
α-Aminophosphonates, -Phosphinates, and -Phosphine Oxides as Extraction and Precipitation Agents for Rare Earth Metals, Thorium, and Uranium: A Review


Abstract
:1. Introduction
Scope of the Review
2. The Short History of α-Aminophosphonates, -Phosphinates, and -Phosphine Oxides as Extraction and Precipitation Agents
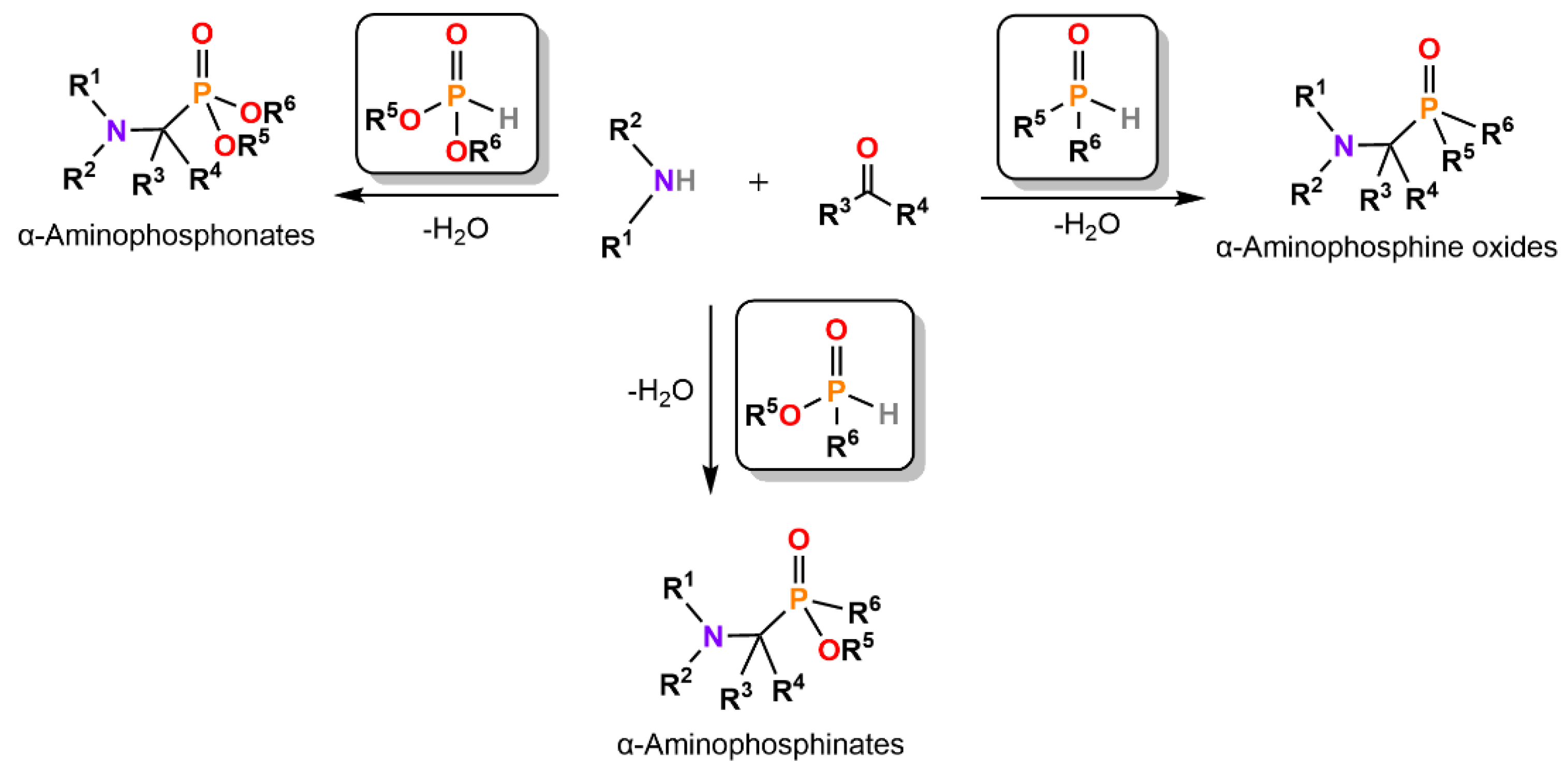
3. Synthesis of α-Aminophosphonates, -Phosphinates, and -Phosphine Oxides
Dimerization of the Synthesized α-Amino-Functionalized Organophosphorus Compounds
4. Characterization of the Extracted Metal Complexes by IR
4.1. Acidic α-Aminophosphonates
4.2. Neutral α-Aminophosphonates
4.3. α-Aminophosphine Oxides and Acidic α-Aminophosphinates
5. Composition of Extracted and Precipitated Complexes
6. Extraction Ability of α-Aminophosphonates, -Phosphinates, and -Phosphine Oxides towards REs and Actinoids
6.1. α-Aminophosphonates
6.2. α-Aminophosphinates
6.3. α-Aminophosphine Oxides
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xie, F.; Zhang, T.A.; Dreisinger, D.; Doyle, F. A Critical Review on Solvent Extraction of Rare Earths from Aqueous Solutions. Miner. Eng. 2014, 56, 10–28. [Google Scholar] [CrossRef]
- Kislik, V.S. Solvent Extraction: Classical and Novel Approaches, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2011; ISBN 978-0-444-53778-2. [Google Scholar]
- Batchu, N.K.; Li, Z.; Verbelen, B.; Binnemans, K. Structural Effects of Neutral Organophosphorus Extractants on Solvent Extraction of Rare-Earth Elements from Aqueous and Non-Aqueous Nitrate Solutions. Sep. Purif. Technol. 2021, 255, 117711. [Google Scholar] [CrossRef]
- Yudaev, P.A.; Kolpinskaya, N.A.; Chistyakov, E.M. Organophosphorous Extractants for Metals. Hydrometallurgy 2021, 201, 105558. [Google Scholar] [CrossRef]
- Kuang, S.; Liao, W. Progress in the Extraction and Separation of Rare Earths and Related Metals with Novel Extractants: A Review. Sci. China Technol. Sci. 2018, 61, 1319–1328. [Google Scholar] [CrossRef]
- Raju, C.S.K.; Subramanian, M.S. DAPPA Grafted Polymer: An Efficient Solid Phase Extractant for U(VI), Th(IV) and La(III) from Acidic Waste Streams and Environmental Samples. Talanta 2005, 67, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Veliscek-Carolan, J.; Hanley, T.L.; Luca, V. Zirconium Organophosphonates as High Capacity, Selective Lanthanide Sorbents. Sep. Purif. Technol. 2014, 129, 150–158. [Google Scholar] [CrossRef]
