
An Experimental Design Approach to Quantitative Expression for Quality Control of a Multicomponent Antidiabetic Formulation by the HILIC Method

 ,
,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
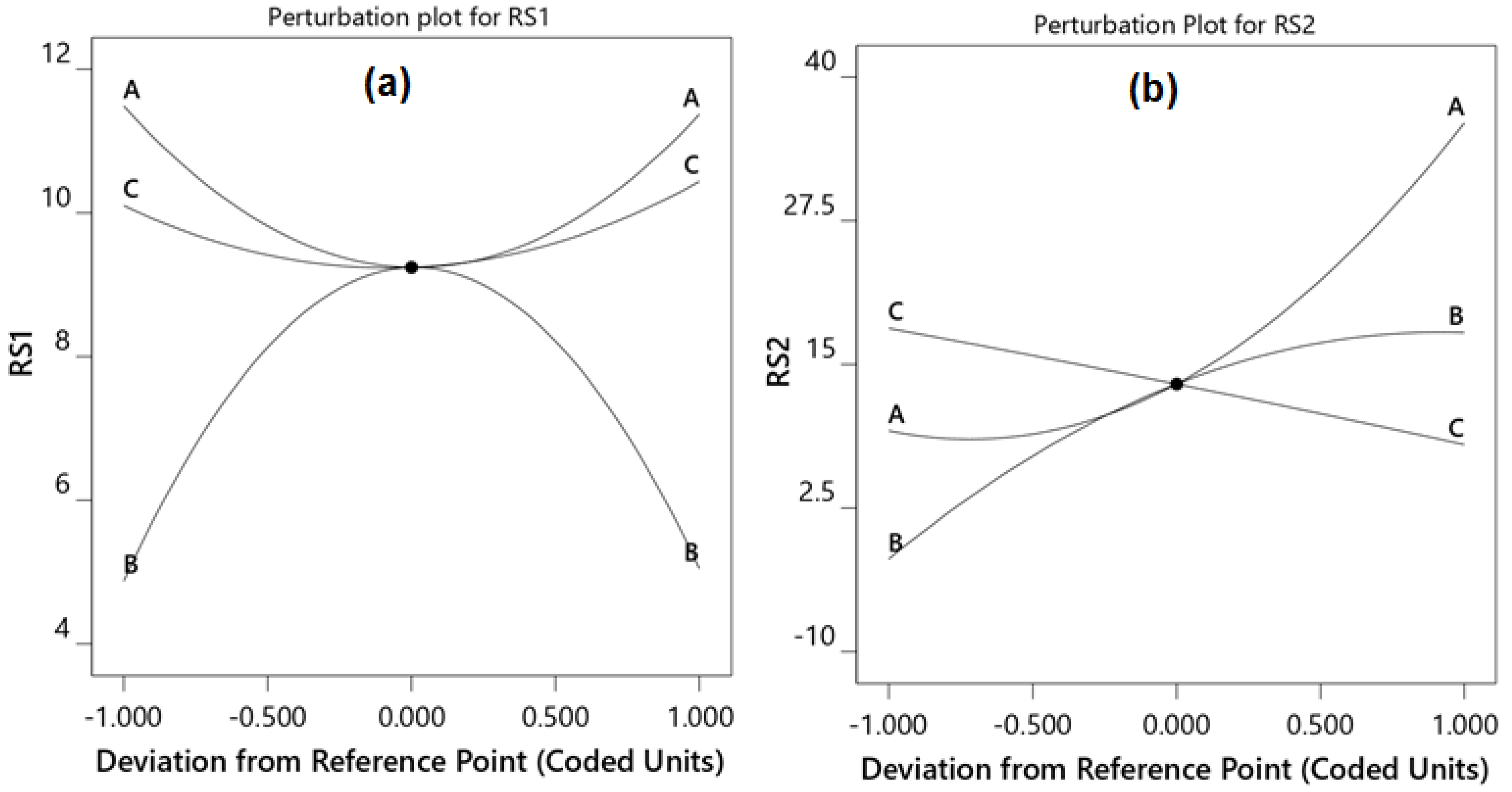
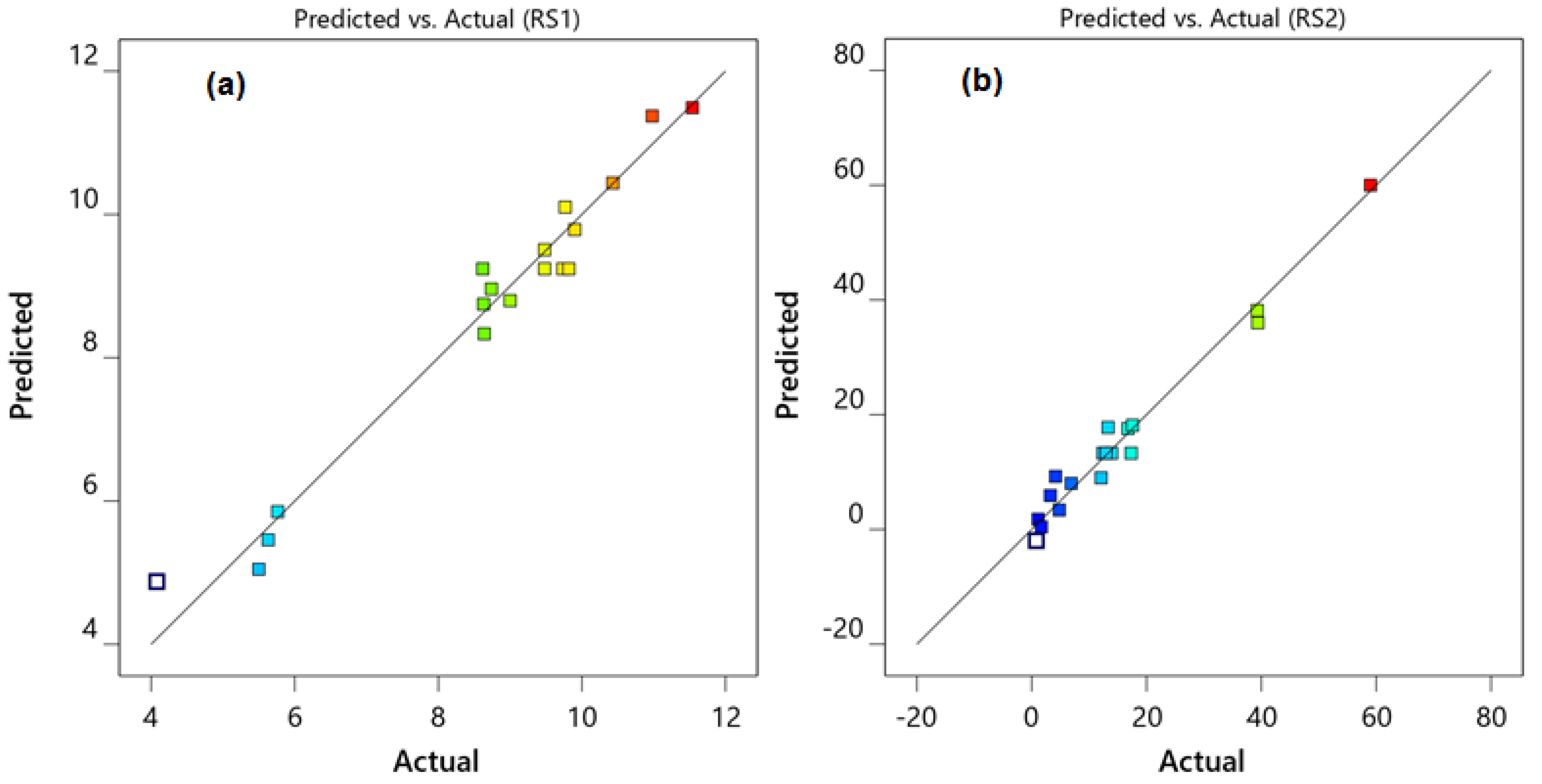
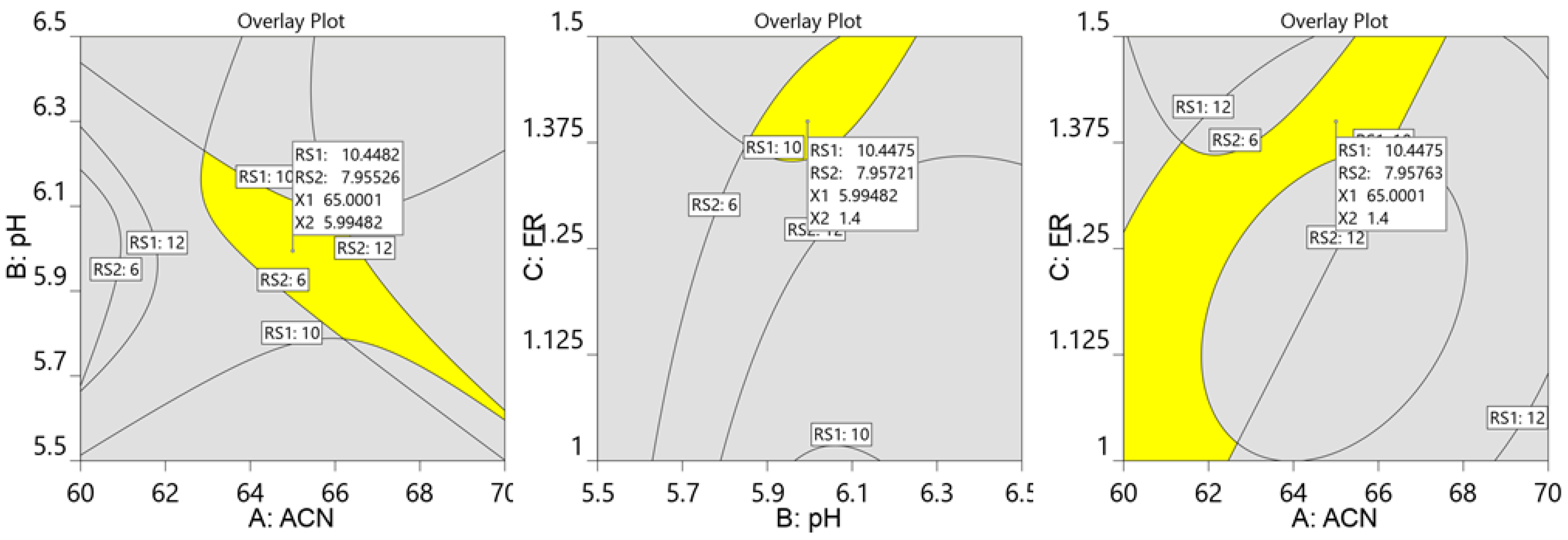
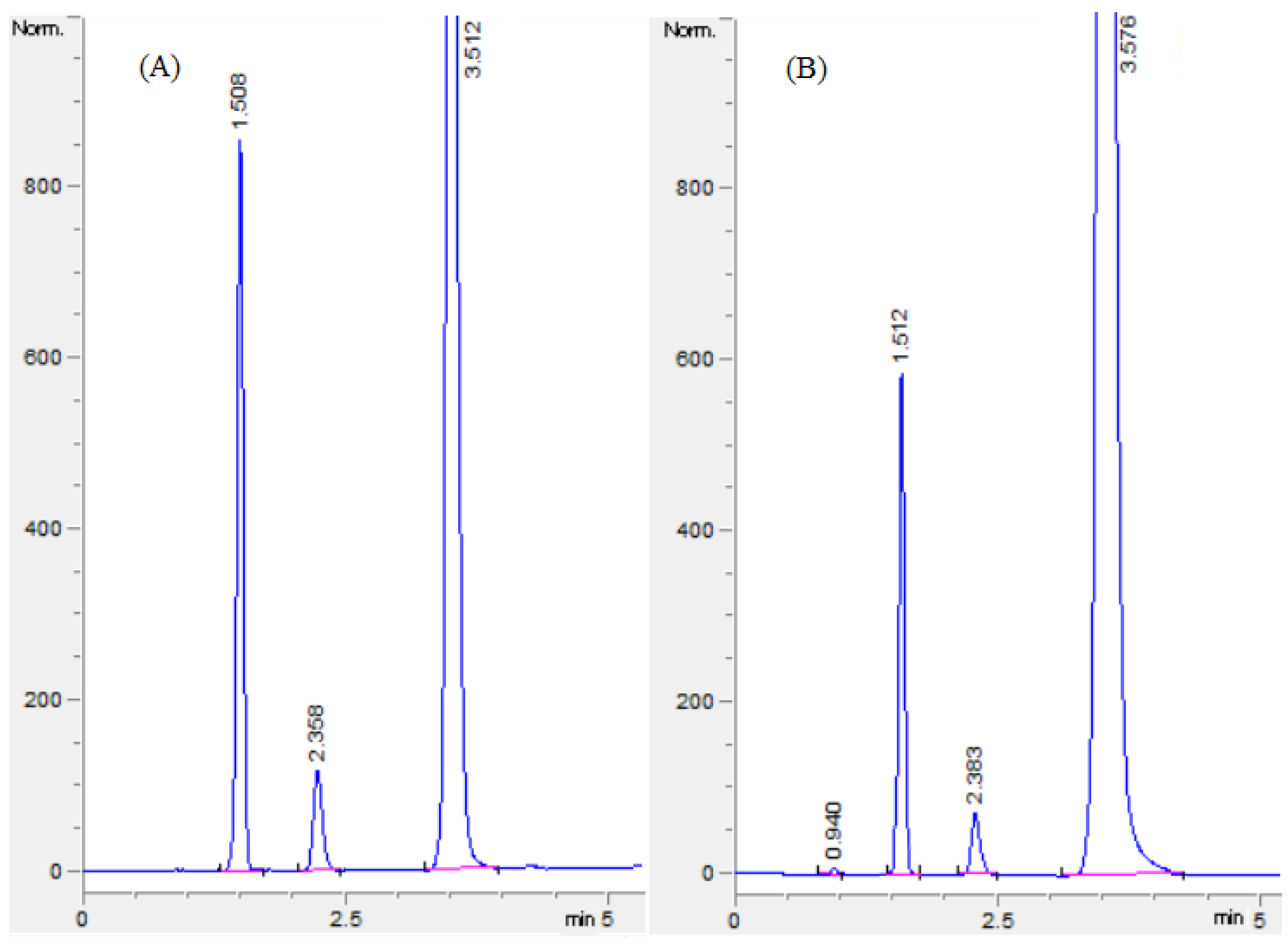
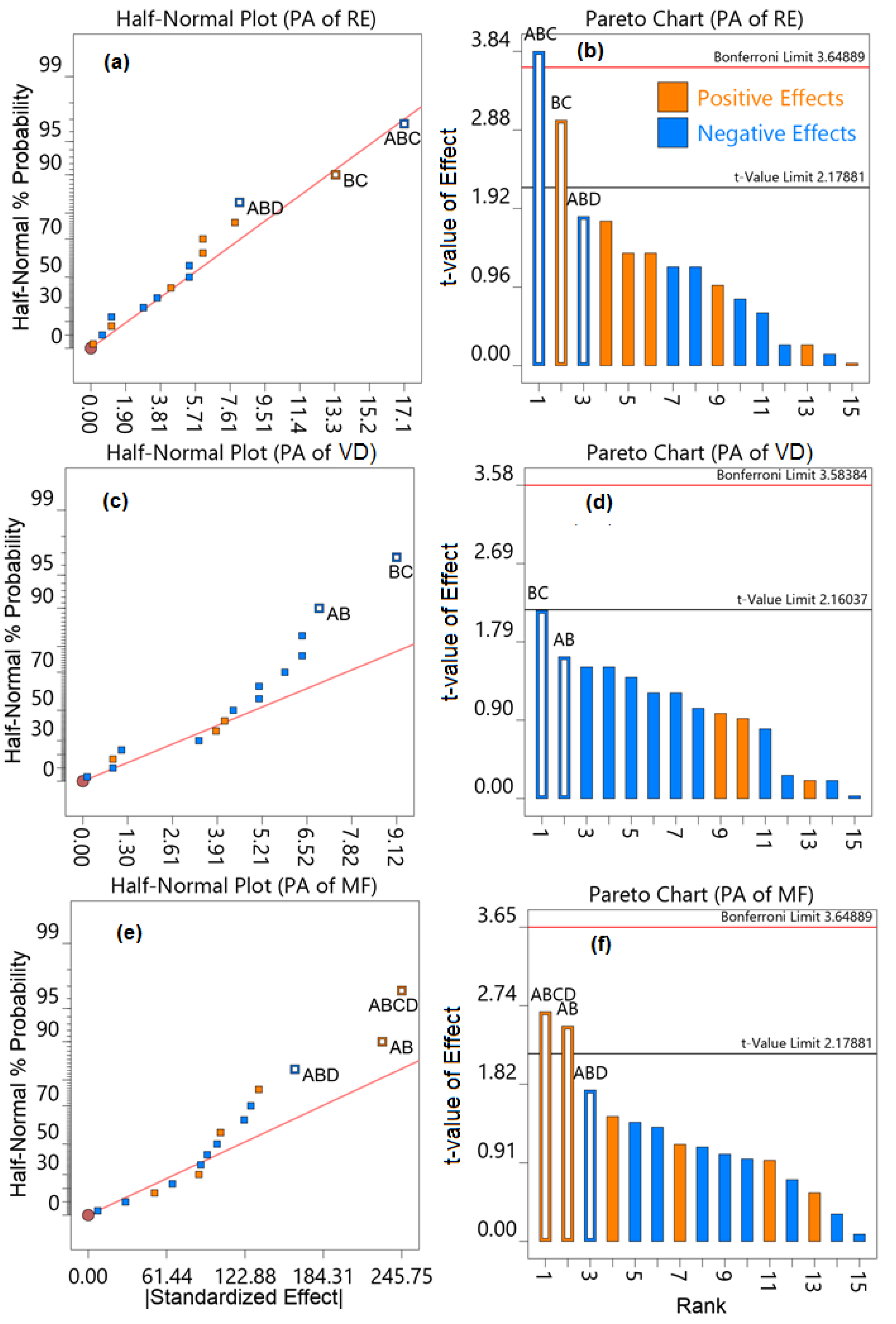
2.1. Optimization of Chromatographic Conditions
2.2. Validation of HILIC Procedure
2.2.1. System Suitability Tests
2.2.2. Linearity of Calibration Curve
2.2.3. Sensitivity
2.2.4. Precision and Accuracy
2.2.5. Selectivity
2.2.6. Stability of Solutions
2.2.7. Robustness
2.3. Application of the HPLC Method for the Analysis of Formulation
3. Materials and Methods
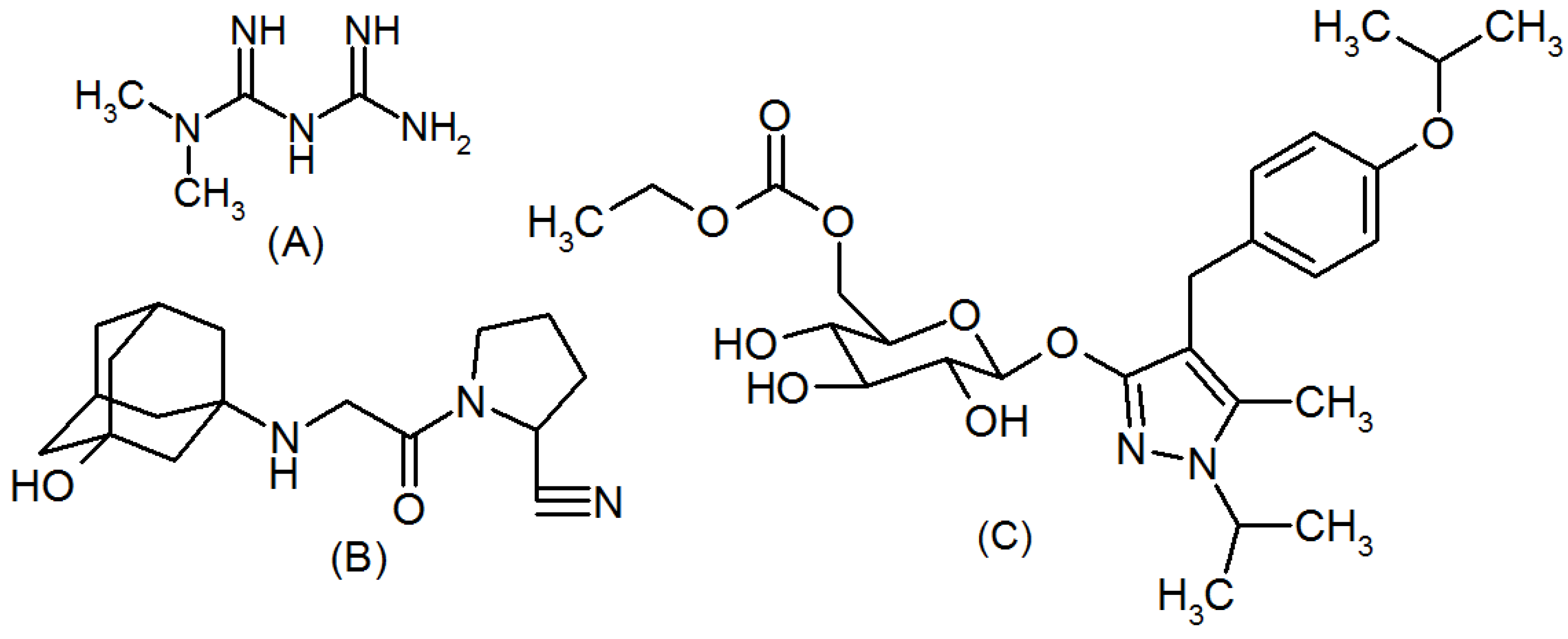
3.1. Materials
3.2. Instrumentation and Software
3.3. Preparation of Primary Stock and Working Standard Solutions
3.4. Preparation of Sample Solution
3.5. Chromatographic Procedure
3.6. Optimization by Central Composite Design
3.7. Validation of the Chromatographic Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chatterjee, S.; Davies, M.; Khunti, K. Type 2 diabetes. Lancet 2017, 389, 2239–2251. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef] [PubMed]
- Mouhayyar, C.E.; Riachy, R.; Khalil, A.B.; Eid, A.; Azar, S. SGLT2 Inhibitors, GLP-1 Agonists, and DPP-4 Inhibitors in Diabetes and Microvascular Complications: A Review. Int. J. Endocrinol. 2020, 2020, 1762164. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; Puckett, C.; Triplitt, C.; Maggs, D.; Adams, J.; Cersosimo, E.; DeFronzo, R.A. Initial combination therapy with metformin, pioglitazone and exenatide is more effective than sequential add-on therapy in subjects with new-onset diabetes. Results from the Efficacy and Durability of Initial Combination Therapy for Type 2 Diabetes (EDICT): A randomized trial. Diabetes Obes. Metab. 2015, 17, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes—2020 American Diabetes Association. Diabetes Care 2020, 43 (Suppl. S1), S98–S110; [Google Scholar] [CrossRef] [Green Version]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.J.; Wilcock, C.; Scarpello, J.H. Metformin and the intestine. Diabetologia 2008, 51, 1552–1553. [Google Scholar] [CrossRef] [Green Version]
- Al-Janabi, K.W.; Mahmood, A.K.; Luaibi, H.M. Determination of the Dissociation Constants of Metformin from a Second Derivative UV Spectrum. Int. J. Res. Pharm. Sci. 2020, 11, 790–796. [Google Scholar] [CrossRef]
