational Studies of Cyclic Opioid Peptidomimetics Containing β3-Lysine

 , ,
, ,  and
and
Abstract
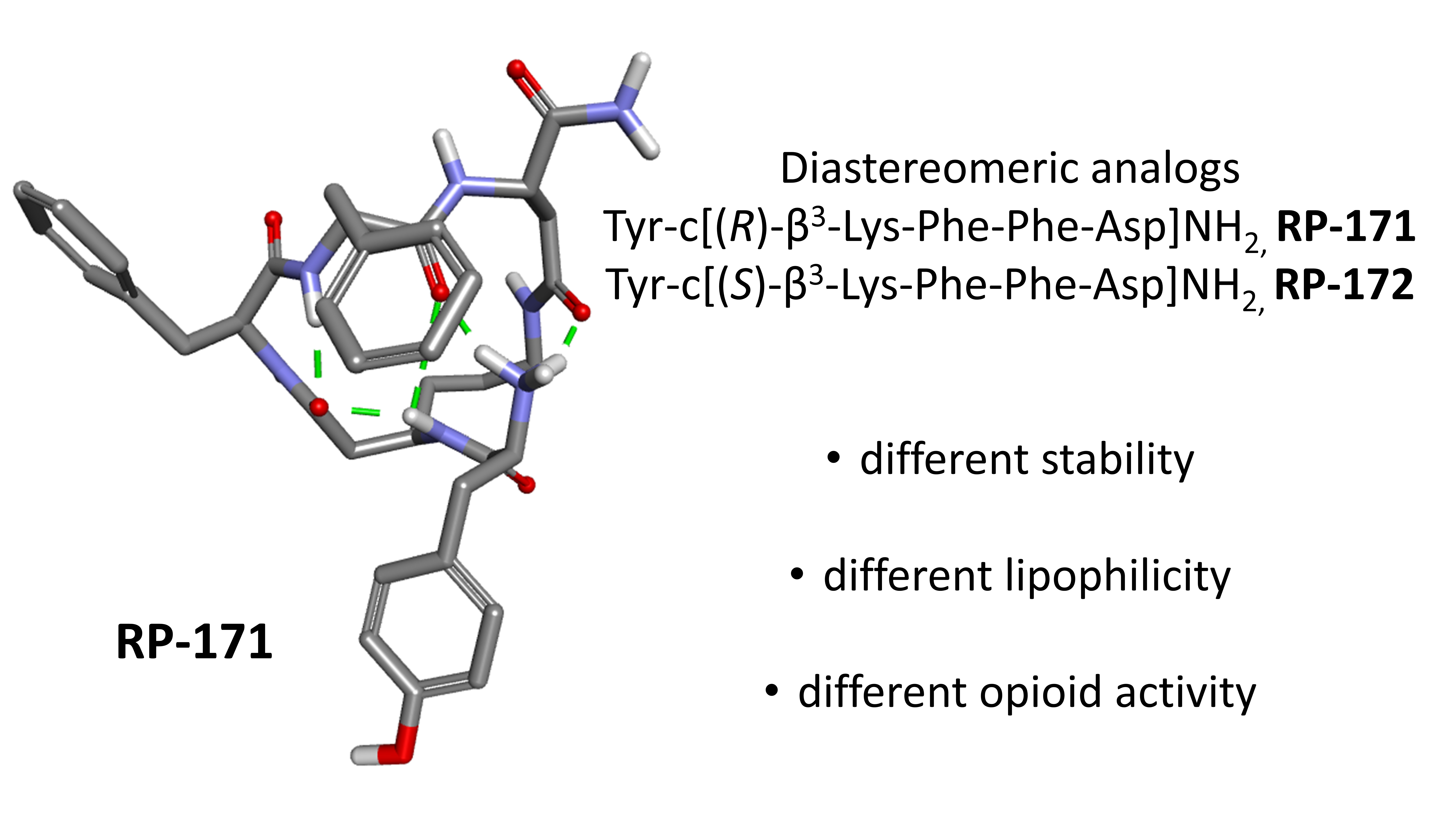
:
1. Introduction
2. Results
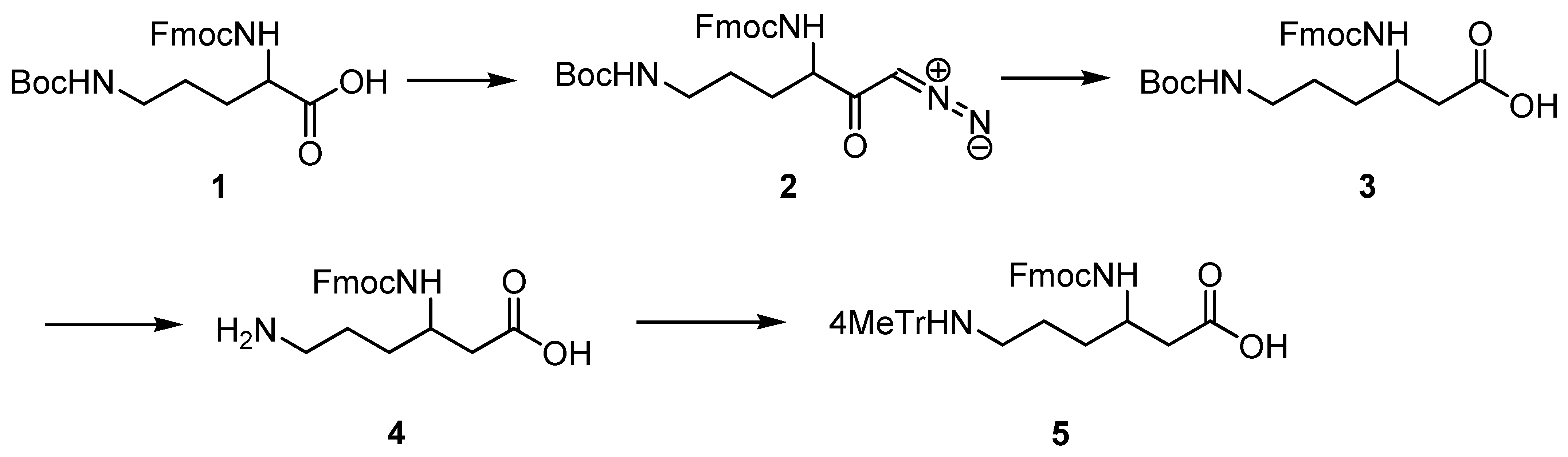
2.1. Synthesis of Protected (R)- and (S)-β3-Lys
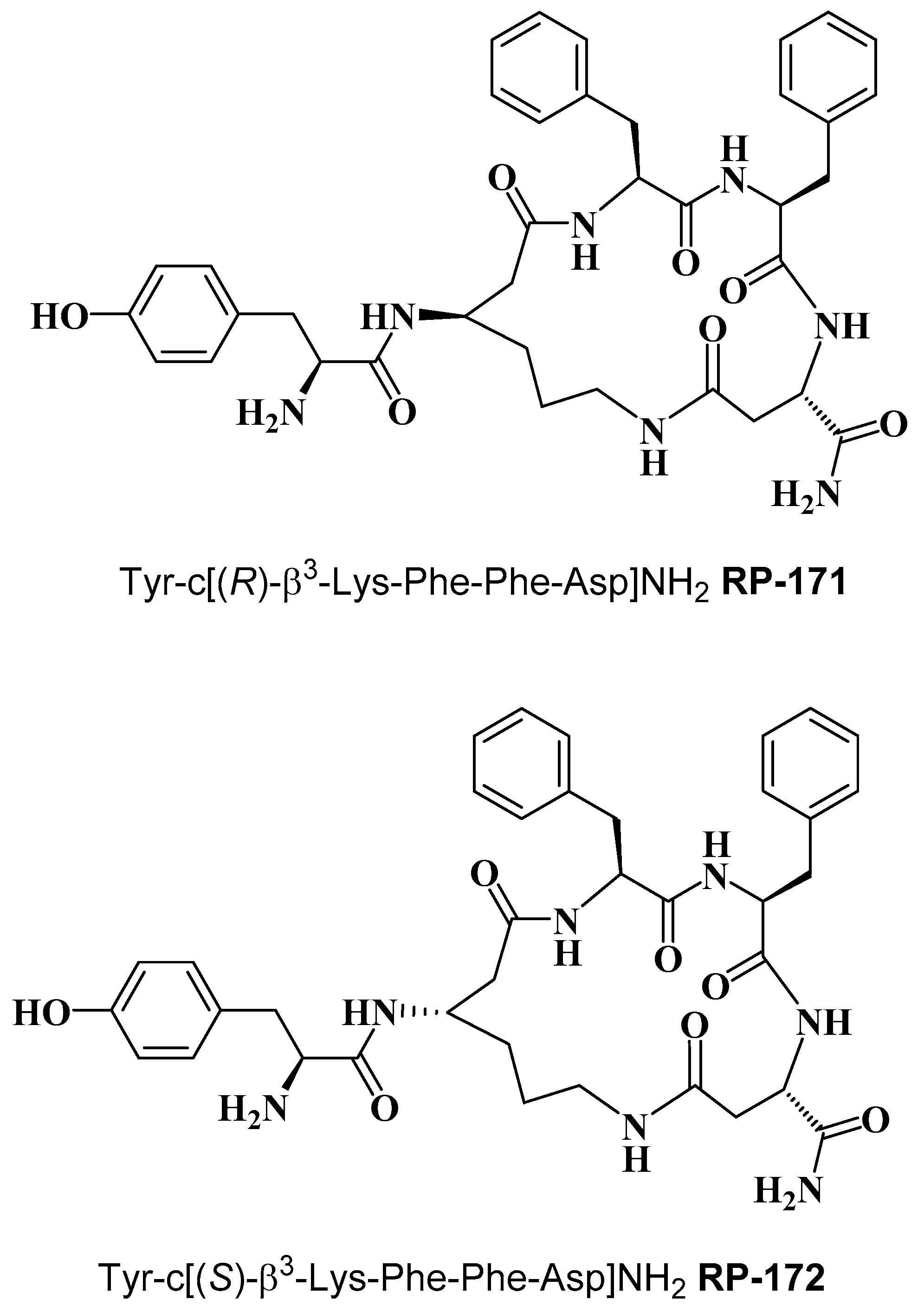
2.2. Synthesis of Cyclopeptides
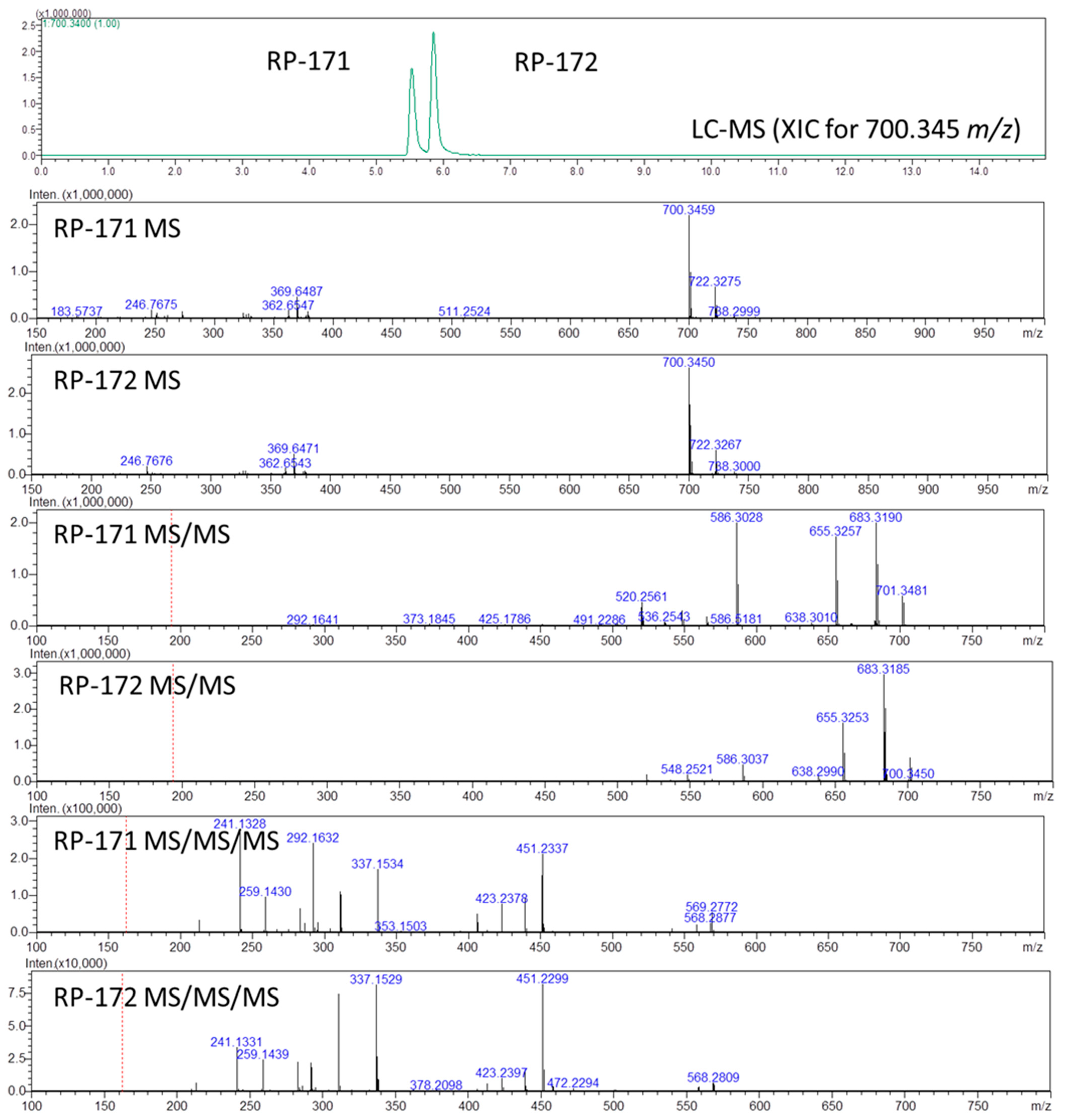
2.3. LC-MS, LC-MSn, and Quantum Chemical Calculation Studies
2.4. Receptor Binding and Functional Activity
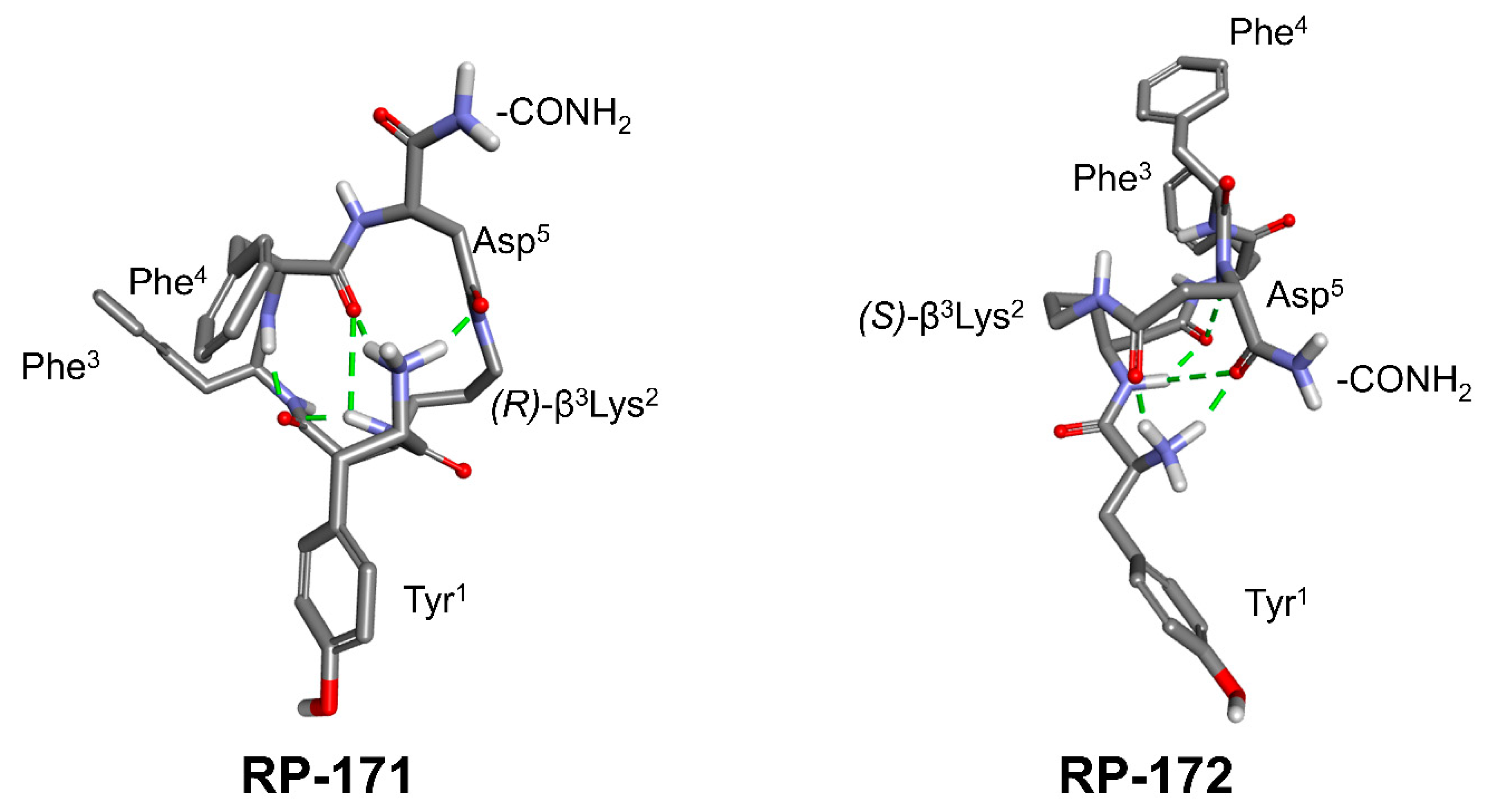
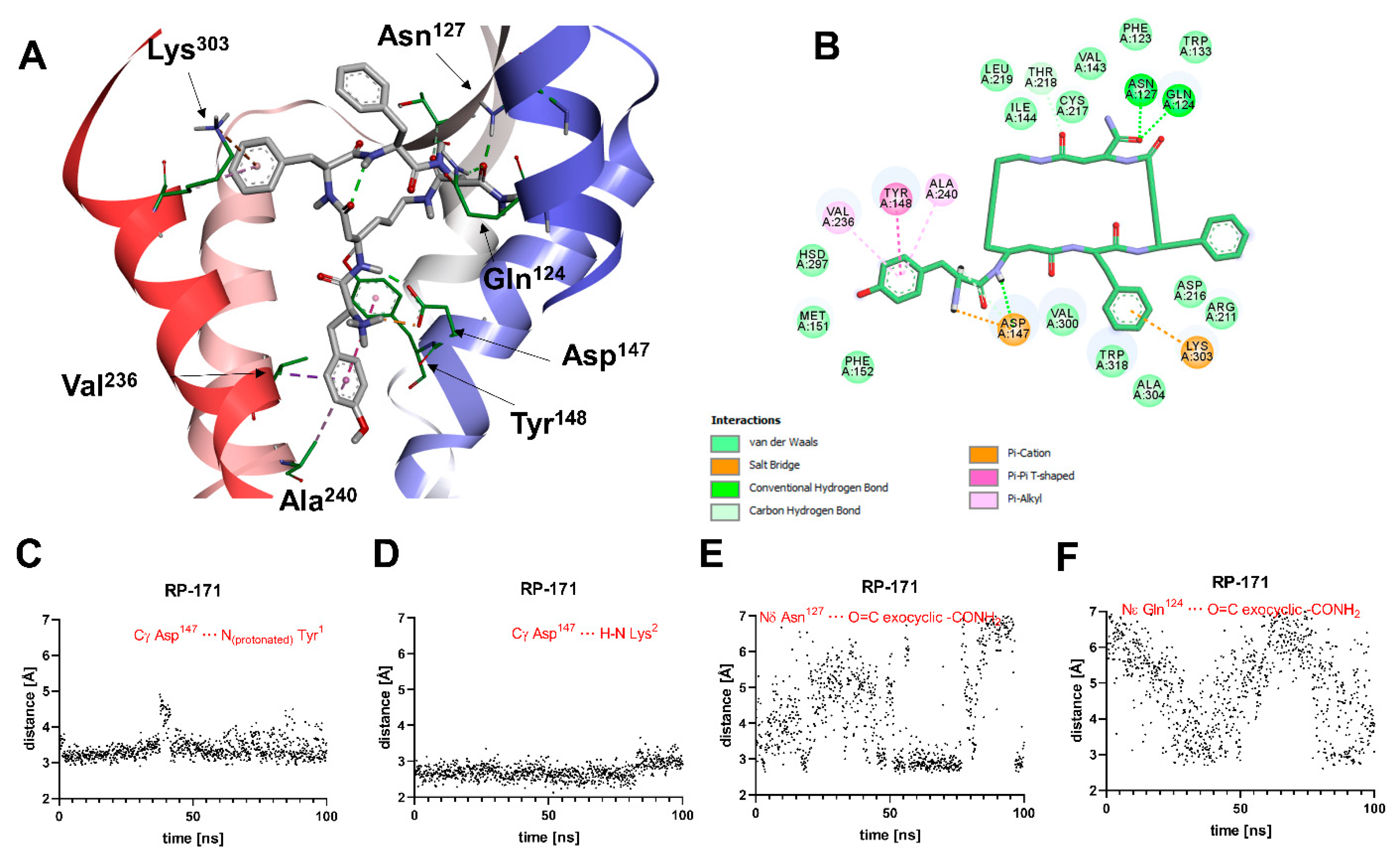
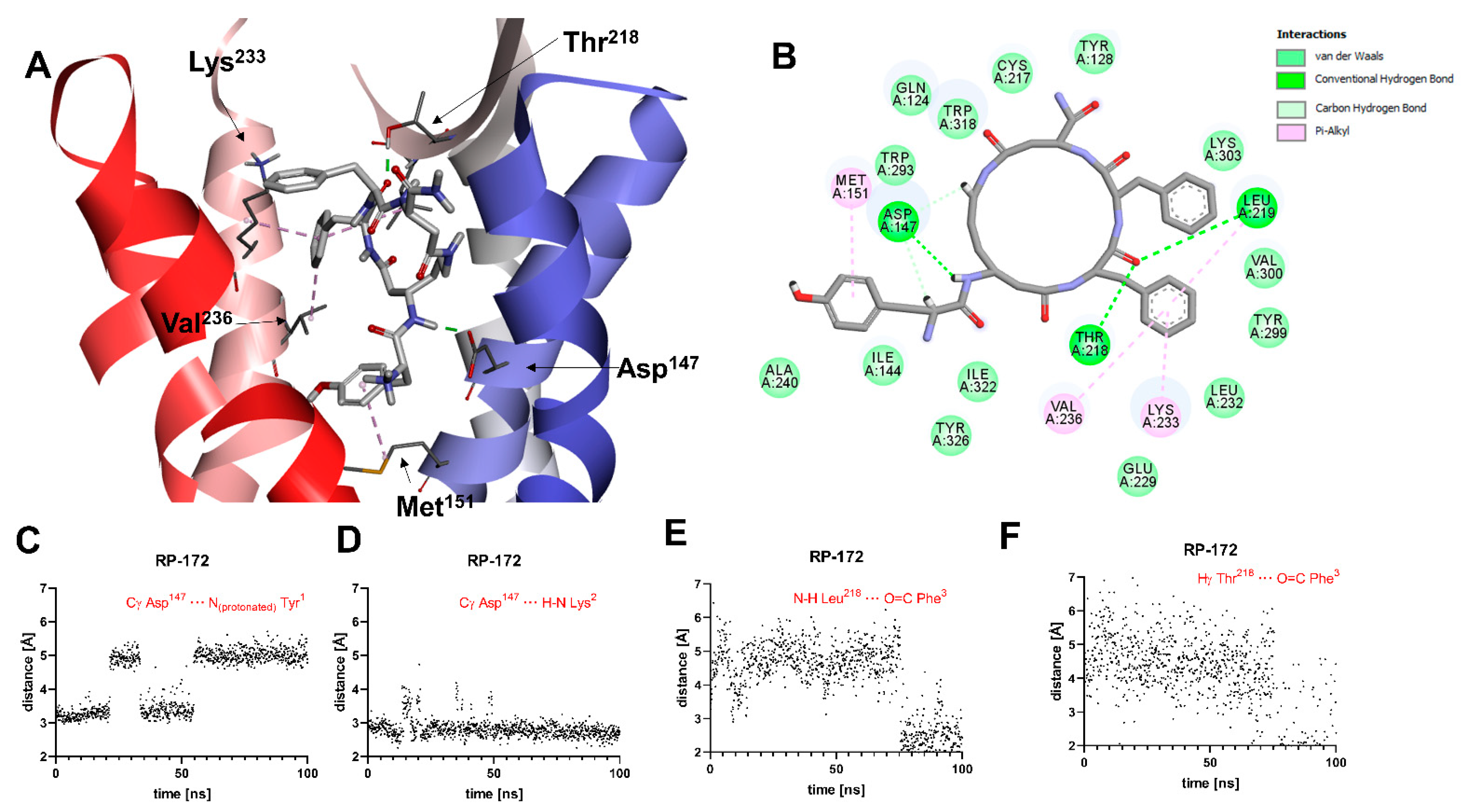
2.5. Molecular Modeling
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis of Fmoc-Protected (R)- and (S)-β3-Lys(Mtt)
4.3. Peptide Synthesis
4.4. Opioid Receptor Binding Assays
4.5. Calcium Mobilization Assay
4.6. Quantum Chemical Calculations
4.7. Molecular Docking
4.8. Molecular Dynamics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janecka, A.; Fichna, J.; Janecki, T. Opioid receptors and their ligands. Curr. Top. Med. Chem. 2004, 4, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fichna, J.; Janecka, A.; Costentin, J.; Do Rego, J.C. The endomorphin system and its evolving neurophysiological role. Pharmacol. Rev. 2007, 59, 88–123. [Google Scholar] [CrossRef]
- Liu, W.X.; Wang, R. Endomorphins: Potential roles and therapeutic indications in the development of opioid peptide analgesic drugs. Med. Res. Rev. 2012, 32, 536–580. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.H.; Wang, B.; Kou, Z.Z.; Bai, Y.; Chen, T.; Dong, Y.L.; Li, H.; Li, Y.Q. Endomorphins: Promising Endogenous Opioid Peptides for the Development of Novel Analgesics. Neurosignals 2017, 25, 98–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.Z.; Yang, W.J.; Wang, X.F.; Wang, M.M.; Zhang, Y.; Gu, N.; Wang, C.L. The spinal anti-allodynic effects of endomorphin analogs with C-terminal hydrazide modification in neuropathic pain model. Peptides 2020, 134, 170407. [Google Scholar] [CrossRef] [PubMed]
- Piekielna, J.; Perlikowska, R.; Gach, K.; Janecka, A. Cyclization in opioid peptides. Curr. Drug Targets 2013, 14, 798–816. [Google Scholar] [CrossRef] [PubMed]
- Vu, Q.N.; Young, R.; Sudhakar, H.K.; Gao, T.; Huang, T.; Tan, Y.S.; Lau, Y.H. Cyclisation strategies for stabilising peptides with irregular conformations. RSC Med. Chem. 2021, 12, 887–901. [Google Scholar] [CrossRef]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Stefanucci, A.; Dimmito, M.P.; Molnar, G.; Streicher, J.M.; Novellino, E.; Zengin, G.; Mollica, A. Developing Cyclic Opioid Analogues: Fluorescently Labeled Bioconjugates of Biphalin. ACS Med. Chem. Lett. 2020, 11, 720–726. [Google Scholar] [CrossRef]
- Leitgeb, B. Structural investigation of endomorphins by experimental and theoretical methods: Hunting for the bioactive conformation. Chem. Biodivers. 2007, 4, 2703–2724. [Google Scholar] [CrossRef]
- Janecka, A.; Gentilucci, L. Cyclic endomorphin analogs in targeting opioid receptors to achieve pain relief. Future Med. Chem. 2014, 6, 2093–2101. [Google Scholar] [CrossRef]