- Galhoum, A.A.; Elshehy, E.A.; Tolan, D.A.; El-Nahas, A.M.; Taketsugu, T.; Nishikiori, K.; Akashi, T.; Morshedy, A.S.; Guibal, E. Synthesis of Polyaminophosphonic Acid-Functionalized Poly(Glycidyl Methacrylate) for the Efficient Sorption of La(III) and Y(III). Chem. Eng. J. 2019, 375, 121932. [Google Scholar] [CrossRef]
- Virtanen, E.J.; Perämäki, S.; Helttunen, K.; Väisänen, A.; Moilanen, J.O. Alkyl-Substituted Aminobis(Phosphonates)—Efficient Precipitating Agents for Rare Earth Elements, Thorium, and Uranium in Aqueous Solutions. ACS Omega 2021, 6, 23977–23987. [Google Scholar] [CrossRef]
- Flett, D.S. Solvent Extraction in Hydrometallurgy: The Role of Organophosphorus Extractants. J. Organomet. Chem. 2005, 690, 2426–2438. [Google Scholar] [CrossRef]
- Cherkasov, R.A.; Garifzyanov, A.R.; Leont’ev, S.V.; Davletshin, R.R.; Koshkin, S.A. Extraction of Scandium Ions by New Aminophosphinyl Extractants. Russ. J. Gen. Chem. 2009, 79, 2599–2605. [Google Scholar] [CrossRef]
- Guanming, Q.; Xikum, L.; Tai, Q.; Haitao, Z.; Honghao, Y.; Ruiting, M. Application of Rare Earths in Advanced Ceramic Materials. J. Rare Earths 2007, 25, 281–286. [Google Scholar] [CrossRef]
- Gschneidner, K.A. Rare Earth Alloys: A Critical Review of the Alloy Systems of the Rare Earth, Scandium, and Yttrium Metals; Books on Demand: Norderstedt, Germany, 1961; ISBN 9780598747457. [Google Scholar]
- Bünzli, J.-C.G. Lanthanide Photonics: Shaping the Nanoworld. Trends Chem. 2019, 1, 751–762. [Google Scholar] [CrossRef]
- Roesky, P.W. (Ed.) Molecular Catalysis of Rare-Earth Elements; Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 2010; Volume 137, ISBN 978-3-642-12810-3. [Google Scholar]
- McCallum, R.W.; Lewis, L.; Skomski, R.; Kramer, M.J.; Anderson, I.E. Practical Aspects of Modern and Future Permanent Magnets. Annu. Rev. Mater. Res. 2014, 44, 451–477. [Google Scholar] [CrossRef]
- Alves Dias, P.; Bobba, S.; Carrara, S.; Plazzotta, B. The Role of Rare Earth Elements in Wind Energy and Electric Mobility: An Analysis of Future Supply/Demand Balances; Publications Office of the European Union: Luxembourg, 2020. [Google Scholar]
- Neodymium—2022 Data—2012-2021 Historical—2023 Forecast—Price—Quote—Chart. Available online: https://tradingeconomics.com/commodity/neodymium (accessed on 3 May 2022).
