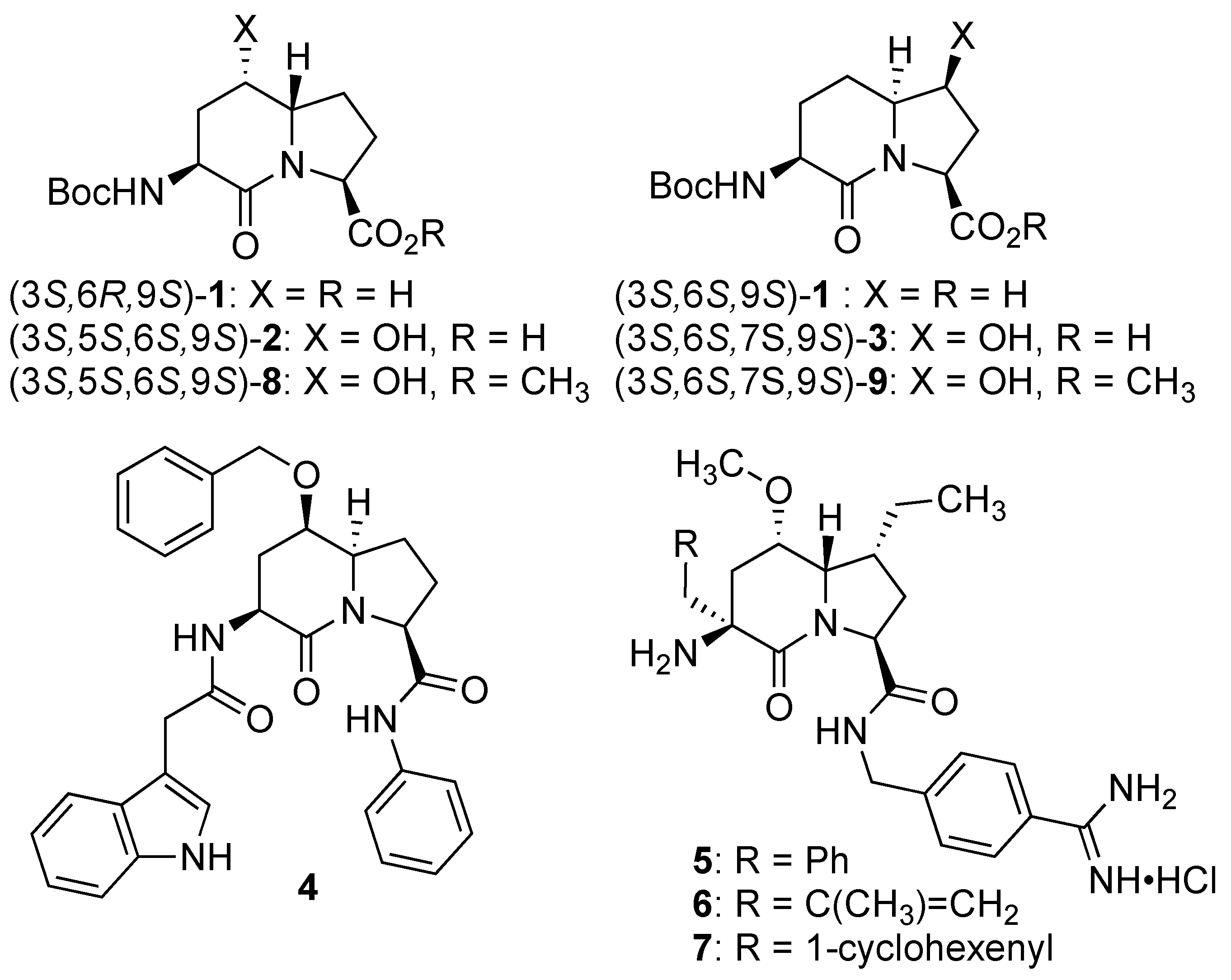
2. Results and Discussion
Initially, 5- and 7-hydroxy indolizidine-2-one
N-(Boc)amino esters
8 and
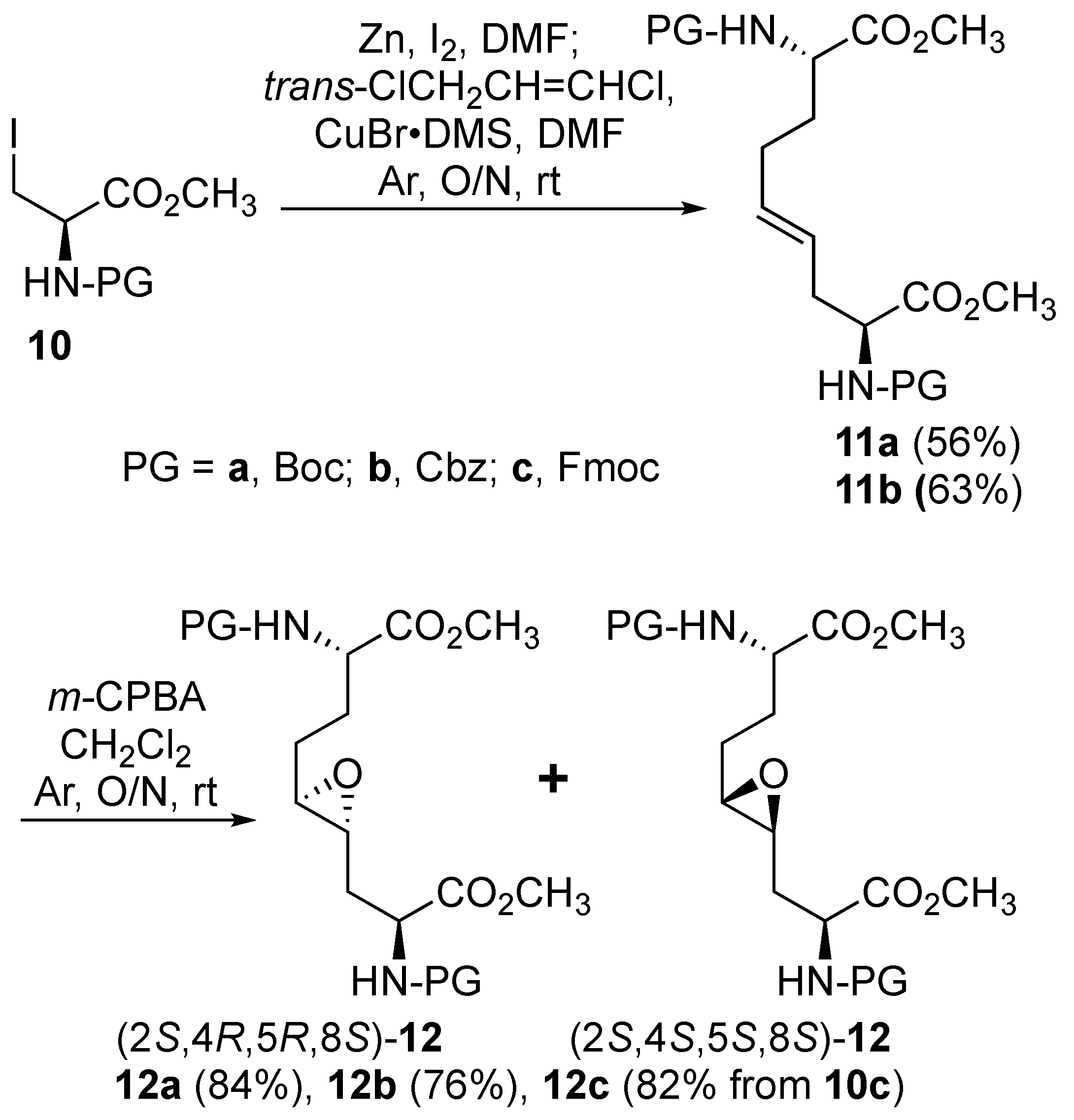
9 were pursued by pathways featuring a ring opening of 4-oxiranyl-2,8-diaminoazelates. Oxiranes
12a–
c were synthesized by epoxidation of ∆
4-2,8-diaminoazelates
11a–
c, which were respectively prepared from (
E)-1,3-dichloroprop-1-ene by copper catalyzed S
N2′ additions of zincates derived from methyl β-iodo alaninates
12a–
c protected with Boc [
25], Cbz [
26], and Fmoc groups (
Scheme 1) [
27]. Although the 15 Hz coupling constant suggested the formation of the
E-
trans olefins
11a and
11b, without the corresponding
Z-
cis isomer, NOESY experiments were performed to confirm the double-bond geometry. The
E-geometry of olefins
11a and
11b was ascertained by NOESY experiments in which the long-range through-space transfer of magnetization was observed, respectively, between the vinyl C4 (5.38 and 5.35 ppm) and allylic C6 protons (2.09 and 2.07 ppm) and between the vinyl C5 (5.51 and 5.48 ppm) and allylic C3 protons (2.47 and 2.50 ppm) (
Scheme 1). No nuclear Overhauser effect was observed between the two vinyl protons nor between the two sets of allylic protons.