- Borics, A.; Tóth, G. Structural comparison of mu-opioid receptor selective peptides confirmed four parameters of bioactivity. J. Mol. Graph. Model. 2010, 28, 495–505. [Google Scholar] [CrossRef]
- Perlikowska, R.; do-Rego, J.C.; Cravezic, A.; Fichna, J.; Wyrebska, A.; Toth, G.; Janecka, A. Synthesis and biological evaluation of cyclic endomorphin-2 analogs. Peptides 2010, 31, 339–345. [Google Scholar] [CrossRef]
- Janecka, A.; Fichna, J.; Kruszynski, R.; Sasaki, Y.; Ambo, A.; Costentin, J.; do-Rego, J.C. Synthesis and antinociceptive activity of cyclic endomorphin-2 and morphiceptin analogs. Biochem. Pharmacol. 2005, 71, 188–195. [Google Scholar] [CrossRef]
- Perlikowska, R.; Piekielna, J.; Gentilucci, L.; De Marco, R.; Cerlesi, M.C.; Calo, G.; Artali, R.; Tömböly, C.; Kluczyk, A.; Janecka, A. Synthesis of mixed MOR/KOR efficacy cyclic opioid peptide analogs with antinociceptive activity after systemic administration. Eur. J. Med. Chem. 2016, 109, 276–286. [Google Scholar] [CrossRef]
- Fichna, J.; Perlikowska, R.; Wyrębska, A.; Gach, K.; Piekielna, J.; do-Rego, J.C.; Toth, G.; Kluczyk, A.; Janecki, T.; Janecka, A. Effect of 2′,6′-dimethyl-L-tyrosine (Dmt) on pharmacological activity of cyclic endomorphin-2 and morphiceptin analogs. Bioorg. Med. Chem. 2011, 19, 6977–6981. [Google Scholar] [CrossRef]
- Piekielna, J.; Kluczyk, A.; Gentilucci, L.; Cerlesi, M.C.; Calo’, G.; Tomböly, C.; Łapiński, K.; Janecki, T.; Janecka, A. Ring size in cyclic endomorphin-2 analogs modulates receptor binding affinity and selectivity. Org. Biomol. Chem. 2015, 13, 6039–6046. [Google Scholar] [CrossRef] [Green Version]