- Pyrzynska, K.; Kubiak, A.; Wysocka, I. Application of Solid Phase Extraction Procedures for Rare Earth Elements Determination in Environmental Samples. Talanta 2016, 154, 15–22. [Google Scholar] [CrossRef]
- Binnemans, K.; Jones, P.T.; Blanpain, B.; Van Gerven, T.; Yang, Y.; Walton, A.; Buchert, M. Recycling of Rare Earths: A Critical Review. J. Clean. Prod. 2013, 51, 1–22. [Google Scholar] [CrossRef]
- Binnemans, K.; Jones, P.T.; Blanpain, B.; Van Gerven, T.; Pontikes, Y. Towards Zero-Waste Valorisation of Rare-Earth-Containing Industrial Process Residues: A Critical Review. J. Clean. Prod. 2015, 99, 17–38. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, H.; Guo, F.; Meng, S.; Li, D. Synergistic Extraction and Recovery of Cerium(IV) and Fluorin from Sulfuric Solutions with Cyanex 923 and Di-2-Ethylhexyl Phosphoric Acid. Sep. Purif. Technol. 2008, 63, 348–352. [Google Scholar] [CrossRef]
- Zhu, Z.; Pranolo, Y.; Cheng, C.Y. Separation of Uranium and Thorium from Rare Earths for Rare Earth Production—A Review. Miner. Eng. 2015, 77, 185–196. [Google Scholar] [CrossRef]
- Humphrey, U.E.; Khandaker, M.U. Viability of Thorium-Based Nuclear Fuel Cycle for the next Generation Nuclear Reactor: Issues and Prospects. Renew. Sustain. Energy Rev. 2018, 97, 259–275. [Google Scholar] [CrossRef]
- Kurniawan, T.A.; Othman, M.H.D.; Singh, D.; Avtar, R.; Hwang, G.H.; Setiadi, T.; Lo, W. Technological Solutions for Long-Term Storage of Partially Used Nuclear Waste: A Critical Review. Ann. Nucl. Energy 2022, 166, 108736. [Google Scholar] [CrossRef]
- Alexandratos, S.D.; Zhu, X. Polymer-Supported Aminomethylphosphinate as a Ligand with a High Affinity for U(VI) from Phosphoric Acid Solutions: Combining Variables to Optimize Ligand–Ion Communication. Solvent Extr. Ion Exch. 2016, 34, 290–295. [Google Scholar] [CrossRef]
- Imam, E.A.; El-Tantawy El-Sayed, I.; Mahfouz, M.G.; Tolba, A.A.; Akashi, T.; Galhoum, A.A.; Guibal, E. Synthesis of α-Aminophosphonate Functionalized Chitosan Sorbents: Effect of Methyl vs Phenyl Group on Uranium Sorption. Chem. Eng. J. 2018, 352, 1022–1034. [Google Scholar] [CrossRef]
- Cheira, M.F. Synthesis of Aminophosphonate-Functionalised ZnO/Polystyrene-Butadiene Nanocomposite and Its Characteristics for Uranium Adsorption from Phosphoric Acid. Int. J. Environ. Anal. Chem. 2021, 101, 1710–1734. [Google Scholar] [CrossRef]