Previously, epoxidations of
N-Boc and
N-Cbz allyl- and homoallyl-glycine esters with
m-chloroperbenzoic acid (
m-CPBA) in dichloromethane had given 1:1 diastereomeric mixtures of the corresponding oxiranes, which were inseparable by chromatography [
28,
29,
30]. The C3-protons of benzyl (2
S,4
RS)-2-(Boc)amino-3-(2-oxiranyl)propionate was reported to exhibit a doubling of signals in the
1H NMR spectrum [
28]. The appearance of multiple sets of signals for the two possible isomers was similarly observed in the spectra of inseparable epoxide diastereomers
12a–
c and validated by COSY spectra of the Cbz and Fmoc analogs
12b and
12c in which through-bond couplings between two sets of C3-protons with two overlapping downfield α-(C2)-proton signals were observed. Oxiranes
12a–
c were thus obtained as 1:1 diastereomeric mixtures, which were used in the subsequent chemistry.
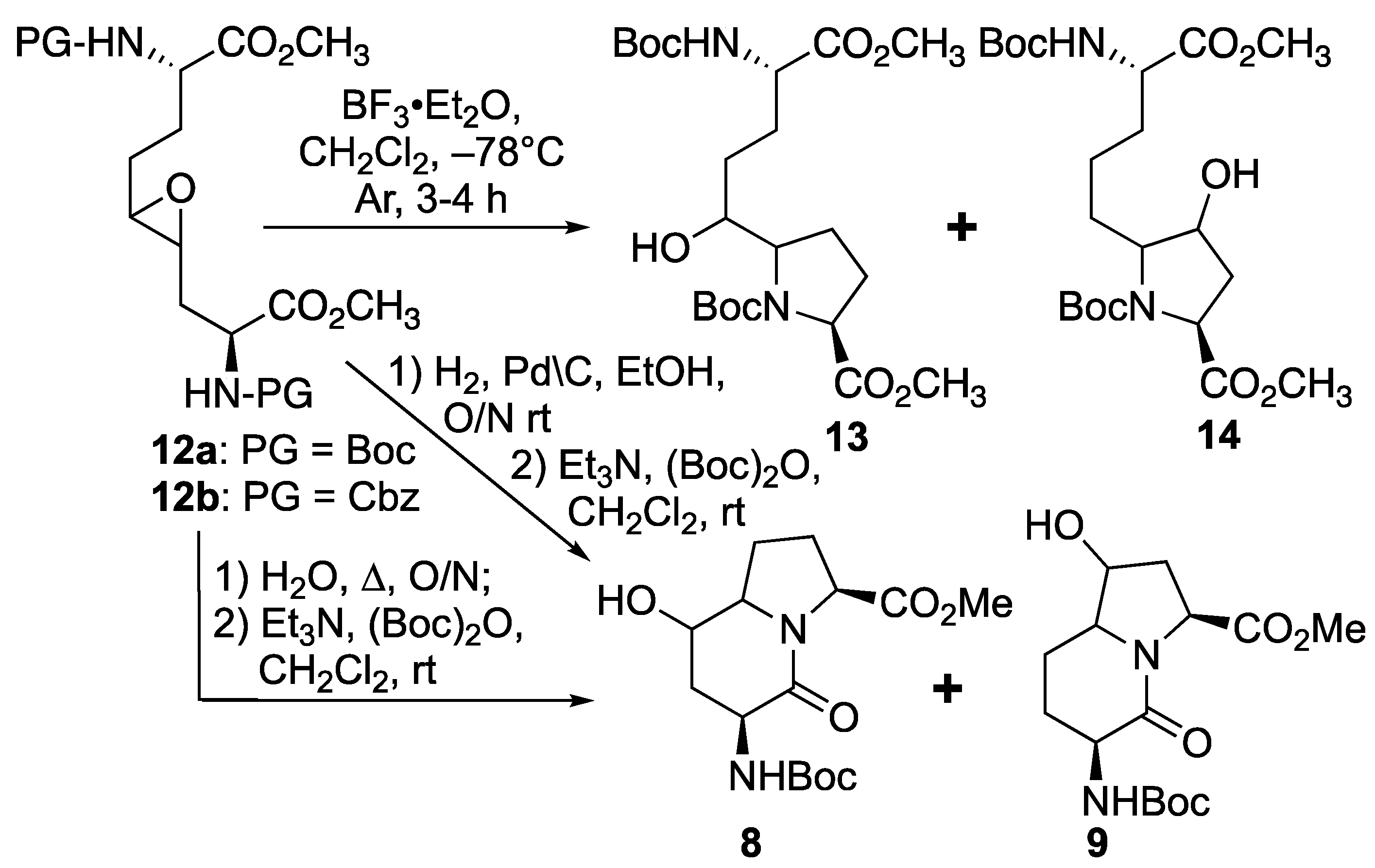
Based on the successful synthesis of 6-hydroxymethyl I
2aa diastereomers in which 5-hydroxymethyl prolines were prepared from a related C2 symmetric oxirane using Lewis-acid activation with BF
3·Et
2O in DCM at −78 °C [
31], similar conditions were employed for the intramolecular ring-opening of epoxide
12a (
Scheme 2). Multiple isomers of the material with a molecular ion corresponding to proline
13 and hydroxyproline
14 were obtained from oxirane
12a likely by
endo and
exo ring openings by the attack of the two different carbamate-protected nitrogen [
28,
32,
33]. Considering that the isomeric mix could be due, in part, to carbocation intermediates formed under the Lewis acid conditions, a method to remove the Boc group without the ring opening of the epoxide was attempted featuring heating oxirane
12a in water at reflux [
34]. Deprotection of the Boc group, intramolecular epoxide ring opening, and lactam formation all occurred upon treating
12a with boiling water. Amine protection with di-
tert-butyl dicarbonate and triethyl amine in dichloromethane, however, afforded four isomers of 5- and 7-hydroxy I
2aa esters
8 and
9, which were observed by LCMS in a 1:1:1:1 ratio. Employing Cbz-protected epoxide
12b, hydrogenolytic cleavage of the carbamate using hydrogen and palladium-on-carbon in ethanol commenced an epoxide ring opening and lactam formation sequence, which was followed by Boc protection as described above to afford four isomers of
8 and
9, which were observed in a 1:5:5:1 ratio by HPLC. The improvement in selectivity may be due to a favored
exo-
tet-like ring opening of the epoxide diastereomers by the free amine, which when generated at a lower temperature reacted to favor the proline instead of the hydroxyproline counterparts [
32,
33]. In spite the possibility of improved regioselectivity in the oxirane ring opening, the route (
Scheme 2) was, however, deemed inefficient due to the complications engendered from the lack of diastereomeric selectivity in the epoxidation of olefins
11.