- Joshi, H.; Khristi, A. Absorbance ratio method development and validation for the simultaneous estimation of teneligliptin hydrobromide hydrate and metformin hydrochloride in tablet dosage form. Int. Res. J. Pharm. 2018, 9, 47–55. [Google Scholar] [CrossRef]
- Majithia, R.H.; Khodadiya, A.; Patel, V.B. Spectrophotometric method development and validation for simultaneous estimation of Anagliptin and Metformin HCl BY Q—Absorption ratio method in synthetic mixture. Heliyon 2020, 6, e03855. [Google Scholar] [CrossRef]
- Mrunal, C.S.; Annapurna, M.M. Simultanous determination of metformin and teneligliptin by liquid chromatography in tablets. Asian J. Pharm. 2018, 12, 736–739. [Google Scholar] [CrossRef]
- Gurav, S.B.; Manish, N. Development and Validation of Novel Stability- Indicating LC Method for the Determination of Saxagliptin and Metformin. Indian J. Pharm. Educ. Res. 2020, 54, s350–s357. [Google Scholar] [CrossRef]
- Chaudhari, K.; Wang, J.; Xu, Y.; Winters, A.; Wang, L.; Dong, X.; Cheng, E.Y.; Liu, R.; Yang, S.-H. Determination of metformin bio-distribution by LC-MS/MS in mice treated with a clinically relevant paradigm. PLoS ONE 2020, 15, e0234571. [Google Scholar] [CrossRef]
- Vella, A. Mechanism of action of DPP-4 inhibitors—New insights. J. Clin. Endocrinol. Metab. 2012, 97, 2626–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baksh, S.N.; Segal, J.B.; McAdams-DeMarco, M.; Kalyani, R.R.; Alexander, G.C.; Ehrhardt, S. Dipeptidyl peptidase-4 inhibitors and cardiovascular events in patients with type 2 diabetes, without cardiovascular or renal disease. PLoS ONE 2020, 15, e0240141. [Google Scholar] [CrossRef] [PubMed]
- Kawanami, D.; Takashi, Y.; Takahashi, H.; Motonaga, R.; Tanabe, M. Renoprotective Effects of DPP-4 Inhibitors. Antioxidants 2021, 10, 246. [Google Scholar] [CrossRef]
- Bhavadasan, K.; Davis, A.M.; Kolanthavel, B. Impact of Dipeptidyl Peptidase-4 Inhibitors on Glycemic Control and Cardiovascular Safety with Adherence: An Overview. Int. J. Diabetes Metab. 2019, 25, 90–99. [Google Scholar] [CrossRef]
- Joshi, S.S.; Singh, T.; Newby, D.E.; Singh, J. Sodium-glucose co-transporter 2 inhibitor therapy: Mechanisms of action in heart failure. Heart 2021, 107, 1032–1038. [Google Scholar] [CrossRef]
- Dayoub, L.A.; Amali, F. Development of a new visible Spectrophotometric analytical method for determination of Vildagliptin in bulk and Pharmaceutical dosage forms. Res. J. Pharm. Technol. 2020, 13, 2807–2810. [Google Scholar] [CrossRef]
