
In Silico Identification of Potential Sites for a Plastic-Degrading Enzyme by a Reverse Screening through the Protein Sequence Space and Molecular Dynamics Simulations


Abstract
:1. Introduction
2. Results
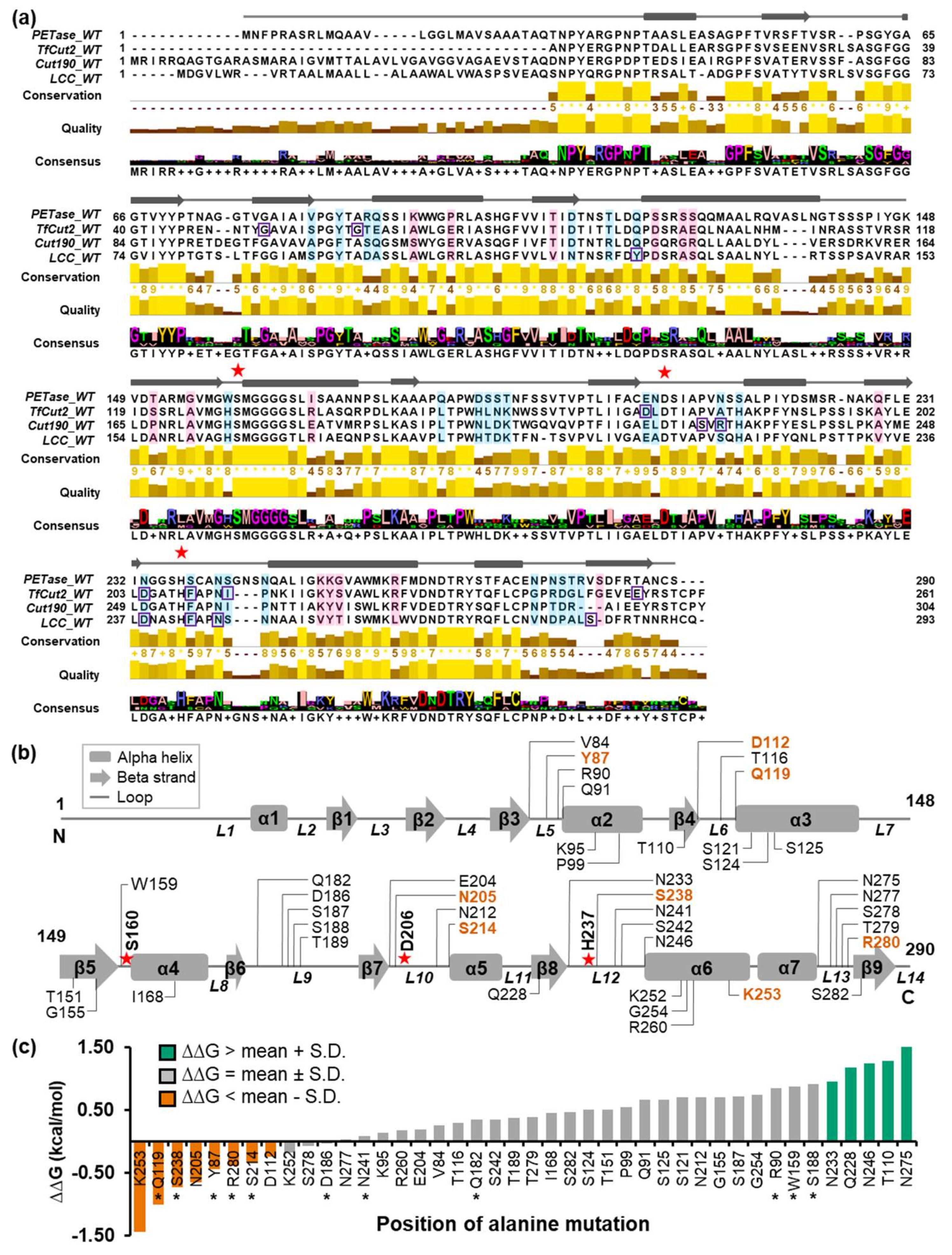
2.1. Search for Candidate Mutation Sites
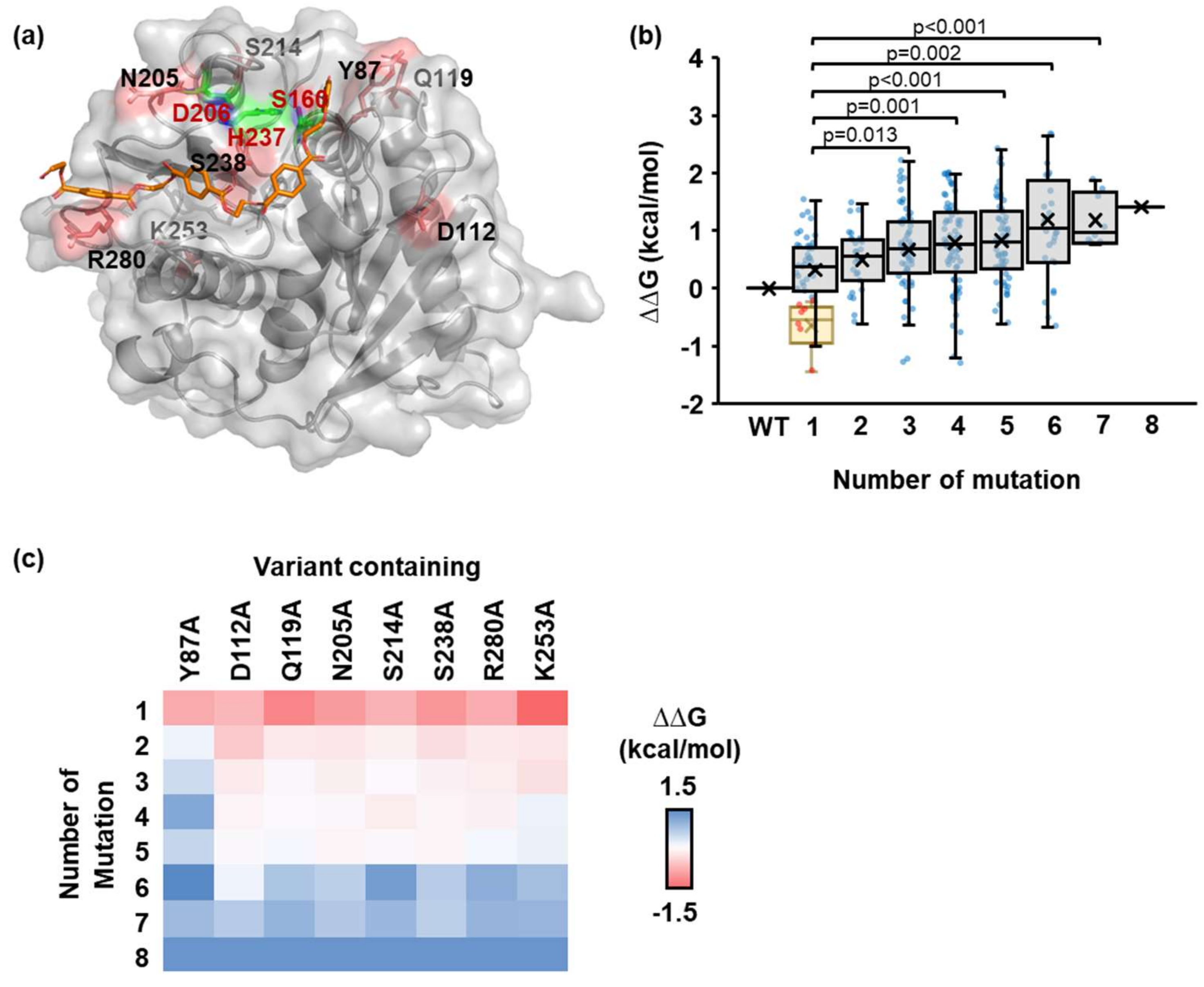
2.2. Identification of the Number and Position of Mutation Sites
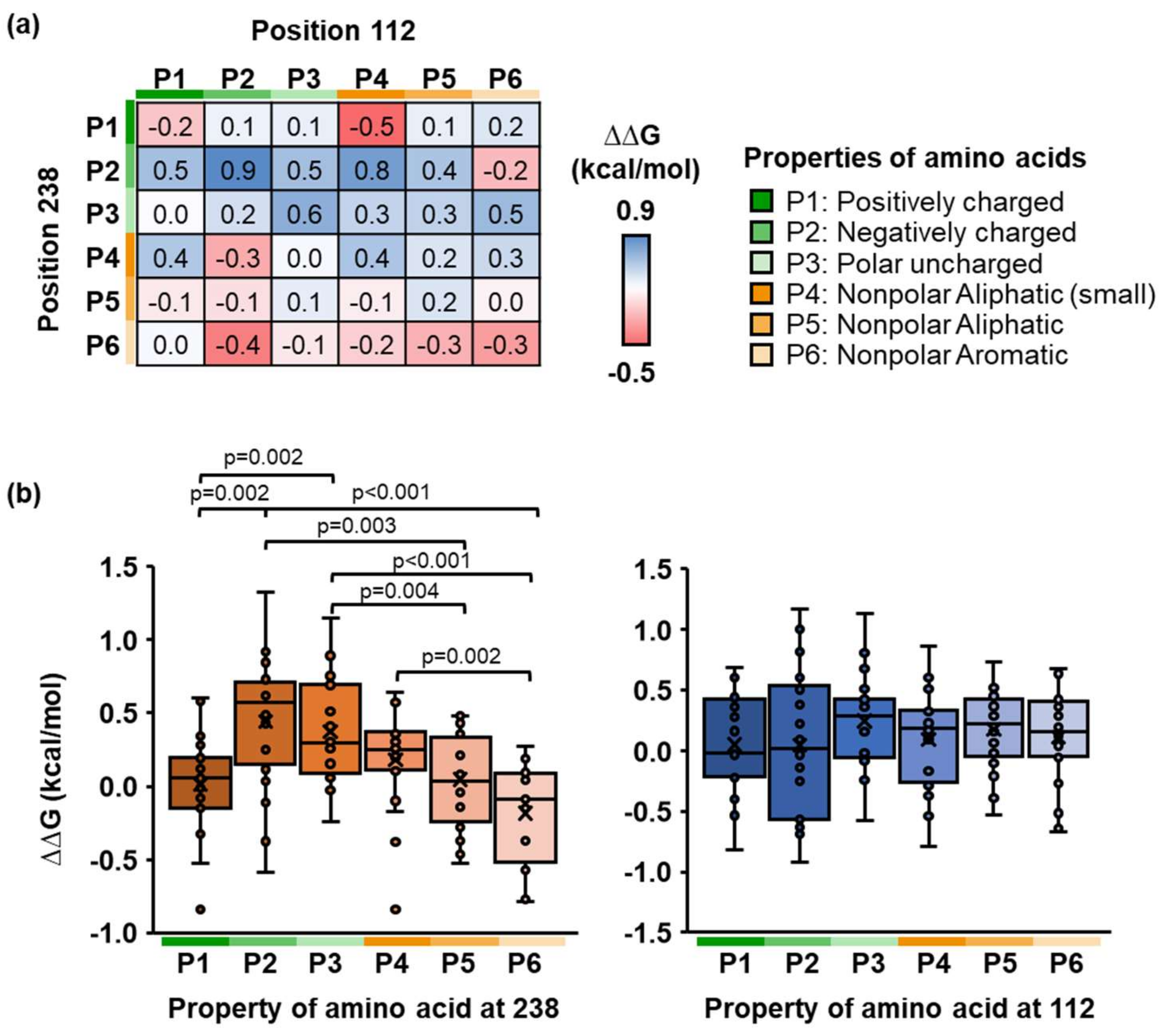
2.3. Identification of Physicochemical Properties of Side Chains of PETase
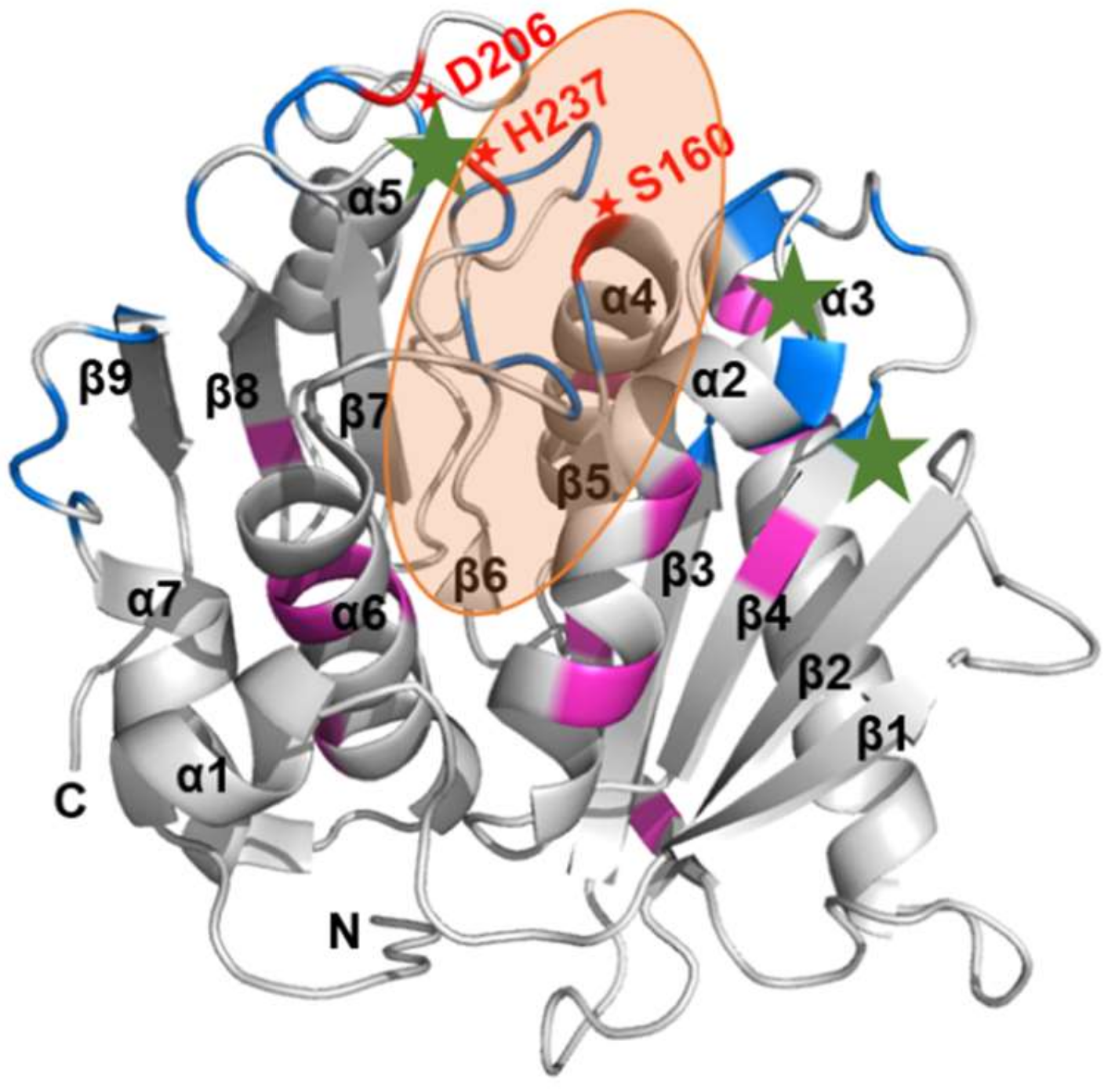
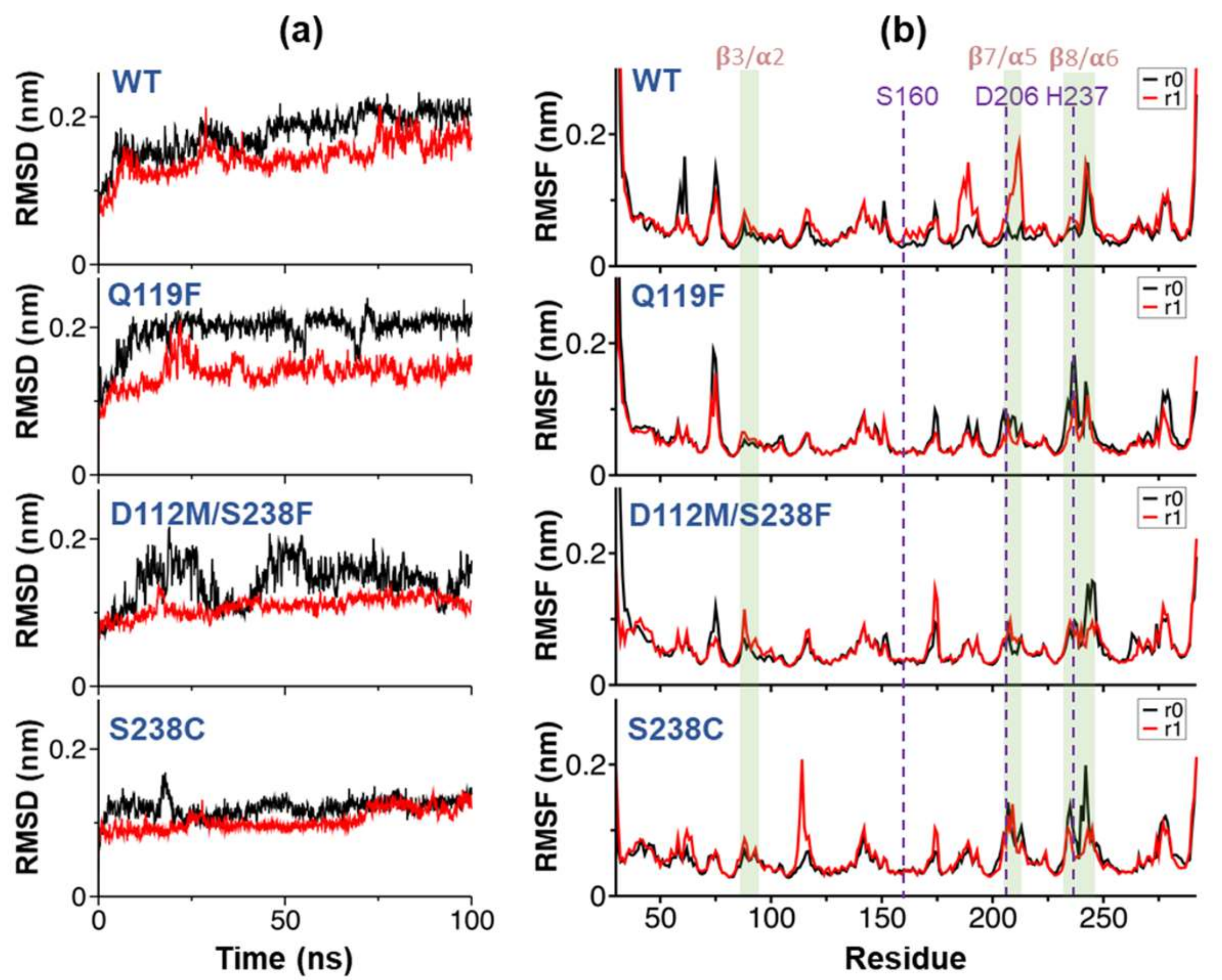
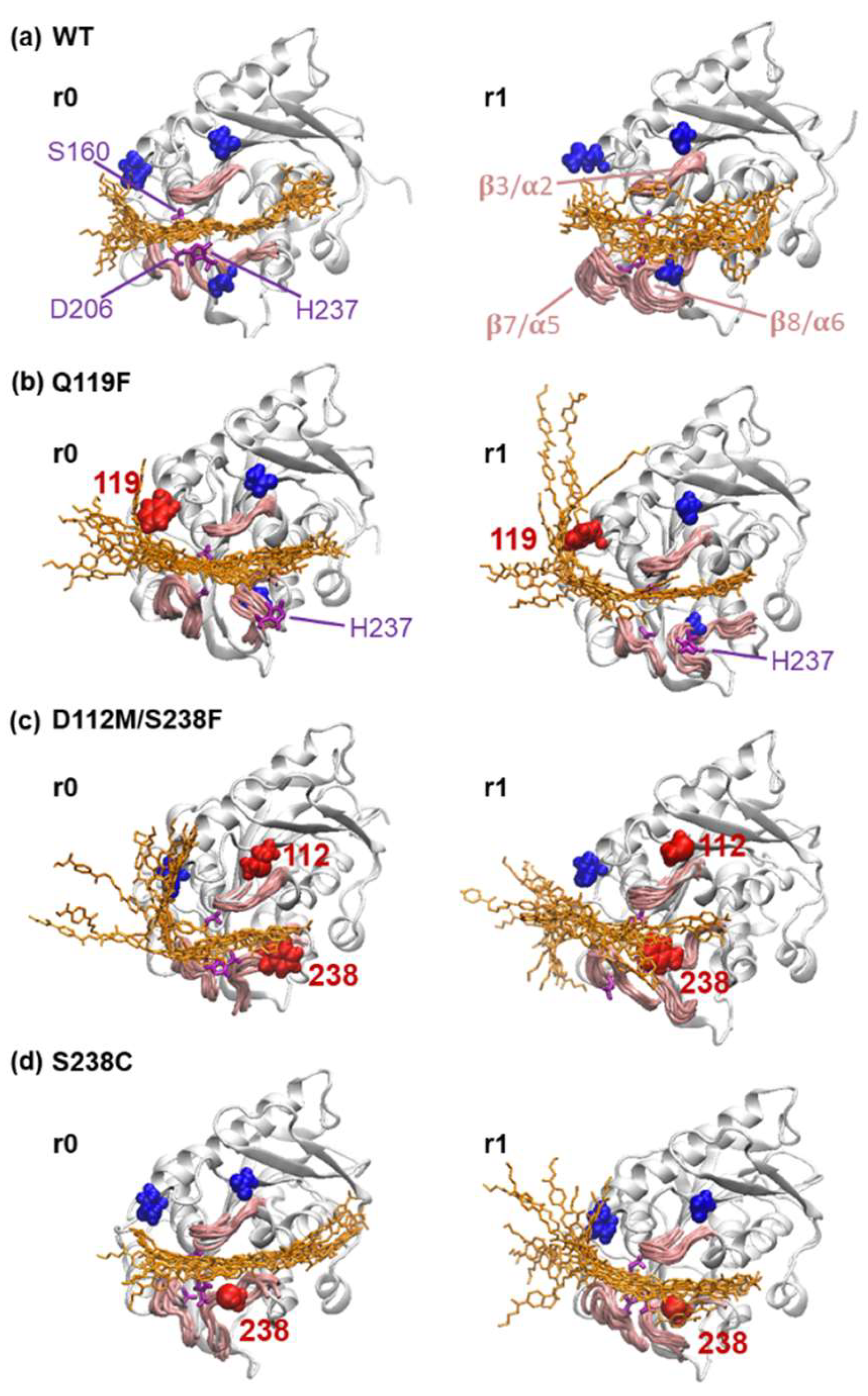
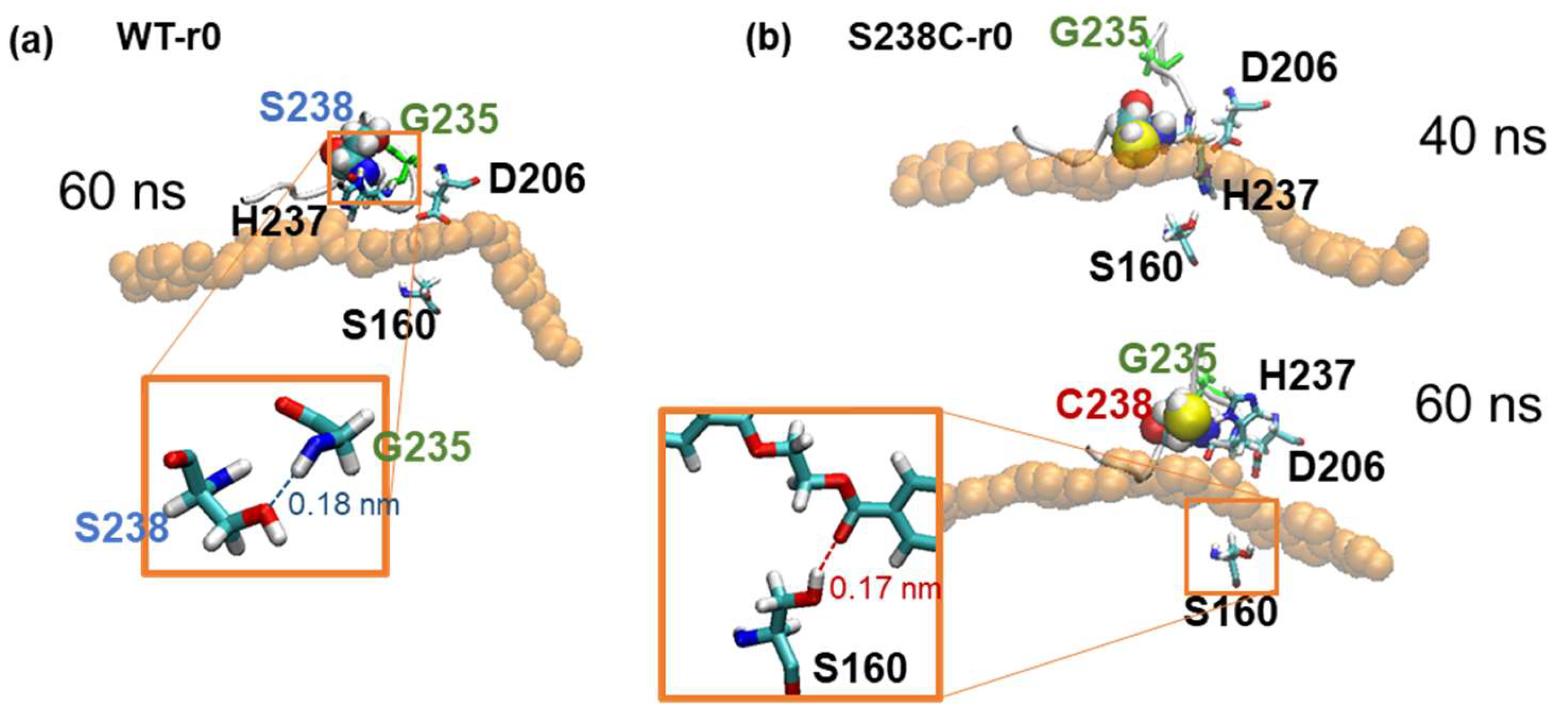
2.4. Substrate Binding Visualizations from MD Trajectories of PETase and Variants
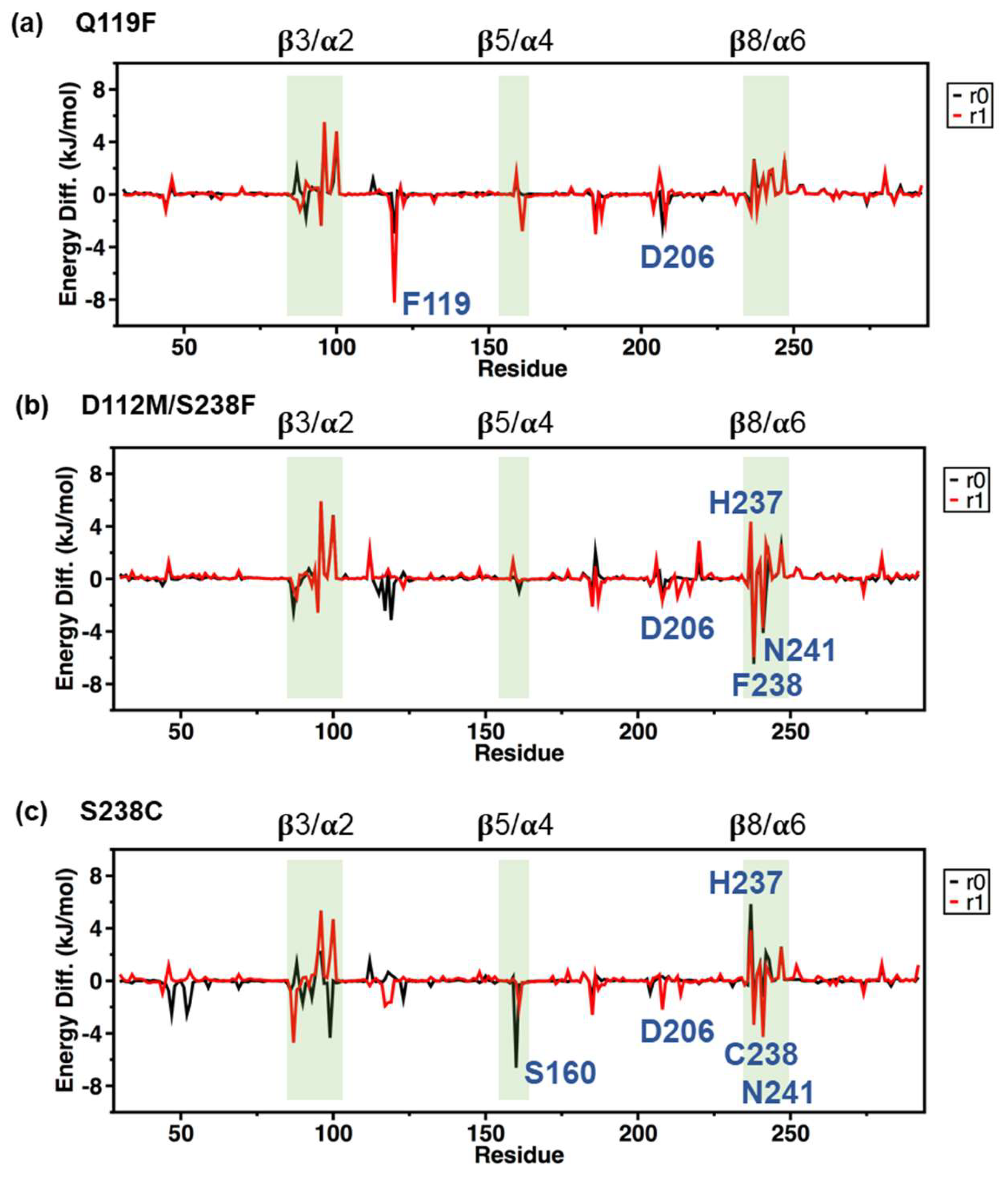
2.5. MM/PBSA Analysis on the Local Contribution of Amino Acids
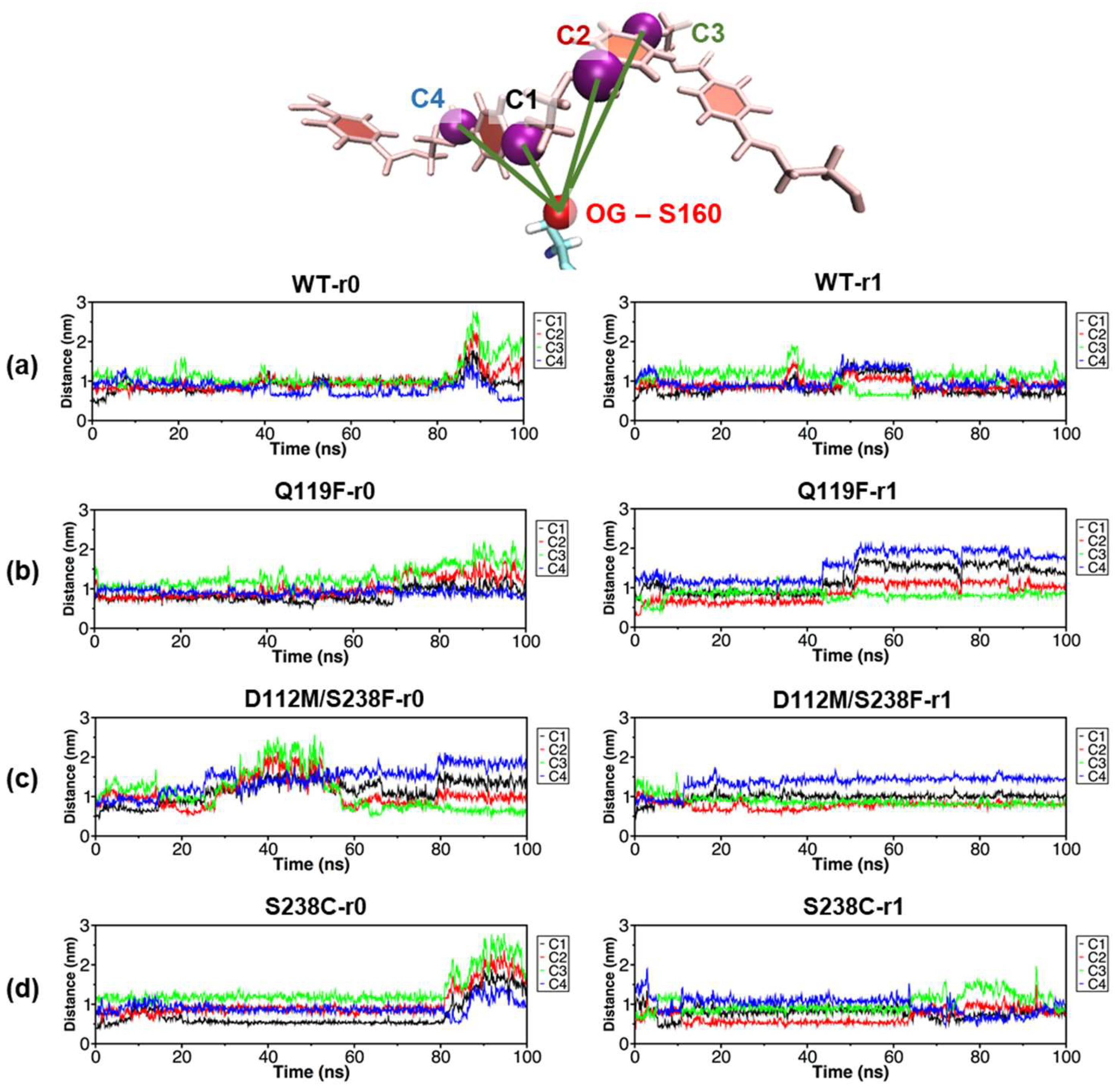
2.6. Local Effects Induced by the S238C Mutation on the PET Substrate Binding
3. Discussion
4. Materials and Methods
4.1. Multiple Sequence Alignment
4.2. Preparation of Enzyme Structure for Molecular Docking
4.3. Molecular Docking
4.4. Candidate Mutation Selection
4.5. Alanine Scanning Mutagenesis
4.6. In Silico Site-Saturation Mutagenesis
4.7. Molecular Dynamics Simulations and Analysis
4.8. Binding Free Energy Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Awaja, F.; Pavel, D. Recycling of PET. Eur. Polym. J. 2005, 41, 1453–1477. [Google Scholar] [CrossRef]
- Samak, N.A.; Jia, Y.; Sharshar, M.M.; Mu, T.; Yang, M.; Peh, S.; Xing, J. Recent advances in biocatalysts engineering for polyethylene terephthalate plastic waste green recycling. Environ. Int. 2020, 145, 106144. [Google Scholar] [CrossRef] [PubMed]
- Webb, H.K.; Arnott, J.; Crawford, R.J.; Ivanova, E.P. Plastic degradation and its environmental implications with special reference to poly (ethylene terephthalate). Polymers 2013, 5, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Huang, J.; Liang, X.; Wei, M.; Liang, F.; Feng, D.; Xu, C.; Xian, M.; Zou, H. Production and waste treatment of polyesters: Application of bioresources and biotechniques. Crit. Rev. Biotechnol. 2022, 1–18. [Google Scholar] [CrossRef]