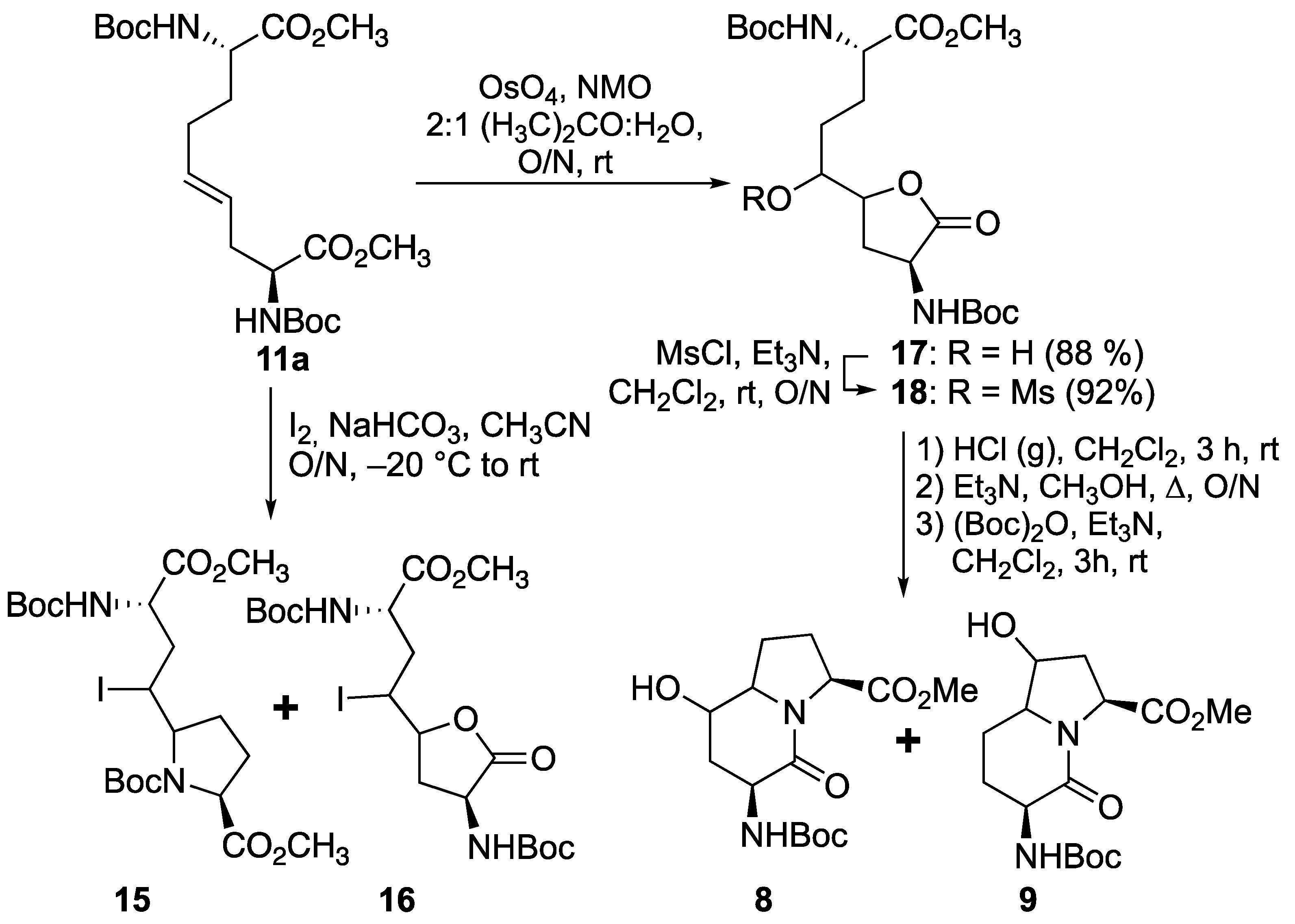
Prompted by earlier success using transannular iodolactamization to prepare azabicyclo[X.Y.0]alkan-2-one ring systems [
15,
35], and related iodoamination protocols for preparing iodomethyl pyrrolidines and piperidines [
36,
37,
38], ∆
4-diaminoazelate
11a was subjected to iodine and NaHCO
3 at −20 °C (
Scheme 3). The ring opening of the iodonium intermediate by one of the two carbamate-protected nitrogen appeared to be a method for selectively obtaining proline
15 instead of the azetidine counterpart; however, a mixture of diastereomeric iodolactones
16 was also produced as a competing side product. Considering the lactone as a potential means for differentiating between the two carboxylates, dihydroxylation of ∆
4-diaminoazelate
11a was performed using osmium tetroxide and
N-methylmorpholine
N-oxide (NMO) in aqueous acetone to provide hydroxy lactone
17 as a mixture of diasteromers [
39]. Mesylate
18 was obtained by methanesulfonation of hydroxy lactone
17 using methanesulfonyl chloride and triethylamine in dichloromethane. Mesylate
18 was converted to hydroxy I
2aa analogs
8 and
9 by a three-step sequence featuring proline formation after Boc group removal with HCl gas bubbles in dichloromethane, lactam cyclization upon treatment of the hydrochloride salt with triethylamine in methanol at reflux, and amine protection with di-
tert-butyl decarbonate in dichloromethane. The HPLC chromatogram of the products from this sequence exhibited four peaks with molecular ions corresponding to 5- and 7-hydroxy Boc-I
2aa-OMe isomers
8 and
9 (
Scheme 3) in a 1:1:1:1 ratio.
Different mixtures of 5- and 7-hydroxy Boc-I
2aa-OMe diastereomers
8 and
9 likely arose from a combination of a lack of facial selectivity in the epoxidation and the dihydroxylation of olefin
11 and competing nucleophilic attack from both nitrogen of diamino azelate epoxide
12 and methanesulfonate
18. The loss of stereochemical integrity may also arise from competing S
N1 processes due to the epoxide ring opening prior to pyrrolidine formation. Intrigued by the production of iodolactone
16 as a side product from the iodoamination strategy, an iodolactonization approach was considered because of the high facial selectivity achieved on simpler γ,δ-unsaturated carboxylic acids [
24,
40,
41].
After saponification of diester
11a with lithium hydroxide in aqueous dioxane, dicarboxylic acid
19 was treated with cesium carbonate and iodine in an ice-cold acetonitrile solution (
Scheme 4). Analysis by LCMS demonstrated a major peak with a molecular ion corresponding to lactone
20. Subsequent treatment with iodomethane and potassium carbonate in DMF furnished the corresponding methyl ester tetrahydrofuran-2-one (1′
R,5
S)-
16 after chromatography in 55% yield from diacid acid
19. Attempts to perform the iodolactonization without a base gave a product mostly from the loss of Boc protection. Employing the same three-step sequence described above to convert methane sulfonate
18 into esters
8 and
9, iodide (1′
R,5
S)-
16 was transformed into separable 5- and 7-hydroxy I
2aa esters (5
S,6
S)-
8 and (6
S,7
S)-
9 in 42% and 34% overall yields, respectively. Subsequent saponification of esters (5
S,6
S)-
8 and (6
S,7
S)-
9 gave, respectively, the acids (5
S,6
S)-
2 and (6
S,7
S)-
3 in 64% and 78% yields.