- Kumari, B.; Khansili, A. Analytical Method Development and Validation of UV-visible Spectrophotometric Method for the Estimation of Vildagliptin in Gastric Medium. Drug Res. 2020, 70, 417–423. [Google Scholar] [CrossRef]
- Moneeb, M.S. Spectrophotometric and spectrofluorimetric methods for the determination of saxagliptin and vildagliptin in bulk and pharmaceutical preparations. Bull. Fac. Pharmacy Cairo Univ. 2013, 51, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Patil, K.R.; Deshmukh, T.A.; Patil, V.R. A stability indicating HPTLC method development and validation for analysis of Vildagliptin as bulk drug and from its pharmaceutical dosage form. Int. J. Pharm. Sci. Res. 2020, 11, 2310–2316. [Google Scholar] [CrossRef]
- Attimarad, M.; Sreeharsha, N.; Aldhubaib, B.E.; Nair, A.B.; Venugopala, K.N. Determination on Vildagliptin in Rat Plasma by Capillary Electrophoresis Tandem Mass Spectrometry: It’s Application to Pharmacokinetic Study. Indian J. Pharma. Edu. Res. 2017, 51, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Giordani, C.F.A.; Campanharo, S.; Wingert, N.R.; Bueno, L.M.; Manoel, J.W.; Garcia, C.V.; Volpato, N.M.; Iop, G.D.; Mello, P.D.A.; Flores, E.M.D.M.; et al. UPLC-ESI/Q-TOF MS/MS Method for Determination of Vildagliptin and its Organic Impurities. J. Chromatogr. Sci. 2020, 58, 718–725. [Google Scholar] [CrossRef]
- Altunkaynak, Y.; Yavuz, Ö.; Levent, A. Firstly electrochemical examination of vildagliptin at disposable graphite sensor: Sensitive determination in drugs and human urine by square-wave voltammetry. Microchem. J. 2021, 170, 106653. [Google Scholar] [CrossRef]
- Nelinson, D.S.; Sosa, J.M.; Chilton, R.J. SGLT2 inhibitors: A narrative review of efficacy and safety. J. Osteopath. Med. 2021, 121, 229–239. [Google Scholar] [CrossRef]
- Tentolouris, A.; Vlachakis, P.; Tzeravini, E.; Eleftheriadou, I.; Tentolouris, N. SGLT2 Inhibitors: A Review of Their Antidiabetic and Cardioprotective Effects. Int. J. Environ. Res. Public Health 2019, 16, 2965. [Google Scholar] [CrossRef] [Green Version]
- Bonora, B.M.; Avogaro, A.; Fadini, G.P. Extraglycemic Effects of SGLT2 Inhibitors: A Review of the Evidence. Diabetes Metab. Syndr. Obes. 2020, 13, 161–174. [Google Scholar] [CrossRef] [Green Version]