- Karigar, C.S.; Rao, S.S. Role of microbial enzymes in the bioremediation of pollutants: A review. Enzyme Res. 2011, 2011, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fojan, P.; Jonson, P.H.; Petersen, M.T.N.; Petersen, S.B. What distinguishes an esterase from a lipase: A novel structural approach. Biochimie 2000, 82, 1033–1041. [Google Scholar] [CrossRef]
- Rastogi, M.; Nandal, M.; Khosla, B. Microbes as vital additives for solid waste composting. Heliyon 2020, 6, e03343. [Google Scholar] [CrossRef]
- Jadaun, J.S.; Bansal, S.; Sonthalia, A.; Rai, A.K.; Singh, S.P. Biodegradation of Plastics for Sustainable Environment. Bioresour. Technol. 2022, 347, 126697. [Google Scholar] [CrossRef]
- Bhatt, P.; Bhandari, G.; Bhatt, K.; Maithani, D.; Mishra, S.; Gangola, S.; Bhatt, R.; Huang, Y.; Chen, S. Plasmid-mediated catabolism for the removal of xenobiotics from the environment. J. Hazard. Mater. 2021, 420, 126618. [Google Scholar] [CrossRef]
- Chen, S.; Tong, X.; Woodard, R.W.; Du, G.; Wu, J.; Chen, J. Identification and characterization of bacterial cutinase. J. Biol. Chem. 2008, 283, 25854–25862. [Google Scholar] [CrossRef] [Green Version]
- Kawai, F.; Oda, M.; Tamashiro, T.; Waku, T.; Tanaka, N.; Yamamoto, M.; Mizushima, H.; Miyakawa, T.; Tanokura, M. A novel Ca2+-activated, thermostabilized polyesterase capable of hydrolyzing polyethylene terephthalate from Saccharomonospora viridis AHK190. Appl. Microbiol. Biotechnol. 2014, 98, 10053–10064. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, S.; Yamato, S.; Kanaya, E.; Kim, J.-J.; Koga, Y.; Takano, K.; Kanaya, S. Isolation of a novel cutinase homolog with polyethylene terephthalate-degrading activity from leaf-branch compost by using a metagenomic approach. Appl. Environ. Microbiol. 2012, 78, 1556–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Wang, D.; Wei, N. Enzyme discovery and engineering for sustainable plastic recycling. Trends Biotechnol. 