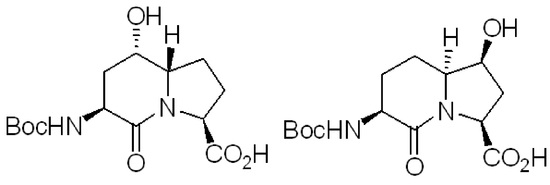
Assignment of Regio-Chemistry and Stereochemistry of 5- and 7-Hydroxy I2aa Esters
The configuration of the ring fusion and hydroxyl group carbons of the 5- and 7-hydroxy I
2aa esters
8 and
9, as well as the alcohol position on the ring system, were all assigned based on two-dimensional NMR spectroscopic experiments. The locations of the indolizidine-2-one ring protons were initially assigned by COSY experiments in which through-bond couplings were used to trace the sequence from the downfield shifted carbamate NH to the C9 hydrogen. Subsequently, heteronuclear single quantum coherence (HSQC) spectroscopy was used to correlate the protons linked to similar carbons. The β-protons on the same face as the C3 carbamate and C9 carboxylate appeared generally up-field of their α-counterparts due to anisotropic effects caused by the latter functional groups [
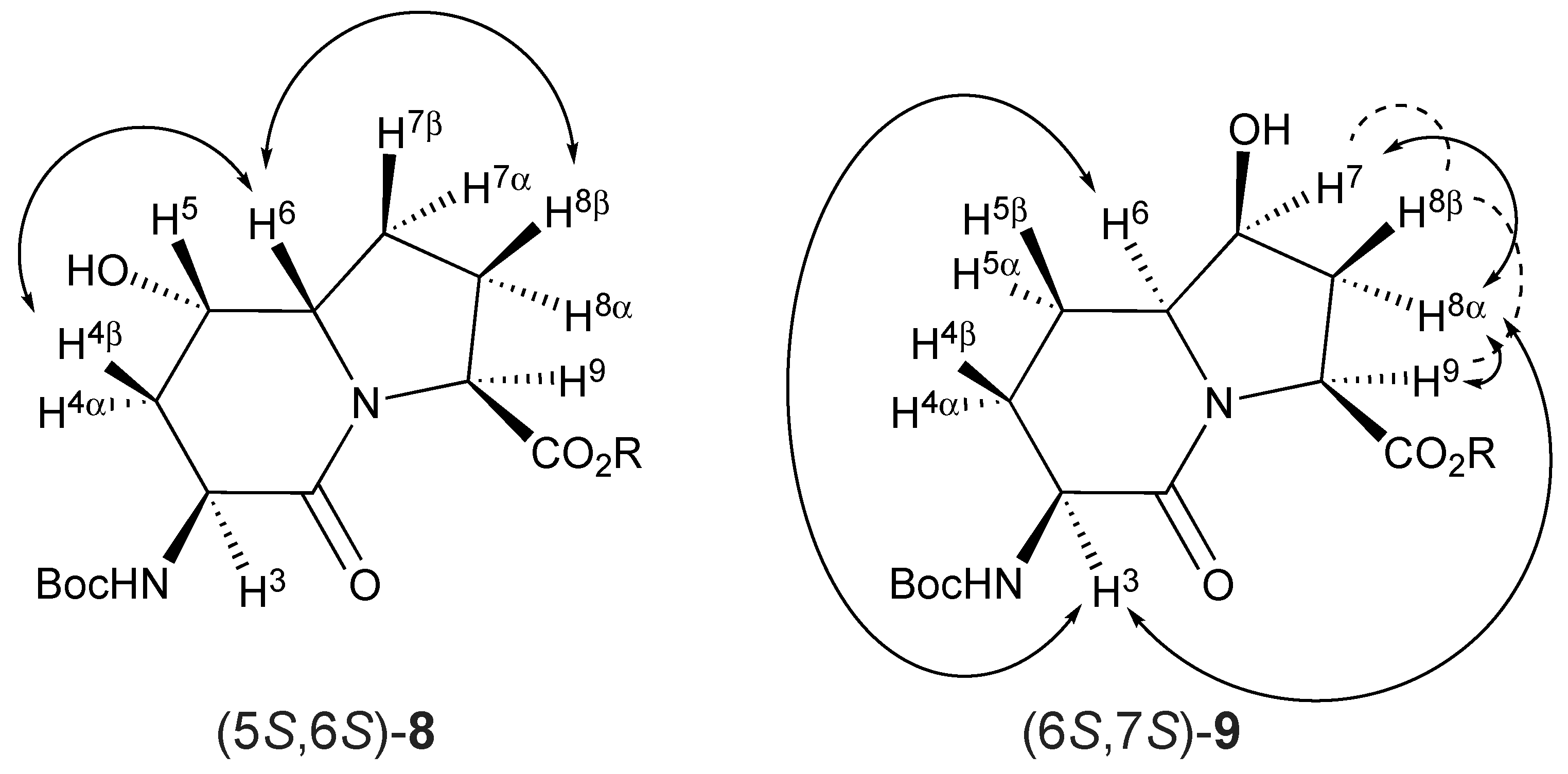
42]. Finally, relative configurations were ascertained (
Figure 2) based on NOESY experiments in which the observed through-space transfers of magnetization were used to correlate the stereochemical assignments.
The ring fusion protons (3.88 and 3.74 ppm) of 5- and 7-hydroxy Boc-I
2aa-OMe (5
S,6
S)-
8 and (6
S,7
S)-
9 were respectively assigned the
S stereochemistry based on nuclear Overhauser effects (nOe) with the C4β and C8β protons (1.99 and 1.84 ppm) and with the C3 proton (4.13 ppm,
Figure 2). No long-range through-space transfer of magnetization was observed for the protons on the alcohol-bearing carbons. In the case of (6
S,7
S)-
9, the relative nOe between the C7 proton was stronger for the C8α proton (2.35 ppm) compared to that of the C8β proton (2.15 ppm). The stereochemical assignments for Boc-(7-OH)I
2aa-OMe (6
S,7
S)-
9 were confirmed by X-ray analysis as discussed below.
The configurations of the hydroxyl group in Boc-(5-OH)I
2aa-OMe (5
S,6
S)-
8 and the iodolactone of tetrahydrofuran-2-one (1′
R,5
S)-
16 were based on the latter serving as a common intermediate for both the former and Boc-(7-OH)I
2aa-OMe (6
S,7
S)-
9. The stereochemistry of the ring-fusion and alcohol carbons are respectively derived from the inversion on nitrogen attack of the iodide and retention on the lactone opening during synthesis of the bicycle. Although the order of attack of the iodine and carboxylate may proceed by a traditional iodonium intermediate (
Scheme 4) [
24], and by a more concerted nucleophile-assisted alkene activation mechanism [
43], the stereochemical outcome of iodolactone (1′
R,5
S)-
20 arises from the attack of iodine by the face of the olefin on the opposite side of the proximal carboxylate of ∆
4-azelate
19 (
Scheme 4).