- Galhoum, A.A.; Eisa, W.H.; El-Tantawy El-Sayed, I.; Tolba, A.A.; Shalaby, Z.M.; Mohamady, S.I.; Muhammad, S.S.; Hussien, S.S.; Akashi, T.; Guibal, E. A New Route for Manufacturing Poly(Aminophosphonic)-Functionalized Poly(Glycidyl Methacrylate)-Magnetic Nanocomposite—Application to Uranium Sorption from Ore Leachate. Environ. Pollut. 2020, 264, 114797. [Google Scholar] [CrossRef] [PubMed]
- Jagodić, V.; Herak, M.J. Synthesis and Physical Properties of a Novel Aminophosphonic Acid as an Extracting Agent for Metals. J. Inorg. Nucl. Chem. 1970, 32, 1323–1332. [Google Scholar] [CrossRef]
- Jagodić, V.; Herak, M.J.; Šipalo, B.; Radošević, J. Solvent Extraction Study of Lanthanum and Europium by Acidic Esters of Aminophosphonic Acids. J. Inorg. Nucl. Chem. 1971, 33, 2651–2659. [Google Scholar] [CrossRef]
- Herak, M.J.; Jagodić, V. Distribution and Dimerization of Organophosphorus Extractants and Their Extraction Efficiency in Different Solvents. J. Inorg. Nucl. Chem. 1973, 35, 995–1001. [Google Scholar] [CrossRef]
- Radošević, J.; Jagodić, V.; Herak, M.J. Extraction and Complex Formation of Cerium(III) and Praseodymium(III) with Monoesters of Phosphonic Acids. J. Inorg. Nucl. Chem. 1977, 39, 2053–2056. [Google Scholar] [CrossRef]
- Ziganshina, A.Y.; Kazakova, E.H.; Fedorenko, S.V.; Mustafina, A.R.; Konovalov, A.I. Aminomethylphosphonate Derivatives of Tetramethylcalix[4]Resorcinolarene. Synthesis and Some Extraction Properties in Relation to the Lantane Ions. Russ. J. Gen. Chem. 2001, 71, 1422–1425. [Google Scholar] [CrossRef]
- Fedorenko, S.V.; Mustafina, A.R.; Kazakova, E.K.; Pod’yachev, S.N.; Kharitonova, N.I.; Pudovik, M.A.; Konovalov, A.I.; Tananaev, I.G.; Myasoedov, B.F. Sodium Picrate Effect on Extraction of Lanthanum and Lutetium by Aminophosphonate Calix[4]Resorcinarenes. Russ. Chem. Bull. 2003, 52, 562–566. [Google Scholar] [CrossRef]
- Cherkasov, R.A.; Garifzyanov, A.R.; Bazanova, E.B.; Davletshin, R.R.; Leont’eva, S.V. Liquid Extraction of Some Rare Earth Elements with Aminomethylphosphine Oxides. Russ. J. Gen. Chem. 2012, 82, 33–42. [Google Scholar] [CrossRef]
- Garifzyanov, A.R.; Leont’ev, S.V.; Davletshina, N.V.; Voloshin, A.V.; Cherkasov, R.A. Synthesis, Transport, and Ionophore Properties of α,ω-Biphosphorylated Azapodands: VIII. Solvent Extraction of the Metal Ions with N,N′-Bis(Dioctylphosphorylmethyl)-1,8-Diamino-3,6-Dioxaoctane. Russ. J. Gen. Chem. 2013, 83, 1997–2004. [Google Scholar] [CrossRef]