2022, 40, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Then, J.; Wei, R.; Oeser, T.; Gerdts, A.; Schmidt, J.; Barth, M.; Zimmermann, W. A disulfide bridge in the calcium binding site of a polyester hydrolase increases its thermal stability and activity against polyethylene terephthalate. FEBS Open Bio 2016, 6, 425–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, R.; Oeser, T.; Schmidt, J.; Meier, R.; Barth, M.; Then, J.; Zimmermann, W. Engineered bacterial polyester hydrolases efficiently degrade polyethylene terephthalate due to relieved product inhibition. Biotechnol. Bioeng. 2016, 113, 1658–1665. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Guo, Z.-Y.; Wang, L.; Yan, Z.-F.; Jin, C.-X.; Huang, Q.-S.; Kong, D.-M.; Rao, D.-M.; Wu, J. Directional-path modification strategy enhances PET hydrolase catalysis of plastic degradation. J. Hazard. Mater. 2022, 433, 128816. [Google Scholar] [CrossRef]
- Oda, M.; Yamagami, Y.; Inaba, S.; Oida, T.; Yamamoto, M.; Kitajima, S.; Kawai, F. Enzymatic hydrolysis of PET: Functional roles of three Ca2+ ions bound to a cutinase-like enzyme, Cut190*, and its engineering for improved activity. Appl. Microbiol. Biotechnol. 2018, 102, 10067–10077. [Google Scholar] [CrossRef]
- Tournier, V.; Topham, C.M.; Gilles, A.; David, B.; Folgoas, C.; Moya-Leclair, E.; Kamionka, E.; Desrousseaux, M.-L.; Texier, H.; Gavalda, S. An engineered PET depolymerase to break down and recycle plastic bottles. Nature 2020, 580, 216–219. [Google Scholar] [CrossRef]
- Wei, R.; Zimmermann, W. Microbial enzymes for the recycling of recalcitrant petroleum-based plastics: How far are we? Microb. Biotechnol. 2017, 10, 1308–1322. [Google Scholar] [CrossRef] [Green Version]
- Bollinger, A.; Thies, S.; Knieps-Grünhagen, E.; Gertzen, C.; Kobus, S.; Höppner, A.; Ferrer, M.; Gohlke, H.; Smits, S.H.J.; Jaeger, K.-E. A novel polyester hydrolase from the marine bacterium Pseudomonas aestusnigri–structural and functional insights. Front. Microbiol. 2020, 11, 114. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Hiraga, K.; Takehana, T.; Taniguchi, I.; Yamaji, H.; Maeda, Y.; Toyohara, K.; Miyamoto, K.; Kimura, Y.; Oda, K. A bacterium that degrades and assimilates poly (ethylene terephthalate). Science 2016, 351, 1196–1199. [Google Scholar] [CrossRef] [PubMed]