- Piekielna, J.; Perlikowska, R.; do-Rego, J.C.; do-Rego, J.L.; Cerlesi, M.C.; Calo, G.; Kluczyk, A.; Łapiński, K.; Tömböly, C.; Janecka, A. Synthesis of mixed opioid affinity cyclic endomorphin-2 analogues with fluorinated phenylalanines. ACS Med. Chem. Lett. 2015, 6, 579–583. [Google Scholar] [CrossRef] [Green Version]
- Matthews, J.L.; Braun, C.; Guibourdenche, C.; Overhand, M.; Seebach, D. Enantioselective Synthesis of β-Amino Acids; Juaristi, E., Ed.; Wiley-VCH: Weinheim, Germany, 1996; pp. 105–126. [Google Scholar]
- Fang, Z.; Baghdady, Y.Z.; Schug, K.A.; Chowdhury, S.M. Evaluation of different stationary phases in the separation of inter-cross-linked peptides. J. Proteome Res. 2019, 18, 1916–1925. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Caers, J.; Peymen, K.; Suetens, N.; Temmerman, L.; Janssen, T.; Schoofs, L.; Beets, I. Characterization of G protein-coupled receptors by a fluorescence-based calcium mobilization assay. J. Vis. Exp. 2014, 89, e51516. [Google Scholar] [CrossRef] [Green Version]
- Camarda, V.; Calo’, G. Chimeric G proteins in fluorimetric calcium assays: Experience with opioid receptors. Methods Mol. Biol. 2013, 937, 293–306. [Google Scholar]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the µ-opioid receptor–Gi protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Konishi, M.; Nishio, M.; Saitoh, K.; Miyaki, T.; Oki, T.; Kawaguchi, H. Cispentacin, a new antifungal antibiotic. I. Production, isolation, physico-chemical properties and structure. J. Antibiot. (Tokyo) 1989, 42, 1749–1755. [Google Scholar] [CrossRef] [Green Version]