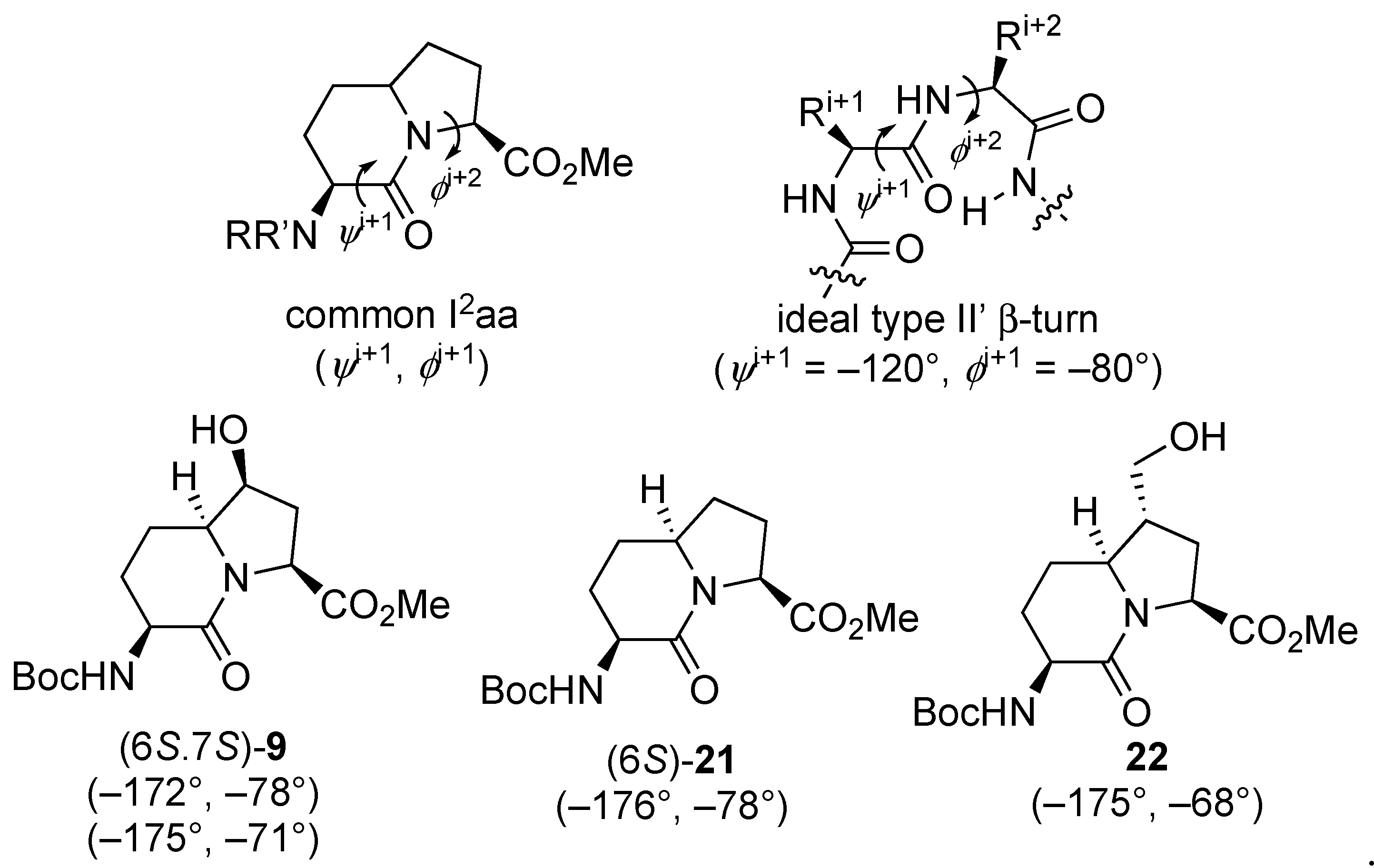
The relative configurational assignments for 7-hydroxy Boc-I
2aa-OMe (6
S,7
S)-
9 were confirmed by X-ray analysis of crystals grown from a dichloromethane-in-hexanes mixture (
Figure 3). Two conformers differing primarily by the carbamate orientation were present in the unit cell and connected by an intermolecular hydrogen bond from the 7-hydroxyl group donor to the lactam carbonyl oxygen acceptor. Examination of the backbone dihedral angles embedded in the I
2aa ring system (
ψi+1 −172° and
ϕi+2 −78°;
ψi+1 −175° and
ϕi+2 −71°) of the conformers in the X-ray structure of the 7-hydroxy analog (6
S,7
S)-
9 indicated a close relation to those of the central residues of an ideal type II’ β-turn (
ψi+1 –120° and
ϕi+2 −80°) [
44], and to that of the methyl ester of the parent I
2aa counterpart (6
S)-
21 (
ψi+1 −176° and
ϕi+2 −78°,
Figure 4) [
45]. Relative to the values in the crystal structure of Boc-I
2aa-OMe (6
S)-
21, the
ϕi+2 dihedral angle was apparently less influenced by the smaller 7β-hydroxy substituent than the 7α-hydroxymethyl substituent in Boc-(7-HOCH
2)I
2aa-OMe (
22,
ψi+1 −175° and
ϕi+2 −68°) [
11].
3. Materials and Methods
Anhydrous solvents (CH
3CN, DMF, (CH
3)
2CO, CH
2Cl
2, and CH
3OH) were obtained by passage through solvent filtration systems (GlassContour, Irvine, CA, USA). All reagents from commercial sources were used as received: Iodine was purchased from Aldrich (USA) and solvents were obtained from Fisher Chemical. The
N-(Boc)-, (Cbz)-, and (Fmoc)-3-iodo-
l-alanine methyl esters
10a–
c were respectively prepared according to the literature methods reported in references [
25,
26,
27]. Purification by silica gel chromatography was performed on 230−400 mesh silica gel; analytical thin-layer chromatography (TLC) was performed on silica gel 60 F254 (aluminum sheet) and visualized by UV absorbance or staining with KMnO
4. Melting points are reported in degree Celsius (°C), uncorrected and obtained using a Mel-Temp melting point apparatus equipped with a thermometer on the sample that was placed in a capillary tube. Spectroscopic
1H and
13C NMR experiments were recorded at room temperature (298 K) in CDCl
3 (7.26/77.16 ppm), DMSO-
d6 (2.5/39.56), and CD
3OD (3.31/49.0 ppm) on Bruker AV (500/125, and 700/175 MHz) instruments using an internal solvent as the reference. Spectra are presented in the
Supplementary Materials. Chemical shifts are reported in parts per million (ppm), and coupling constant (
J) values in Hertz (Hz). Abbreviations for peak multiplicities are s (singlet), d (doublet), t (triplet), q (quadruplet), q (quintuplet), m (multiplet), and br (broad). Certain
13C NMR chemical shift values were extracted from HSQC spectra. High-resolution mass spectrometry (HRMS) data were obtained on an LC-MSD instrument in electrospray ionization (ESI-TOF) mode by the Centre Régional de Spectrométrie de Masse de l’Université de Montréal. Either protonated molecular ions [M + H]
+ or sodium adducts [M + Na]
+ were used for empirical formula confirmation. Infrared spectra were recorded in the neat on a Perkin Elmer Spectrometer FT-IR instrument, and are reported in reciprocal centimeters (cm
−1). The X-ray structure was solved using a Bruker Venture Metaljet diffractometer by the Laboratoire de diffraction des rayons X de l’Université de Montréal. Specific rotations [α]
D were measured at 25 °C at the specified concentrations (
c in g/100 mL) using a 0.5 dm cell on a PerkinElmer Polarimeter 589 instrument and expressed using the general formula [α]
D25 = (100 × α)/(d ×
c).
3.1. (3S,5S,6S,9S)-3-N-(Boc)amino-5-hydroxy-indolizin-2-one-9-carboxylic Acid [(3S,5S,6S,9S)-2]
A 0 °C solution of ester (3S,5S,6S,9S)-8 (15 mg, 0.046 mmol) in 1,4-dioxane (0.5 mL) was treated with a 1N solution of LiOH (1.9 mg, 0.046 mmol, 1 equiv.). The cooling bath was removed. The reaction mixture was warmed to room temperature with stirring overnight, at which time TLC indicated the consumption of the starting material. The volatiles were evaporated under reduced pressure. The residue was partitioned between H2O (5 mL) and ethyl acetate (5 mL). The aqueous phase was acidified with 1 N HCl to pH 3 and extracted with ethyl acetate (3 × 10 mL). The organic extractions were combined, dried with Na2SO4, filtered, and concentrated under vacuum to afford (3S,5S,6S,9S)-2 (9 mg, 64%) as a white foam; [α]D25 –10.2 (c 0.32, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.39 (s, br, 1H), 4.71 (s, 1H), 4.39 (s, br, 1H), 4.29–4.28 (m, 1H), 3.84–3.80 (m, 1H), 2.6–2.52 (m, 1H), 2.39-2.33 (m, 2H), 2.26-2.20 (m, 1H), 2.05-2.02 (m, 1H), 2.0-1.95 (m, 1H), 1.67-1.63 (m, 1H), 1.47 (s, 9H); 13C{1H} NMR (125 MHz, CDCl3) δ 172.0, 167.3, 147.3, 80.5, 64.0, 60.0, 35.2, 32.0, 30.0, 28.3, 26.1, 23.0; FT-IR (neat) νmax 3328, 2919, 1702, 1521, 1449, 1362, 1208, 1166, 1050, 1031 cm−1; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C14H22N2O6Na 337.1370, found 337.1374.