- Scheen, A.J. Pharmacokinetic Characteristics and Clinical Efficacy of an SGLT2 Inhibitor Plus DPP-4 Inhibitor Combination Therapy in Type 2 Diabetes. Clin. Pharmacokinet. 2017, 56, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Lingvay, I. Sodium Glucose Cotransporter 2 and Dipeptidyl Peptidase-4 Inhibition: Promise of A Dynamic Duo. Endocr. Pract. 2017, 23, 831–840. [Google Scholar] [CrossRef]
- Sigafoos, J.F.; Bowers, G.D.; Castellino, S.; Culp, A.G.; Wagner, D.S.; Reese, M.J.; Humphreys, J.E.; Hussey, E.K.; Semmes, R.L.C.; Kapur, A.; et al. Assessment of the drug interaction risk for remogliflozin etabonate, a sodium-dependent glucose cotransporter-2 inhibitor: Evidence from in vitro, human mass balance, and ketoconazole interaction studies. Drug Metab. Dispos. 2012, 40, 2090–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tayade, A.B.; Patil, A.S.; Shirkhedkar, A.A. Development and Validation of Zero Order UV-Spectrophotometric Method by Area under Curve Technique and High Performance Thin Layer Chromatography for the Estimation of Remogliflozin Etabonate in Bulk and In-House Tablets. Invent. Rapid Pharm. Anal. Qual. Assur. 2019, 3, 1–5. [Google Scholar]
- Bhatkar, T.; Badkhal, A.V.; Bhajipale, N.S. Stability Indicating RP-HPLC Method Development and Validation for the Estimation of Remogliflozin Etabonate in Bulk and Pharmaceutical Dosage Form. Int. J. Pharm. Res. 2020, 12, 4197–4207. [Google Scholar] [CrossRef]
- Shah, D.A.; Gondalia, I.I.; Patel, V.B.; Mahajan, A.; Chhalotiya, U.K. Stability indicating liquid chromatographic method for the estimation of remogliflozin etabonate. J. Chem. Metrol. 2020, 14, 125–132. [Google Scholar] [CrossRef]
- Abdel-Ghany, M.; Abdel-Aziz, O.; Ayad, M.F.; Tadros, M.M. Validation of Different Spectrophotometric Methods for Determination of Vildagliptin and Metformin in Binary Mixture. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 2014, 125, 175–182. [Google Scholar] [CrossRef]
- Shakoor, A.; Ahmed, M.; Ikram, R.; Hussain, S.; Tahir, A.; Jan, B.M.; Adnan, A. Stability-indicating RP-HPLC method for simultaneous determination of metformin hydrochloride and vildagliptin in tablet and biological samples. Acta Chromatogr. 2020, 32, 39–43. [Google Scholar] [CrossRef]
- Attimarad, M.; Sreeharsha, N.; Aldhubaib, B.E.; Nair, A.B. Simultaneous Determination of Metformin and Three Gliptins in Pharmaceutical Formulations Using RP HPLC: Application to Stability Studies on Linagliptin Tablet Formulation. Indian J. Pharma. Edu. Res. 2014, 48, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Attimarad, M. Multivariate optimization of a capillary zone electrophoresis assay method for simultaneous quantification of metformin and vildagliptin from a formulation. J. Liq. Chromatogr. Relat. Technol. 2016, 39, 401–407. [Google Scholar] [CrossRef]