- Austin, H.P.; Allen, M.D.; Donohoe, B.S.; Rorrer, N.A.; Kearns, F.L.; Silveira, R.L.; Pollard, B.C.; Dominick, G.; Duman, R.; Omari, K.E.; et al. Characterization and engineering of a plastic-degrading aromatic polyesterase. Proc. Natl. Acad. Sci. USA 2018, 115, E4350–E4357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, S.; Cho, I.J.; Seo, H.; Son, H.F.; Sagong, H.Y.; Shin, T.J.; Choi, S.Y.; Lee, S.Y.; Kim, K.J. Structural insight into molecular mechanism of poly(ethylene terephthalate) degradation. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; He, L.; Wang, L.; Li, T.; Li, C.; Liu, H.; Luo, Y.; Bao, R. Protein crystallography and site-direct mutagenesis analysis of the poly(Ethylene terephthalate) hydrolase petase from Ideonella sakaiensis. ChemBioChem 2018, 19, 1471–1475. [Google Scholar] [CrossRef]
- Han, X.; Liu, W.; Huang, J.W.; Ma, J.; Zheng, Y.; Ko, T.P.; Xu, L.; Cheng, Y.S.; Chen, C.C.; Guo, R.T. Structural insight into catalytic mechanism of PET hydrolase. Nat. Commun. 2017, 8, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Fecker, T.; Galaz-Davison, P.; Engelberger, F.; Narui, Y.; Sotomayor, M.; Parra, L.P.; Ramírez-Sarmiento, C.A. Active Site Flexibility as a Hallmark for Efficient PET Degradation by I. sakaiensis PETase. Biophys. J. 2018, 114, 1302–1312. [Google Scholar] [CrossRef] [Green Version]
- Rauwerdink, A.; Kazlauskas, R.J. How the same core catalytic machinery catalyzes 17 different reactions: The serine-histidine-aspartate catalytic triad of α/β-hydrolase fold enzymes. ACS Catal. 2015, 5, 6153–6176. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Yue, Y.; Zheng, M.; Li, Y.; Zhang, Q.; Wang, W. Is PETase-and Is MHETase-Catalyzed Cascade Degradation Mechanism toward Polyethylene Terephthalate. ACS Sustain. Chem. Eng. 2021, 9, 9823–9832. [Google Scholar] [CrossRef]
- Jerves, C.; Neves, R.P.P.; Ramos, M.J.; da Silva, S.; Fernandes, P.A. Reaction Mechanism of the PET Degrading Enzyme PETase Studied with DFT/MM Molecular Dynamics Simulations. ACS Catal. 2021, 11, 11626–11638. [Google Scholar] [CrossRef]
- da Costa, C.H.S.; Dos Santos, A.M.; Alves, C.N.; Martí, S.; Moliner, V.; Santana, K.; Lameira, J. Assessment of the PETase conformational changes induced by poly (ethylene terephthalate) binding. Proteins Struct. Funct. Bioinform. 2021, 89, 1340–1352. [Google Scholar] [CrossRef]