![Molecules 27 00067 i001]()
3.2. (3S,6S,7S,9S)-3-N-(Boc)amino-7-hydroxy-indolizin-2-one-9-carboxylic Acid [(3S,6S,7S,9S)-3]
A 0 °C solution of ester (3S,6S,7S,9S)-9 (150 mg, 0.46 mmol) in 1,4-dioxane (5 mL) was treated with a 1N solution of LiOH (19.2 mg, 0.46 mmol, 1 equiv.). The cooling bath was removed. The reaction mixture was warmed to room temperature with stirring for 3 h, at which time TLC indicated the consumption of the starting material. The volatiles were evaporated under reduced pressure. The residue was partitioned between H2O (10 mL) and ethyl acetate (5 mL). The aqueous phase was acidified with 1 N HCl to pH 3 and extracted with ethyl acetate (3 × 10 mL). The organic extractions were combined, dried with Na2SO4, filtered, and concentrated under vacuum to afford (3S,6S,7S,9S)-3 (112 mg, 78%) as a white solid: mp 105–106 °C; [α]D25 –19.13 (c 0.23, CHCl3); 1H NMR (500 MHz, CD3OD): δ 4.460–4.43 (dd, J = 9.3, 4.3 Hz, 1H), 4.26–4.24 (m, 1H), 4.22–4.17 (m, 1H), 3.76–3.72 (m, 1H), 2.47–2.41 (m, 1H), 2.21–2.18 (d, J = 14.2 Hz, 1H), 2.15–2.07 (m, 2H), 1.87–1.82 (m, 2H), 1.48 (s, 9H); 13C{1H} NMR (125 MHz, CD3OD) δ 174.0, 170.0, 156.6, 79.1, 71.2, 62.3, 57.2, 50.0, 37.0, 27.3, 27.0, 19.0; FT-IR (neat) νmax 3325, 2922, 1697, 1523, 1451, 1365, 1211, 1162, 1055, 1032 cm−1; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C14H22N2O6Na 337.1370, found 337.1374.
![Molecules 27 00067 i002]()
3.3. Methyl (3S,5S,6S,9S)-3-N-(Boc)amino)-5-hydroxy-indolizin-2-one-9-carboxylate and (3S,6S,7S,9S)-3-N-(Boc)amino)-7-hydroxy-indolizin-2-one-9-carboxylate [(3S,5S,6S,9S)-8 and (3S,6S,7S,9S)-9]
A solution of (1′R,5S)-1′-iodo-tetrahydrofuran-2-one (1′R,5S)-16 (1.0 g, 1.8 mmol) in dichloromethane (20 mL) was treated with HCl gas bubbles for 2-3 h, when TLC indicted complete consumption of the starting carbamate and LCMS analysis indicated a new peak RT = 0.7 min (C18 column, 10:90 CH3CN:H2O) with a molecular ion of [M + H]+ m/z 357. The reaction mixture was evaporated to a residue, which was dissolved in MeOH (5 mL), treated with triethylamine (545 mg, 5.4 mmol, 3 equiv.), and heated at reflux using an oil bath overnight, when LCMS indicated a new peak RT = 0.68 min (eluent C18 column, 10:90 CH3CN:H2O) with the molecular ion [M + H]+ m/z. The volatiles were evaporated under reduced pressure. The residue was dissolved in dichloromethane (10 mL), treated with (Boc)2O (0.14 g, 0.63 mmol, 1.2 equiv.), and stirred for 3 h, when TLC indicated two new spots and LCMS indicated a new peak RT = 5.0 min (C18 column, 10:90 CH3CN:H2O). The volatiles were removed under reduced pressure. The residue was purified by flash column chromatography using 60–80% EtOAc in hexanes as eluent.
![Molecules 27 00067 i003]()
The first to elute was Boc-(7-HO)I2aa-OMe (3S,6S,7S,9S)-9 (200 mg, 34%) as a white solid: mp 138–140 °C; Rf = 0.47, (100% EtOAc twice eluted, visualized with KMnO4); [α]D25 −28.2 (c 0.85, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.14 (s, br, NH), 4.46–4.44 (dd, J = 10 Hz, 1H), 4.22–4.18 (m, 1H), 4.15–4.12 (m, 1H), 3.84 (s, 3H), 3.76–3.71 (m, 1H), 3.61–3.58 (d, J = 15 Hz, OH), 2.4–2.33 (m, 2H), 2.29–2.22 (m, 1H), 2.15–2.12 (dt, J = 14.5, 0.9 Hz, 1H), 2.0–1.93 (m, 1H), 1.77–1.69 (m, 1H), 1.47 (s, 9H); 13C{1H} NMR (125 MHz, CDCl3) δ 175.5, 170.1, 156.0, 80.0, 73.0, 61.5, 57.1, 53.2, 51.0, 36.3, 28.3, 27.1, 19.2; FT-IR (neat) νmax 3357, 2979, 1693, 1636, 1518, 1437, 1392, 1365, 1249, 1165, 1099, 1063, 1005 cm−1 HRMS (ESI-TOF) m/z [M + Na]+ calcd for C15H24N2O6Na 351.1526 found 351.1522.
Next to elute was Boc-(5-HO)I2aa-OMe (3S,5S,6S,9S)-8 (250 mg, 42%) as a white solid: mp 75–77 °C; Rf = 0.3 (100% EtOAc, twice eluted, visualized with KMnO4); [α]D25 –12.6 (c 0.75, CHCl3). 1H NMR (500 MHz, CDCl3) δ 5.25 (s, br, NH), 4.50–4.44 (m, 2H), 4.27 (s, 1H), 3.89-3.86 (m, 1H), 3.77 (s, 3H), 2.74–2.68 (m, 1H), 2.44–2.40 (m, 1H), 2.11–2.04 (m, 2H), 2.00–1.97 (m, 1H), 1.95–1.92 (m, 1H), 1.88–1.83 (m, 1H), 1.45 (s, 9H); 13C{1H} NMR (125 MHz, CDCl3) δ 173.0, 168.0, 156.2, 80.0, 64.0, 63.3, 58.2, 52.3, 47.4, 36.0, 28.3, 28.0, 27.0; FT-IR (neat) νmax 3360, 2983, 1702, 1633, 1518, 1438, 1395, 1250, 1162, 1102, 1002 cm−1; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C15H24N2O6Na 351.1526 found 351.1522.