- Attimarad, M.; Nair, A.B.; Sreeharsha, N.; Al-Dhubiab, B.E.; Venugopala, K.N.; Shinu, P. Development and Validation of Green UV Derivative Spectrophotometric Methods for Simultaneous Determination Metformin and Remogliflozin from Formulation: Evaluation of Greenness. Int. J. Environ. Res. Public Health 2021, 18, 448. [Google Scholar] [CrossRef]
- Attimarad, M.; Elgorashe, R.E.E.; Subramaniam, R.; Islam, M.M.; Venugopala, K.N.; Nagaraja, S.; Balgoname, A.A. Development and Validation of Rapid RP-HPLC and Green Second-Derivative UV Spectroscopic Methods for Simultaneous Quantification of Metformin and Remogliflozin in Formulation using Experimental Design. Separations 2020, 7, 5925. [Google Scholar] [CrossRef]
- Tammisetty, M.; Challa, B.R.; Puttagunta, S.B. A novel analytical method for the simultaneous estimation of remogliflozin and metformin hydrochloride by UPLC/PDA in bulk and formulation. Application to the estimation of product traces. Turk. J. Pharm. Sci. 2020, 39699, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Attimarad, M.; Venugopala, K.N.; Al-Dhubiab, B.E.; Elgorashe, R.E.E.; Shafi, S. Development of Ecofriendly Derivative Spectrophotometric Methods for the Simultaneous Quantitative Analysis of Remogliflozin and Vildagliptin from Formulation. Molecules 2021, 26, 6160. [Google Scholar] [CrossRef] [PubMed]
- Mandale, D.A.; Shah, C.; Jatt, R. Development and Validation of Novel RP-HPLC Method for the Simultaneous Determination of Remogliflozin and Vildagliptin in Bulk and in synthetic Mixture. J. Pharm. Res. Int. 2021, 33, 338–349. [Google Scholar] [CrossRef]
- Attimarad, M.; Venugopala, K.N.; Islam, M.M.; Shafi, S.; Altaysan, A.I. Rapid Simultaneous Quantitative Analysis of Hypoglycemic agents by RP HPLC: Development, Validation and Application to Medicine. Indian J. Pharm. Edu. Res. 2022, 56, 564–572. [Google Scholar] [CrossRef]
- Sai Prudhvi, N.; Venkateswarlu, B.S.; Kumudhavalli, M.V.; Muruganantham, V. Novel stability indicating LC-MS/MS method for the simultaneous estimation of remogliflozin etabonate and vildagiption human plasma. J. Med. Pharm. Allied. Sci. 2021, 10, 1655. [Google Scholar] [CrossRef]