- Liao, W.; Zhang, Z.; Li, Y.; Wu, G.; Lu, Y. CN105734286A—Method for Separating Cerium-Fluoride and Thorium. China Patent CN105734286A, 11 December 2014. [Google Scholar]
- Lu, Y.; Zhang, Z.; Li, Y.; Liao, W. Extraction and Recovery of Cerium(IV) and Thorium(IV) from Sulphate Medium by an α-Aminophosphonate Extractant. J. Rare Earths 2017, 35, 34–40. [Google Scholar] [CrossRef]
- Wei, H.; Li, Y.; Zhang, Z.; Xue, T.; Kuang, S.; Liao, W. Selective Extraction and Separation of Ce (IV) and Th (IV) from RE(III) in Sulfate Medium Using Di(2-Ethylhexyl)-N-Heptylaminomethylphosphonate. Solvent Extr. Ion Exch. 2017, 35, 117–129. [Google Scholar] [CrossRef]
- Kuang, S.; Zhang, Z.; Li, Y.; Wu, G.; Wei, H.; Liao, W. Selective Extraction and Separation of Ce(IV) from Thorium and Trivalent Rare Earths in Sulfate Medium by an α-Aminophosphonate Extractant. Hydrometallurgy 2017, 167, 107–114. [Google Scholar] [CrossRef]
- Kuang, S.; Zhang, Z.; Li, Y.; Wei, H.; Liao, W. Extraction and Separation of Heavy Rare Earths from Chloride Medium by α-Aminophosphonic Acid HEHAPP. J. Rare Earths 2018, 36, 304–310. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, Z.; Li, Y.; Bian, X.; Liao, W. Solvent Extraction and Separation of Rare Earths from Chloride Media Using α-Aminophosphonic Acid Extractant HEHAMP. Solvent Extr. Ion Exch. 2018, 36, 136–149. [Google Scholar] [CrossRef]
- Wei, H.; Li, Y.; Kuang, S.; Zhang, Z.; Liao, W. Separation of Trivalent Rare Earths from Chloride Medium Using Solvent Extraction with Heptylaminomethyl Phosphonic Acid 2-Ethylhexyl Ester (HEHHAP). Hydrometallurgy 2019, 188, 14–21. [Google Scholar] [CrossRef]
- Le, W.; Kuang, S.; Zhang, Z.; Wu, G.; Li, Y.; Liao, C.; Liao, W. Selective Extraction and Recovery of Scandium from Sulfate Medium by Cextrant 230. Hydrometallurgy 2018, 178, 54–59. [Google Scholar] [CrossRef]
- Kuang, S.; Zhang, Z.; Li, Y.; Wei, H.; Liao, W. Synergistic Extraction and Separation of Rare Earths from Chloride Medium by the Mixture of HEHAPP and D2EHPA. Hydrometallurgy 2017, 174, 78–83. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, Y.; Kuang, S.; Zhang, Z.; Bian, X.; Liao, W. Synergistic Extraction of Heavy Rare Earths by Mixture of α-Aminophosphonic Acid HEHAMP and HEHEHP. J. Rare Earths 2019, 37, 422–428. [Google Scholar] [CrossRef]
- Wei, H.; Li, Y.; Zhang, Z.; Liao, W. Synergistic Solvent Extraction of Heavy Rare Earths from Chloride Media Using Mixture of HEHHAP and Cyanex272. Hydrometallurgy 2020, 191, 105240. [Google Scholar] [CrossRef]