3.4. Dimethyl (2S,4E,8S)-∆4-2,8-(di-N-(Boc)amino)azelate (11a)
In a 250-mL round bottom flask, fitted with a three-way stopcock, CuBr•DMS (1.22 g, 0.006 mol, 0.13 equiv.) was weighed, dried gently with a heat gun under vacuum until the powder changed color from white to light green, placed under argon, treated with dry DMF (30 mL), followed by (E)-1,3-dichloroprop-1-ene (2.5 g, 0.023 mol, 0.5 equiv.). In a Schlenk tube, zinc (8.9 g, 0.14 mol, 3 equiv.) and iodine (0.35 g, 0.0014 mol, 0.03 equiv.) were mixed under an argon atmosphere, and thrice heated under vacuum with a heat gun for 10 min and cooled under a flush of argon. A solution of N-(Boc)-3-iodo-l-alanine methyl ester 10a (15 g, 0.046 mol) in dry DMF (30 mL) was added to the Schlenk tube and stirred for 1h, when TLC analysis confirmed the consumption of the iodide (Rf = 0.7, 30% EtOAc in hexanes) and formation of the organozinc reagent (Rf = 0.2, 30% EtOAc in hexanes). Stirring was stopped, the excess zinc powder was allowed to settle, and the supernatant was transferred dropwise via a syringe with care to minimize the transfer of zinc into the flask containing the copper catalyst. After stirring at rt overnight, TLC indicated a new spot (Rf = 0.48, 40% EtOAc in hexanes) and the reaction mixture was diluted with ethyl acetate (150 mL), stirred for 15 min, and filtered through a silica gel pad. The filtrate was treated with water (100 mL), transferred into a separatory funnel, and diluted with ethyl acetate (50 mL). The organic phase was washed successively with 1 M Na2S2O3 (2 × 100 mL), water (4 × 100 mL), and brine (2 × 100 mL), dried over Na2SO4, filtered, and evaporated. The volatiles were removed under reduced pressure to afford a residue that was purified by chromatography using 25–30% EtOAc in hexanes as the eluent. Evaporation of the collected fractions gave azelate 11a (11.4 g, 56%) as a colorless liquid: Rf = 0.48 (2:3 EtOAc/Hexanes, visualized with KMnO4); [α]D25 +25.2 (c 1.04, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.54–5.48 (dt, J = 15, 5 Hz, 1H), 5.39–5.34 (dt, J = 15, 5 Hz, 1H), 5.25–5.24 (d, J = 5.0 Hz, 1H), 5.03–5.01 (d, J = 10 Hz, 1H), 4.40–4.37 (m, 1H), 4.34–4.30 (m, 1H), 3.76 (s, 3H), 3.75 (s, 3H), 2.52–2.43 (m, 2H), 2.12–2.07 (m, 2H), 1.90–1.84 (m, 1H), 1.71–1.67 (m, 1H), 1.47 (s, 9H), 1.46 (s, 9H); 13C{1H} (125 MHz, CDCl3) δ 173.3, 173.0, 155.3, 155.2, 133.1, 125.5, 79.95, 79.84, 53.2, 53.0, 52.3, 52.2, 35.6, 32.4, 28.4, 28.3, 23.2; FT-IR (neat) νmax 3363, 2977, 1698, 1508, 1437, 1391, 1365, 1247, 1211, 1157, 1103, 1050, 1021 cm−1; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C21H36N2O8Na 467.2363 found 467.2359.
![Molecules 27 00067 i004]()
3.5. Dimethyl (2S,4E,8S)-∆4-2,8-(di-N-(Cbz)amino)azelate (11b)
Diamino azelate 11b with Cbz protection was synthesized according to the protocol described for the synthesis of Boc counterpart 11a using N-(Cbz)-3-iodo-l-alanine methyl ester 10b (8.0 g, 0.02 mmol) and isolated as a colorless liquid (3.5 g, 63%): Rf = 0.30 (2:3 E.A/Hexanes, visualized by UV); [α]D25 +15.9 (c 1.09, CHCl3); 1H NMR (500 MHz, CD3OD): δ 7.40–7.31(m, 10H), 5.57–5.55 (d, J = 10 Hz, 1H), 5.52–5.45 (dt, J = 15, 5 Hz, 1H), 5.38–5.33 (dt, J = 15, 5 Hz, 1H), 5.29–5.27 (d, J = 10Hz, 1H), 5.16–5.11(m, 4H), 4.48–4.44 (m, 1H), 4.42–4.37 (m, 1H), 3.76 (s, 3H), 3.75 (s, 3H) 2.58–2.46 (m, 2H), 2.13–2.01 (m 2H), 1.94–1.82 (m, 1H), 1.74–1.67 (m, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ 172.2, 156.0, 136.2, 132.3, 128.57, 128.54, 128.52, 128.46, 128.25, 128.22, 128.16, 128.13, 125.32, 67.1, 67.0, 54.0, 53.0, 52.4, 52.3, 35.4, 32.4, 32.2, 28.2; FT-IR (neat) νmax 3332, 2953, 1699, 1521, 1437, 1341, 1207, 1050 cm−1; HRMS (ESI-TOF) m/z [M + H]+ calcd for C27H33N2O8 513.2231, found 513.2234.
3.6. Dimethyl (2S,4RS,5RS,8S)-2,8-di-N-(Boc)amino-4-oxiranyl-azelate (12a)
A solution of ∆4-di-N-(Boc)aminoazelate 11a (2.0 g, 4.5 mmol) in dichloromethane (DCM, 30 mL) was cooled to 0 °C and treated with m-chloroperoxybenzoic acid (2.0 g, 9.0 mmol, 2.0 equiv.). The ice bath was removed. The suspension was warmed to room temperature with stirring overnight, when TLC showed the complete consumption of olefin 11a (Rf = 0.48, 40% EtOAc in hexanes) and a new polar spot for epoxide 12a (Rf = 0.2, 40% EtOAc in hexanes). The reaction mixture was diluted with DCM (30 mL), transferred to a separatory funnel, and washed sequentially with 1N NaOH (2 × 20 mL), water (20 mL), and brine (20 mL), dried over Na2SO4, filtered, and concentrated under vacuum to a residue that was purified by flash column chromatography using 20% EtOAc in hexanes as the eluent. Evaporation of the collected fractions afforded epoxide 20a (1.75 g, 84%) as colorless oil: Rf = 0.2 (2:3 EtOAc/hexanes, visualized with KMnO4); [α]D25 +2.5 (c 0.81, CHCl3); 1H NMR (500 MHz, CD3OD): δ 4.32–4.26 (m, 1H), 4.18–4.13 (m, 1H), 3.75 (s, 3H), 3.74 (s, 3H), 2.87–2.75 (m, 2H), 1.97–1.90 (m, 2H), 1.80–1.72 (m, 1H), 1.64–1.59 (m, 1H), 1.47 (s, 20H); 13C{1H} NMR (125 MHz, CD3OD) δ 173.2, 173.0, 156.7, 156.6, 79.4, 79.2, 58.0, 57.5, 55.4, 55.3, 53.5, 53.1, 51.51, 51.4, 51.3, 34.0, 27.3; FT-IR (neat) νmax 3326, 2955, 1699, 1523, 1437, 1210, 1045, 912 cm−1; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C21H36N2O9Na 483.2313, found 483.2321.
![Molecules 27 00067 i006]()
3.7. Dimethyl (2S,4RS,5RS,8S)-2,8-di-N-(Cbz)amino-4-oxiranyl-azelate (12b)
Epoxide 12b with Cbz protection was synthesized using the protocol described for the preparation of Boc counterpart 12a using dimethyl ∆4-di-(Cbz)amino azelate 11b (3.2 g, 6.2 mmol) and isolated as a colorless liquid (2.5g, 76%): Rf = 0.21 (2:3 EtOAc/hexanes, visualized by UV); [α]D25 +7.95 (c 0.88, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.40–7.33 (m, 10H), 5.68–5.62 (d, J = 10Hz, 1H), 5.44–5.32 (d, J = 5Hz, 1H), 5.16–5.11 (m, 4H), 4.58–4.11 (m, 1H), 4.45–4.39 (s, 1H), 3.79–3.76 (s, 6H), 2.81–2.70 (m, 2H), 2.25–2.07 (m, 1H), 2.04–1.92 (m, 2H), 1.83–1.75 (m, 1H), 1.71–1.65 (m, 1H), 1.54–1.44 (m, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ 172.5, 172.1, 156.0, 136.2, 128.6, 128.3, 128.2, 67.1, 57.5, 55.3, 55.1, 53.5, 53.2, 53.0, 52.65, 52.57, 52.51, 52.2, 35.0, 30.0, 29.0, 28.0, 27.5; FT-IR (neat) νmax 3332, 2953, 1700, 1521, 1437, 1344, 1208, 1049 cm−1; HRMS (ESI-TOF) m/z [M + H]+ calcd for C27H33N2O9 529.2180, found 529.2190.