- Wilczynska, D.; Kosson, P.; Kwasiborska, M.; Ejchart, A.; Olma, A. Synthesis and receptor binding of opioid peptide analogues containing β3-homo-amino acids. J. Pept. Sci. 2009, 15, 777–782. [Google Scholar] [CrossRef]
- Keresztes, A.; Szucs, M.; Borics, A.; Kövér, K.E.; Forró, E.; Fülöp, F.; Tömböly, C.; Péter, A.; Páhi, A.; Fábián, G.; et al. New endomorphin analogues containing alicyclic β-amino acids: Influence on bioactive conformation and pharmacological profile. J. Med. Chem. 2008, 51, 4270–4279. [Google Scholar] [CrossRef]
- Lesma, G.; Salvadori, S.; Airaghi, F.; Murray, T.F.; Recca, T.; Sacchetti, A.; Balboni, G.; Silvani, A. Structural and biological exploration of Phe3-Phe4-modified endomorphin-2 peptidomimetics. ACS Med. Chem. Lett. 2013, 4, 795–799. [Google Scholar] [CrossRef] [Green Version]
- Mollica, A.; Pinnen, F.; Costante, R.; Locatelli, M.; Stefanucci, A.; Pieretti, S.; Davis, P.; Lai, J.; Rankin, D.; Porreca, F.; et al. Biological active analogues of the opioid peptide biphalin: Mixed α/β3-peptides. J. Med. Chem. 2013, 56, 3419–3423. [Google Scholar] [CrossRef] [Green Version]
- Fraczak, O.; Lasota, A.; Kosson, P.; Lesniak, A.; Muchowska, A.; Lipkowski, A.W.; Olma, A. Biphalin analogs containing β3-Homo-amino acids at the 4,4positions: Synthesis and opioid activity profiles. Peptides 2015, 66, 13–18. [Google Scholar] [CrossRef]
- Fraczak, O.; Lasota, A.; Tymecka, D.; Kosson, P.; Muchowska, A.; Misicka, A.; Olma, A. Synthesis, binding affinities and metabolic stability of dimeric dermorphin analogs modified with β 3-Homo-amino acids. J. Pept. Sci. 2016, 22, 222–227. [Google Scholar] [CrossRef]