- Ma, Y.; Yao, M.; Li, B.; Ding, M.; He, B.; Chen, S.; Zhou, X.; Yuan, Y. Enhanced poly (ethylene terephthalate) hydrolase activity by protein engineering. Engineering 2018, 4, 888–893. [Google Scholar] [CrossRef]
- Son, H.F.; Cho, I.J.; Joo, S.; Seo, H.; Sagong, H.-Y.; Choi, S.Y.; Lee, S.Y.; Kim, K.-J. Rational protein engineering of thermo-stable PETase from Ideonella sakaiensis for highly efficient PET degradation. ACS Catal. 2019, 9, 3519–3526. [Google Scholar] [CrossRef]
- Chen, R. Enzyme engineering: Rational redesign versus directed evolution. Trends Biotechnol. 2001, 19, 13–14. [Google Scholar] [CrossRef]
- Limsakul, P.; Peng, Q.; Wu, Y.; Allen, M.E.; Liang, J.; Remacle, A.G.; Lopez, T.; Ge, X.; Kay, B.K.; Zhao, H.; et al. Directed Evolution to Engineer Monobody for FRET Biosensor Assembly and Imaging at Live-Cell Surface. Cell Chem. Biol. 2018, 25, 370–379.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirillo, V.; Orlando, M.; Tessaro, D.; Pollegioni, L.; Molla, G. An Efficient Protein Evolution Workflow for the Improvement of Bacterial PET Hydrolyzing Enzymes. Int. J. Mol. Sci. 2021, 23, 264. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Anand, P.; Nagarajan, D.; Mukherjee, S.; Chandra, N. ABS–Scan: In silico alanine scanning mutagenesis for binding site residues in protein–ligand complex. F1000Research 2014, 3, 214. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Damborsky, J.; Brezovsky, J. Computational tools for designing and engineering enzymes. Curr. Opin. Chem. Biol. 2014, 19, 8–16. [Google Scholar] [CrossRef]
- Miyakawa, T.; Mizushima, H.; Ohtsuka, J.; Oda, M.; Kawai, F.; Tanokura, M. Structural basis for the Ca2+-enhanced thermostability and activity of PET-degrading cutinase-like enzyme from Saccharomonospora viridis AHK190. Appl. Microbiol. Biotechnol. 2015, 99, 4297–4307. [Google Scholar] [CrossRef] [PubMed]