- Sahu, P.K.; Ramisetti, N.R.; Cecchi, T.; Swain, S.; Patro, C.S.; Panda, J. An overview of experimental designs in HPLC method development and validation. J. Pharm. Biomed. Anal. 2018, 147, 590–611. [Google Scholar] [CrossRef]
- Gurrala, S.; Raj, S.; Subrahmanyam, C.V.S.; Anumolu, P.D. Multivariate optimization of liquid chromatographic conditions for determination of dapagliflozin and saxagliptin, application to an in vitro dissolution and stability studies. Future J. Pharm. Sci. 2021, 7, 85. [Google Scholar] [CrossRef]
- Hemdan, A.; Al-Tannak, N.F.; Mohamed, E.H. Development of a multivariate model with desirability-based optimization for determination of atenolol and hydrochlorothiazide by eco-friendly HPLC method with fluorescence detection. J. Sep. Sci. 2022, 45, 824–831. [Google Scholar] [CrossRef]
- Attimarad, M.; Venugopala, K.N.; SreeHarsha, N.; Aldhubiab, B.E.; Nair, A.B. Validation of rapid RP-HPLC method for concurrent quantification of amlodipine and celecoxib in pure and formulation using an experimental design. Microchem. J. 2020, 152, 104365. [Google Scholar] [CrossRef]
- Jovanović, M.; Rakić, T.; Jančić–Stojanović, B. Theoretical and Empirical Models in Hydrophilic Interaction Liquid chromatography. Instrument. Sci. Technol. 2014, 42, 230–266. [Google Scholar] [CrossRef]
- Kasagicı-Vujanovic, I.; Jancicˇı-Stojanovic, B.; Rakicı, T.; Ivanovicı, D. Design of experiments in optimization and validation of a hydrophilic interaction liquid chromatography method for determination of amlodipine besylate and bisoprolol fumarate. J. Liq. Chromatogr. Relat. Technol. 2015, 38, 919–928. [Google Scholar] [CrossRef]
- Moussa, B.A.; Mahrouse, M.A.; Fawzy, M.G. Application of experimental design in HPLC method optimization and robustness for the simultaneous determination of canagliflozin, empagliflozin, linagliptin, and metformin in tablet. Biomed. Chromatogr. 2021, 35, e5155. [Google Scholar] [CrossRef] [PubMed]
- Mahrousea, M.A.; Lamie, N.T. Experimental design methodology for optimization and robustness determination in ion pair RP-HPLC method development: Application for the simultaneous determination of metformin hydrochloride, alogliptin benzoate and repaglinide in tablets. Microchem. J. 2019, 147, 691–706. [Google Scholar] [CrossRef]
- Verseput, R. Digging into DOE. Selecting the Right Central Composite Design for Response Surface Methodology Applications. Available online: https://www.qualitydigest.com/june01/html/doe.html (accessed on 1 May 2022).