3.8. Dimethyl (2S,4RS,5RS,8S)-2,8-di-N-(Fmoc)amino-4-oxiranyl-azelate (12c)
Dimethyl (2S,4E,8S)-∆4-2,8-(di-N-(Fmoc)amino)azelate (11c) was synthesized using the protocol described for the synthesis of ∆4-di-(Boc)amino azelate 11a from N-(Fmoc)-3-iodo-L-alanine methyl ester (10c, 1.5 g, 0.0022 mol) and isolated as a colorless liquid (0.7 g, 63%): Rf = 0.21 (4:6 ethyl acetate/hexanes, visualized by UV). Epoxidation was performed as described for Boc counterpart 11a using dimethyl (2S,4E,8S)-∆4-2,8-(di-N-(Fmoc)amino)azelate (11c, 600 mg, 0.87 mmol), which gave a colorless solid (500 mg, 82%): mp 89–92 °C; Rf = 0.30 (4:6 EtOAc/hexanes, visualized by UV); [α]D25 +5.5 (c 0.51, CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.79–7.77 (d, J = 10 Hz, 4H) 7.63–7.57 (m, 4H), 7.43–7,40 (m, 4H), 7.34–7.31 (m, 4H), 5.74–5.67 (dd, J = 10, 5 Hz, 1H), 5.48–5.34 (dd, J = 12, 10 Hz, 1H), 4.60–4.51 (m, 2H), 4.46–4.40 (m, 4H), 4.26–4.22 (m, 2H), 3.81 (s, 3H), 3.78 (s, 3H), 2.85–2.73 (m, 2H), 2.16–1.74 (m, 6H); 13C{1H} NMR (125 MHz, CDCl3) δ 172.6, 172.1, 156.0, 143.8, 143.7, 141.3, 130.0, 128.0, 127.1, 125.1, 120.0, 67.2, 67.1, 67.0, 57.4, 55.3, 55.1, 53.2, 52.73, 52.7, 52.6, 52.5, 47.1, 35.0, 30.0, 28.97, 28.9, 27.6, 27.5; FT-IR (neat) νmax 3290, 2952, 1690, 1531, 1448, 1260, 1215, 1085, 1045 cm−1; HRMS (ESI-TOF) m/z [M + H]+ calcd for C41H41N2O9 705.2806, found 705.2819.
![Molecules 27 00067 i008]()
3.9. (2. S,4E,8S)-∆4-2,8-(di-N-(Boc)amino)azelic Acid (19)
A 0 °C solution of dimethyl (2S,4E,8S)-∆4-2,8-(di-N-(Boc)amino)azelate (11a, 500 mg, 1.12 mmol) in 1,4-dioxane (5 mL) was treated with a 1N solution of LiOH (94.4 mg, 2.25 mmol, 2 equiv.). The cooling bath was removed. The reaction mixture was warmed to room temperature with stirring for 3 h, at which time TLC indicated the consumption of the starting material. The volatiles were evaporated under reduced pressure. The residue was partitioned between H2O (10 mL) and EtOAc (5 mL). The aqueous phase was acidified with 1 N HCl to pH 3 and extracted with ethyl acetate (3 × 10 mL). The organic extractions were combined, dried with Na2SO4, filtered, and concentrated under vacuum to afford diacid 19 (430 mg, 92%) as a white solid: mp 71–73 °C; [α]D25 +39.0 (c 0.82, CHCl3); 1H NMR (500 MHz, DMSO-d6): δ 12.42 (s, 2H), 7.08–7.07 (d, J = 5.0 Hz, 1H), 6.97–6.96 (d, J = 5 Hz, 1H), 5.50–5.44 (m, 1H), 5.40–5.35 (m, 1H), 3.90–3.84 (m, 2H), 2.37–2.32 (m, 1H), 2.29–2.23 (m, 1H), 1.71–1.50 (m, 4H), 1.39 (s, 9H), 1.38 (s, 9H); 13C{1H} NMR (125 MHz, DMSO-d6) δ 175.0, 174.0, 156.03, 155.88, 132.2, 127.0, 78.47, 78.41, 60.2, 54.1, 53.3, 34.5, 31.1, 28.68, 28.66; FT-IR (neat) νmax 3697, 2980, 1694, 1507, 1393, 1367, 1245, 1157, 1053, 1033, 1018 cm−1; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C19H32N2O8Na 439.2050, found 439.2070.
![Molecules 27 00067 i009]()
3.10. (1′R,5S)-3-N-(Boc)amino-5-[1′-iodo-4′-N-(Boc)amino-4′-methoxcarbonylbutyl]-tetrahydrofuran-2-one [(1′R,5S)-16]
A 0 °C mixture of carboxylic acid (1′R,5S)-20 (2.1 g, 3.87 mmol) and K2CO3 (800 mg, 5.8 mmol, 1.5 equiv.) in DMF (20 mL) was treated with methyl iodide (820 mg, 5.8 mmol, 1.5 equiv.). The ice bath was removed. After stirring for 2–3 h, the room temperature mixture exhibited a nonpolar spot (2:3 EtOAc/hexanes) by TLC and indicated a new peak at RT = 9.0 min (C18 column, 10:90 CH3CN:H2O) by LCMS analysis, with a molecular ion of [M + Na]+ m/z 579. The reaction mixture was diluted with water and extracted with ethyl acetate (4 × 50 mL). The ethyl acetate layer was washed with water (4 × 50 mL) and brine (2 × 30 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography using 20–30% EtOAc in hexanes as the eluent. Evaporation of the collected fractions gave tetrahydrofuran-2-one (1′R,5S)-16 (1.1g, 55% from diacid 19) as a colorless solid: mp 58–60 °C; Rf = 0.56 (2:3 EtOAc/hexanes, visualized by KMnO4), [α]D25 +13.4 (c 0.68, CHCl3); 1H NMR (500 MHz, CDCl3): δ 5.10–5.08 (d, J = 10Hz, 2H), 4.45–4.41 (m, 1H), 4.37–4.33 (m, 2H), 4.08–4.04 (t, J = 10 Hz, 1H), 3.79 (s, 3H), 3.14–3.09 (m, 1H), 2.23–2.17 (m, 1H), 2.11–2.05 (m, 1H), 1.95–1.87 (m, 2H), 1.78–1.73 (m, 1H), 1.48 (s, 9H), 1,47 (s, 9H); 13C{1H} NMR (125 MHz, CDCl3) δ 174.0, 173.0, 155.3, 130.0, 81.0, 80.2, 79.2, 53.0, 52.5, 52.0, 38.0, 36.0, 32.3, 32.0, 28.31, 28.27; FT-IR (neat) νmax 3281, 2921, 2853, 1801, 1747, 1697, 1674, 1537, 1451, 1368, 1294, 1252, 1213, 1154, 1060, 1029, 1005 cm−1; HRMS (ESI-TOF) m/z [M + Na]+ calcd for C20H33IN2O8Na 579.1173, found 579.1195.
![Molecules 27 00067 i010]()
3.11. (1′R,5S)-3-N-(Boc)amino-5-[1′-iodo-4′-N-(Boc)amino-4′-hydroxcarbonylbutyl]-tetrahydrofuran-2-one [(1′R,5S)-20]
A solution of diacid 19 (1.6 g, 3.8 mmol) in acetonitrile (20 mL) was treated with Cs2CO3 (3.7 g, 11.5 mmol, 3 equiv.), stirred for 15 min, cooled to 0 °C with an ice bath, and treated with iodine (2.93 g, 11.5 mmol, 3 equiv.). The ice bath was removed. After stirring for 3–4 h, the reaction mixture had warmed to room temperature and was observed by LCMS to contain a new peak at RT = 8.1 min (C18 column, 10:90 CH3CN:H2O) with a molecular ion [M + Na]+ m/z 565. The reaction mixture was filtered through a pad of Celite™ and the filter cake was washed with acetonitrile (3 × 30 mL). The filtrate and washings were combined and evaporated under reduced pressure. The residue was partitioned between H2O (50 mL) and EtOAc (25 mL). The aqueous phase was acidified with 1 N HCl to pH 3 and extracted with ethyl acetate (3 × 50 mL). The organic extractions were combined, dried with Na2SO4, filtered, and concentrated under vacuum to afford tetrahydrofuran-2-one (1′R,5S)-20 (2.1 g) as a pale-yellow solid, which was used without further purification.
![Molecules 27 00067 i011]()























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}