- Magliery, T.J.; Regan, L. Sequence variation in ligand binding sites in proteins. BMC Bioinform. 2005, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.; Zhang, C.; Liu, S.; Zhou, Y. Protein binding site prediction using an empirical scoring function. Nucleic Acids Res. 2006, 34, 3698–3707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, V.E.; Hause, R.J.; Fowler, D.M. Analysis of large-scale mutagenesis data to assess the impact of single amino acid substitutions. Genetics 2017, 207, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibarra, A.A.; Bartlett, G.J.; Hegedus, Z.; Dutt, S.; Hobor, F.; Horner, K.A.; Hetherington, K.; Spence, K.; Nelson, A.; Edwards, T.A. Predicting and experimentally validating hot-spot residues at protein–protein interfaces. ACS Chem. Biol. 2019, 14, 2252–2263. [Google Scholar] [CrossRef]
- Weiss, G.A.; Watanabe, C.K.; Zhong, A.; Goddard, A.; Sidhu, S.S. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. USA 2000, 97, 8950–8954. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Boonyaputthikul, H.; Muhammad, A.; Roekring, S.; Rattanarojpong, T.; Khunrae, P.; Sutthibutpong, T. Synergistic effects between the additions of a disulphide bridge and an N-terminal hydrophobic sidechain on the binding pocket tilting and enhanced Xyn11A activity. Arch. Biochem. Biophys. 2019, 672, 108068. [Google Scholar] [CrossRef]
- Muhammad, A.; Khunrae, P.; Sutthibutpong, T. Effects of oligolignol sizes and binding modes on a GH11 xylanase inhibition revealed by molecular modeling techniques. J. Mol. Model. 2020, 26, 1–14. [Google Scholar] [CrossRef]
- Bhatt, P.; Joshi, T.; Bhatt, K.; Zhang, W.; Huang, Y.; Chen, S. Binding interaction of glyphosate with glyphosate oxidoreductase and C–P lyase: Molecular docking and molecular dynamics simulation studies. J. Hazard. Mater. 2021, 409, 124927. [Google Scholar] [CrossRef]
- Herrero Acero, E.; Ribitsch, D.; Steinkellner, G.; Gruber, K.; Greimel, K.; Eiteljoerg, I.; Trotscha, E.; Wei, R.; Zimmermann, W.; Zinn, M.; et al. Enzymatic surface hydrolysis of PET: Effect of structural diversity on kinetic properties of cutinases from Thermobifida. Macromolecules 2011, 44, 4632–4640. [Google Scholar] [CrossRef] [Green Version]