- Buszewski, B.; Noga, S. Hydrophilic interaction liquid chromatography (HILIC)—A powerful separation technique. Anal. Bioanal. Chem. 2012, 402, 231–247. [Google Scholar] [CrossRef] [Green Version]
- Garg, N.K.; Sharma, G.; Singh, B.; Nirbhavane, P.; Katare, O.P. Quality by Design (QbD)-Based Development and Optimization of a Simple, Robust RP-HPLC Method for the Estimation of Methotrexate. J. Liq. Chromatogr. Relat. Technol. 2015, 38, 1629–1637. [Google Scholar] [CrossRef]
- Musters, J.; van den Bos, L.; Kellenbach, E. Applying QbD Principles to Develop a Generic UHPLC Method Which Facilitates Continual Improvement and Innovation Throughout the Product Lifecycle for a Commercial API. Org. Process Res. Dev. 2013, 17, 87–96. [Google Scholar] [CrossRef]
- Attimarad, M.; Venugopala, K.N.; Nair, A.B.; Sreeharsha, N.; Deb, P.K. Experimental Design Approach for Quantitative Expressions of Simultaneous Quantification of Two Binary Formulations Containing Remogliflozin and Gliptins by RP-HPLC. Separations 2022, 9, 23. [Google Scholar] [CrossRef]
- El-zaher, A.A.; Mahrouse, M.A. A validated spectrofluorimetric method for the determination of nifuroxazide through coumarin formation using experimental design. Chem. Cent. J. 2013, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Siregar, C.; Prabaningdyah, N.K.; Choiri, S.; Riyanto, S.; Rohman, A. Optimization of HPLC Using Central Composite Design for Determination of Curcumin and Demethoxycurcumin in Tablet Dosage Form. Dhaka Univ. J. Pharm. Sci. 2017, 16, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Dritsa, V.; Rigas, F.; Doulia, D.; Avramides, E.J.; Hatzianestis, I. Optimization of Culture Conditions for the Biodegradation of Lindane by the Polypore Fungus Ganoderma australe. Water Air Soil Pollut. 2009, 204, 19–27. [Google Scholar] [CrossRef]
- Stojanovic, B.J. Factorial-Based Designs in Liquid Chromatography. Chromatographia 2013, 76, 227–240. [Google Scholar] [CrossRef]
- Kamau, F.N.; Chepkwony, H.K.; Ngugi, J.K.; Debremaeker, D.; Roets, E.; Hoogmartens, J. Isocratic liquid chromatographic method for the analysis of azithromycin and its structurally related substances in bulk samples. J. Chromatogr. Sci. 2002, 40, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, K.G.; Shah, P.M.; Shah, P.A.; Tejal, R. Validated high-performance thin-layer chromatographic (HPTLC) method for simultaneous determination of nadifloxacin, mometasone furoate, and miconazole nitrate cream using fractional factorial design. J. Food Drug Anal. 2016, 24, 610–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ICH, Q2B. Validation of Analytical Procedures: Methodology. In Proceedings of the International Conference on Harmonization, Geneva, Switzerland, 19 May 1997. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pattern-Coded Value | Run | Factor 1: Percentage Acetonitrile | Factor 2: pH | Factor 3: Flow Rate (mL·Min−1) | Resolution between RE and VD | Resolution between VD and MF |
|---|---|---|---|---|---|---|
| + + + | 1 | 70 | 6.5 | 1.4 | 5.76 | 39.29 |
| − − + | 2 | 60 | 5.5 | 1.4 | 9.90 | 4.81 |
| 0 0 0 | 3 | 65 | 6 | 1.2 | 9.74 | 13.32 |
| 0 0 0 | 4 | 65 | 6 | 1.2 | 9.62 | 12.98 |
| 0 0 0 | 5 | 65 | 6 | 1.2 | 9.48 | 13.93 |
| + − − | 6 | 70 | 5.5 | 1 | 8.64 | 16.77 |
| 0 + 0 | 7 | 65 | 6.5 | 1.2 | 5.50 | 13.30 |
| 0 − 0 | 8 | 65 | 5.5 | 1.2 | 4.08 | 0.81 |
| − − − | 9 | 60 | 5.5 | 1 | 5.63 | 1.11 |
| 0 0 0 | 10 | 65 | 6 | 1.2 | 9.82 | 12.41 |
| 0 0 − | 11 | 65 | 6 | 1 | 9.77 | 17.57 |
| − + + | 12 | 60 | 6.5 | 1.4 | 8.74 | 1.69 |
| − + − | 13 | 60 | 6.5 | 1 | 8.63 | 12.09 |
| + 0 0 | 14 | 70 | 6 | 1.2 | 10.98 | 39.38 |
| − 0 0 | 15 | 60 | 6 | 1.2 | 11.54 | 4.15 |
| + + − | 16 | 70 | 6.5 | 1 | 9.48 | 59.01 |
| + − + | 17 | 70 | 5.5 | 1.4 | 9.00 | 3.26 |
| 0 0 + | 18 | 65 | 6 | 1.4 | 10.43 | 7.89 |
| Coefficient Terms | Resolution between RE and VD (RS1) | Resolution between VD and MF (RS2) | ||
|---|---|---|---|---|
| Coefficient Value | p-Value | Coefficient Value | p-Value | |
| Constant | +9.24 | <0.0001 | +1427.372 | <0.0001 |
| ACN a | −0.058 | 0.7451 | −61.984 | <0.0001 |
| pH | +0.086 | 0.6309 | +88.987 | <0.0001 |
| Flow rate | +0.168 | 0.3571 | +337.112 | 0.0026 |
| ACN × pH | −0.529 | 0.0252 | +3.520 | 0.0001 |
| ACN × flow rate | −0.967 | 0.0010 | −3.317 | 0.0327 |
| pH × flow rate | −1.03 | 0.0007 | −25.406 | 0.0837 |
| ACN2 | +2.19 | 0.0002 | +0.365 | 0.0033 |