- Adamska-Bartłomiejczyk, A.; Lipiński, P.F.J.; Piekielna-Ciesielska, J.; Kluczyk, A.; Janecka, A. Pharmacological profile and molecular modeling of cyclic opioid analogues incorporating various phenylalanine derivatives. Chem. Med. Chem. 2020, 15, 1322–1329. [Google Scholar] [CrossRef]
- Tymecka, D.; Lipiński, P.F.J.; Kosson, P.; Misicka, A. β2-Homo-Amino Acid Scan of µ-Selective Opioid Tetrapeptide TAPP. Molecules 2020, 25, 2461. [Google Scholar] [CrossRef]
- Seebach, D.; Beck, A.K.; Bierbaum, D.J. The world of beta- and gamma-peptides comprised of homologated proteinogenic amino acids and other components. Chem. Biodivers. 2004, 1, 1111–1239. [Google Scholar] [CrossRef]
- Torino, D.; Mollica, A.; Pinnen, F.; Lucente, G.; Feliciani, F.; Davis, P.; Lai, J.; Ma, S.W.; Porreca, F.; Hruby, V.J. Synthesis and evaluation of new endomorphin analogues modified at the Pro(2) residue. Bioorg. Med. Chem. Lett. 2009, 19, 4115–4118. [Google Scholar] [CrossRef] [Green Version]
- Beke, T.; Somlai, C.; Perczel, A. Toward a rational design of beta-peptide structures. J. Comput. Chem. 2006, 27, 20–38. [Google Scholar] [CrossRef]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into µ-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Janecka, A.; Kruszynski, R.; Fichna, J.; Kosson, P.; Janecki, T. Enzymatic degradation studies of endomorphin-2 and its analogs containing N-methylated amino acids. Peptides 2006, 27, 131–135. [Google Scholar] [CrossRef]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Perlikowska, R.; Malfacini, D.; Cerlesi, M.C.; Calo’, G.; Piekielna, J.; Floriot, L.; Henry, T.; do-Rego, J.C.; Tömböly, C.; Kluczyk, A.; et