- Roth, C.; Wei, R.; Oeser, T.; Then, J.; Föllner, C.; Zimmermann, W.; Sträter, N. Structural and functional studies on a thermostable polyethylene terephthalate degrading hydrolase from Thermobifida fusca. Appl. Microbiol. Biotechnol. 2014, 98, 7815–7823. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Song, C.; Gräsing, D.; Schneider, T.; Bielytskyi, P.; Böttcher, D.; Matysik, J.; Bornscheuer, U.T.; Zimmermann, W. Conformational fitting of a flexible oligomeric substrate does not explain the enzymatic PET degradation. Nat. Commun. 2019, 10, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Chen, Y.; Liu, X.; Dong, S.; Tian, Y.; Qiao, Y.; Mitra, R.; Han, J.; Li, C.; Han, X. Computational redesign of a PETase for plastic biodegradation under ambient condition by the GRAPE strategy. ACS Catal. 2021, 11, 1340–1350. [Google Scholar] [CrossRef]
- Lu, H.; Diaz, D.J.; Czarnecki, N.J.; Zhu, C.; Kim, W.; Shroff, R.; Acosta, D.J.; Alexander, B.R.; Cole, H.O.; Zhang, Y. Machine learning-aided engineering of hydrolases for PET depolymerization. Nature 2022, 604, 662–667. [Google Scholar] [CrossRef]
- Sagong, H.-Y.; Kim, S.; Lee, D.; Hong, H.; Lee, S.H.; Seo, H.; Kim, K.-J. Structural and functional characterization of an auxiliary domain-containing PET hydrolase from Burkholderiales bacterium. J. Hazard. Mater. 2022, 429, 128267. [Google Scholar] [CrossRef]
- Nikolaivits, E.; Taxeidis, G.; Gkountela, C.; Vouyiouka, S.; Maslak, V.; Nikodinovic-Runic, J.; Topakas, E. A polyesterase from the Antarctic bacterium Moraxella sp. degrades highly crystalline synthetic polymers. J. Hazard. Mater. 2022, 434, 128900. [Google Scholar] [CrossRef]
- Available online: https://pymol.org/2/support.html?#citing (accessed on 17 March 2022).
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Tanawattanasuntorn, T.; Thongpanchang, T.; Rungrotmongkol, T.; Hanpaibool, C.; Graidist, P.; Tipmanee, V. (−)-Kusunokinin as a potential aldose reductase inhibitor: Equivalency observed via AKR1B1 dynamics simulation. ACS Omega 2020, 6, 606–614. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Stroet, M.; Caron, B.; Visscher, K.M.; Geerke, D.P.; Malde, A.K.; Mark, A.E. Automated topology builder version 3.0: Prediction of solvation free enthalpies in water and hexane. J. Chem. Theory Comput. 2018, 14, 5834–5845. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Martoňák, R.; Laio, A.; Parrinello, M. Predicting crystal structures: The Parrinello-Rahman method revisited. Phys. Rev. Lett. 2003, 90, 75503. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PETase Variant | vdW Energy (kJ/mol) | Electrostatic Energy (kJ/mol) | Polar Solvation (kJ/mol) | Apolar Solvation (kJ/mol) | Total Binding Energy (kJ/mol) |