| pH2 | −4.28 | <0.0001 | −22.299 | 0.0357 |
| Flow rate2 | +1.03 | 0.0145 | −2.570 | 0.9640 |
| Parameters | RE | VD | MF |
|---|---|---|---|
| System suitability results | |||
| Retention time ± SD | 1.51 ± 0.021 | 2.35 ± 0.029 | 3.54 ± 0.065 |
| Peak area ± SD | 5765.5 ± 46.84 a | 824.29 ± 7.34 b | 9678.47 ± 85.75 c |
| Resolution ± SD | - | 7.85 ± 0.05 d | 10.40 ± 0.08 e |
| Tailing factor ± SD | 1.12 ± 0.022 | 1.05 ± 0.013 | 1.15 ± 0.023 |
| Theoretical plate ± SD | 7544.16 ± 65.24 | 10,908.64 ± 78.98 | 20,506.24 ± 96.47 |
| Linearity | |||
| Linearity range (µg/mL) | 20–150 | 10–75 | 50–750 |
| Slope | 57.915 | 17.171 | 50.487 |
| Intercept | 48.462 | −46.048 | −237.84 |
| Regression coefficient (r2) | 0.9992 | 0.9988 | 0.9997 |
| Sensitivity | |||
| DL (µg/mL) | 4.73 | 2.81 | 1.48 |
| QL (µg/mL) | 14.34 | 8.54 | 43.88 |
| Drug | Within-Day | Between-Day | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Amount (µg/mL) | Amount Found Mean (n = 3) ± SD | % RSD | % Recovery | % RE | Amount Found Mean (n = 9) ± SD | % RSD | % Recovery | % RE | |
| RE | 20 | 19.86 ± 0.14 | 0.70 | 99.30 | −0.70 | 20.13 ± 0.23 | 1.14 | 100.65 | 0.65 |
| 75 | 74.18 ± 1.05 | 1.42 | 98.91 | −1.09 | 74.25 ± 1.35 | 1.82 | 99.00 | −1.00 | |
| 150 | 148.64 ± 2.33 | 1.57 | 99.09 | −0.91 | 147.92 ± 2.09 | 1.41 | 98.61 | −1.39 | |
| VD | 10 | 10.02 ± 0.12 | 1.20 | 100.20 | 0.20 | 9.86 ± 0.16 | 1.62 | 98.60 | −1.40 |
| 40 | 39.65 ± 0.37 | 0.93 | 99.13 | −0.88 | 39.38 ± 0.69 | 1.75 | 98.45 | −1.55 | |
| 75 | 74.24 ± 0.82 | 1.10 | 98.99 | −1.01 | 74.46 ± 1.13 | 1.52 | 99.28 | −0.72 | |
| MF | 50 | 49.16 ± 0.51 | 1.04 | 98.32 | −1.68 | 49.07 ± 0.52 | 1.06 | 98.14 | −1.86 |
| 350 | 346.73 ± 5.34 | 1.54 | 99.07 | −0.93 | 353.79 ± 4.66 | 1.32 | 101.08 | 1.08 | |
| 750 | 745.04 ± 6.79 | 0.91 | 99.34 | −0.66 | 746.21 ± 7.08 | 0.95 | 99.49 | −0.51 | |
| Label Claim (mg/Tablet) | Amount Taken (µg·mL−1) | Amount Found (µg·mL−1) | % Purity | %RSD |
|---|---|---|---|---|
| Formulation 1 (VD 50 mg + RE 100 mg + MF 500 mg) | VD 20 RE 40 MF 200 | 20.12 39.73 197.58 | 100.60 99.33 98.79 | 1.15 1.48 1.56 |
| Formulation 2 (VD 50 mg + RE 100 mg + MF 1000 mg) | VG 50 RE 100 MF 500 | 49.62 99.17 495.37 | 99.24 99.17 99.07 | 0.93 1.72 1.08 |
| Standard addition method | Recovery % | |||
| Amount of VD added (µg·mL−1) to formulation solution 1 | 10 | 10.01 | 100.10 | 0.83 |
| 20 | 19.84 | 99.20 | 1.44 | |
| 30 | 29.47 | 98.23 | 1.31 | |
| Amount of RE added (µg·mL−1) to formulation solution 1 | 20 | 19.95 | 99.75 | 1.28 |
| 40 | 39.28 | 98.20 | 1.72 | |
| 60 | 59.04 | 98.40 | 0.76 | |
| Amount of MF added (µg·mL−1) to formulation solution 1 | 100 | 98.83 | 98.83 | 1.34 |
| 200 | 198.11 | 99.06 | 1.77 | |
| 400 | 396.45 | 99.11 | 0.68 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Attimarad, M.; Venugopala, K.N.; Chohan, M.S.; David, M.; Molina, E.I.P.; Sreeharsha, N.; Nair, A.B.; Tratrat, C.; Altaysan, A.I.; Balgoname, A.A. An Experimental Design Approach to Quantitative Expression for Quality Control of a Multicomponent Antidiabetic Formulation by the HILIC Method. Molecules 2022, 27, 3135. https://doi.org/10.3390/molecules27103135
Attimarad M, Venugopala KN, Chohan MS, David M, Molina EIP, Sreeharsha N, Nair AB, Tratrat C, Altaysan AI, Balgoname AA. An Experimental Design Approach to Quantitative Expression for Quality Control of a Multicomponent Antidiabetic Formulation by the HILIC Method. Molecules. 2022; 27(10):3135. https://doi.org/10.3390/molecules27103135
Chicago/Turabian StyleAttimarad, Mahesh, Katharigatta Narayanaswamy Venugopala, Muhammad S. Chohan, Marysheela David, Efren II Plaza Molina, Nagaraja Sreeharsha, Anroop Balachandran Nair, Christophe Tratrat, Abdulrahman Ibrahim Altaysan, and Abdulmalek Ahmed Balgoname. 2022. "An Experimental Design Approach to Quantitative Expression for Quality Control of a Multicomponent Antidiabetic Formulation by the HILIC Method" Molecules 27, no. 10: 3135. https://doi.org/10.3390/molecules27103135
APA StyleAttimarad, M., Venugopala, K. N., Chohan, M. S., David, M., Molina, E. I. P., Sreeharsha, N., Nair, A. B., Tratrat, C., Altaysan, A. I., & Balgoname, A. A. (2022). An Experimental Design Approach to Quantitative Expression for Quality Control of a Multicomponent Antidiabetic Formulation by the HILIC Method. Molecules, 27(10), 3135. https://doi.org/10.3390/molecules27103135