al. Pharmacological characterization of endomorphin-2-based cyclic pentapeptides with methylated phenylalanine residues. Peptides 2014, 55, 145–150. [Google Scholar] [CrossRef]
- Wang, S.; Witek, J.; Landrum, G.A.; Riniker, S. Improving conformer generation for small rings and macrocycles based on distance geometry and experimental torsional-angle preferences. J. Chem. Inf. Model. 2020, 60, 2044–2058. [Google Scholar] [CrossRef]
- Williams, J.T.; Ingram, S.L.; Henderson, G.; Chavkin, C.; Von Zastrow, M.; Schulz, S.; Koch, T.; Evans, C.J.; Christie, M.J. Regulation of mu -Opioid Receptors: Desensitization, Phosphorylation, Internalization, and Tolerance. Mol. Pharmacol. 2013, 65, 223–254. [Google Scholar]
- Biovia Discovery Studio Visualizer v.19; Dassault Systèmes: San Diego, CA, USA, 2018.
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Sequence | Ki a [nM] | ||
|---|---|---|---|---|
| MOR | DOR | KOR | ||
| RP-170 | Tyr-c[D-Lys-Phe-Phe-Asp]NH2 | 0.55 ± 0.02 | 198 ± 4.5 | 1.52 ± 0.20 |
| RP-171 | Tyr-c[(R)-β3-Lys-Phe-Phe-Asp]NH2 | 29 ± 4.32 | >1000 | 420 ± 23 |
| RP-172 | Tyr-c[(S)-β3-Lys-Phe-Phe-Asp]NH2 | 950 ± 45 | >1000 | >1000 |
| Peptide | MOR | DOR | KOR | |||
|---|---|---|---|---|---|---|
| pEC50 (CL95%) | α ± SEM | pEC50 (CL95%) | α ± SEM | pEC50 (CL95%) | α ± SEM | |
| Dermorphin | 8.57 ± 0.07 | 1.00 | inactive | inactive | ||
| DPDPE | inactive | 7.23 ± 0.22 | 1.00 | inactive | ||
| Dynorphin A | 6.67 ± 0.50 | 0.83 ± 0.10 | 7.73 ± 0.27 | 9.04 ± 0.05 | 1.00 | |