|---|---|---|---|---|---|
| WT-r0 | −215 ± 35 | −38 ± 27 | 121 ± 25 | −25 ± 4 | −156 ± 39 |
| WT-r1 | −158 ± 53 | −55 ± 26 | 108 ± 44 | −19 ± 6 | −124 ± 62 |
| Q119F-r0 | −160 ± 18 | −32 ± 19 | 94 ± 38 | −20 ± 2 | −118 ± 38 |
| Q119F-r1 | −155 ± 27 | −31 ± 35 | 88 ± 19 | −19 ± 3 | −116 ± 51 |
| D112M/S238F-r0 | −176 ± 45 | −30 ± 25 | 117 ± 24 | −21 ± 6 | −110 ± 70 |
| D112M/S238F-r1 | −159 ± 33 | −32 ± 19 | 116 ± 42 | −18 ± 3 | −93 ± 46 |
| S238C-r0 | −209 ± 36 | −53 ± 35 | 112 ± 31 | −24 ± 4 | −175 ± 40 |
| S238C-r1 | −173 ± 23 | −39 ± 33 | 112 ± 42 | −20 ± 3 | −121 ± 37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charupanit, K.; Tipmanee, V.; Sutthibutpong, T.; Limsakul, P. In Silico Identification of Potential Sites for a Plastic-Degrading Enzyme by a Reverse Screening through the Protein Sequence Space and Molecular Dynamics Simulations. Molecules 2022, 27, 3353. https://doi.org/10.3390/molecules27103353
Charupanit K, Tipmanee V, Sutthibutpong T, Limsakul P. In Silico Identification of Potential Sites for a Plastic-Degrading Enzyme by a Reverse Screening through the Protein Sequence Space and Molecular Dynamics Simulations. Molecules. 2022; 27(10):3353. https://doi.org/10.3390/molecules27103353
Chicago/Turabian StyleCharupanit, Krit, Varomyalin Tipmanee, Thana Sutthibutpong, and Praopim Limsakul. 2022. "In Silico Identification of Potential Sites for a Plastic-Degrading Enzyme by a Reverse Screening through the Protein Sequence Space and Molecular Dynamics Simulations" Molecules 27, no. 10: 3353. https://doi.org/10.3390/molecules27103353
APA StyleCharupanit, K., Tipmanee, V., Sutthibutpong, T., & Limsakul, P. (2022). In Silico Identification of Potential Sites for a Plastic-Degrading Enzyme by a Reverse Screening through the Protein Sequence Space and Molecular Dynamics Simulations. Molecules, 27(10), 3353. https://doi.org/10.3390/molecules27103353