| RP-170 | 8.93 ± 0.05 | 1.00 | inactive | 8.60 ± 0.14 | 1.00 ± 0.03 | |
| RP-171 | 6.87 ± 0.14 | 0.82 ± 0.02 | inactive | 5.99 ± 0.05 | 0.82 ± 0.05 | |
| RP-172 | 5.45 ± 0.91 | 0.45 ± 0.02 | inactive | inactive | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wtorek, K.; Lipiński, P.F.J.; Adamska-Bartłomiejczyk, A.; Piekielna-Ciesielska, J.; Sukiennik, J.; Kluczyk, A.; Janecka, A. Synthesis, Pharmacological Evaluation, and Computational Studies of Cyclic Opioid Peptidomimetics Containing β3-Lysine. Molecules 2022, 27, 151. https://doi.org/10.3390/molecules27010151
Wtorek K, Lipiński PFJ, Adamska-Bartłomiejczyk A, Piekielna-Ciesielska J, Sukiennik J, Kluczyk A, Janecka A. Synthesis, Pharmacological Evaluation, and Computational Studies of Cyclic Opioid Peptidomimetics Containing β3-Lysine. Molecules. 2022; 27(1):151. https://doi.org/10.3390/molecules27010151
Chicago/Turabian StyleWtorek, Karol, Piotr F. J. Lipiński, Anna Adamska-Bartłomiejczyk, Justyna Piekielna-Ciesielska, Jarosław Sukiennik, Alicja Kluczyk, and Anna Janecka. 2022. "Synthesis, Pharmacological Evaluation, and Computational Studies of Cyclic Opioid Peptidomimetics Containing β3-Lysine" Molecules 27, no. 1: 151. https://doi.org/10.3390/molecules27010151
APA StyleWtorek, K., Lipiński, P. F. J., Adamska-Bartłomiejczyk, A., Piekielna-Ciesielska, J., Sukiennik, J., Kluczyk, A., & Janecka, A. (2022). Synthesis, Pharmacological Evaluation, and Computational Studies of Cyclic Opioid Peptidomimetics Containing β3-Lysine. Molecules, 27(1), 151. https://doi.org/10.3390/molecules27010151