- Wang, X.; Du, M.; Liu, H. Synergistic Extraction Study of Samarium(III) from Chloride Medium by Mixtures of Bis(2,4,4-Trimethylpentyl)Phosphinic Acid and 8-Hydroxyquinoline. Sep. Purif. Technol. 2012, 93, 48–51. [Google Scholar] [CrossRef]
- Fu, Y.; Huang, M.; Zhou, Z.; Li, Z.; Liao, W.; Lu, Y. Separation of Trivalent Rare Earths from Nitrate Medium Using Solvent Extraction with a Novel Extractant 2-Ethylhexyl ((2-Ethylhexylamino)Methyl) Phosphonic Acid. J. Rare Earths 2022, 40, 491–500. [Google Scholar] [CrossRef]
- Huang, M.; Fu, Y.; Lu, Y.; Liao, W.; Li, Z. A Novel Extractant Bis(2-Ethylhexyl) ((2-Ethylhexylamino)Methyl) Phosphine Oxide for Cerium(IV) Extraction and Separation from Sulfate Medium. J. Rare Earths 2020, 38, 1330–1336. [Google Scholar] [CrossRef]
- Jagodić, V.; Grdenić, D. Aminophosphonic Acid Mono-Esters as Reagents for Solvent Extraction of Metals. J. Inorg. Nucl. Chem. 1964, 26, 1103–1109. [Google Scholar] [CrossRef]
- Herak, M.J.; Jagodic, V. Solvent Extraction and Separation of Europium(III) and Terbium(III) from Uranium(VI) by Monooctyl Anilinobenzylphosphonate. Croat. Chem. Acta 1964, 36, 51–58. [Google Scholar]
- Jagodić, V.; Herak, M.J.; Radošević, J. Separation of Lanthanum from Barium by Solvent Extraction with the Monooctyl Ester of Anilinobenzylphosphonic Acid. J. Less Common Met. 1968, 15, 371–375. [Google Scholar] [CrossRef]
- Herak, M.J.; Vujičić, N.; Jagodić, V. Separation of Radioactive Yttrium from Strontium by Means of Mono-Octyl Anilinobenzylphosphonate. Mikrochim. Acta 1969, 57, 16–20. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, Z.; Kuang, S.; Wei, H.; Li, Y.; Wu, G.; Geng, A.; Li, Y.; Liao, W. Removal of Thorium and Uranium from Leach Solutions of Ion-Adsorption Rare Earth Ores by Solvent Extraction with Cextrant 230. Hydrometallurgy 2020, 194, 105343. [Google Scholar] [CrossRef]
- Fields, E.K. The Synthesis of Esters of Substituted Amino Phosphonic Acids. J. Am. Chem. Soc. 1952, 74, 1528–1531. [Google Scholar] [CrossRef]
- Kabachnik, M.I.; Medved, T.Y. New Synthesis of Aminophosphonic Acids. Dokl. Akad. Nauk SSSR 1952, 83, 689–692. [Google Scholar]
- Moedritzer, K.; Irani, R.R. The Direct Synthesis of α-Aminomethylphosphonic Acids. Mannich-Type Reactions with Orthophosphorous Acid. J. Org. Chem. 1966, 31, 1603–1607. [Google Scholar] [CrossRef]
- Cherkasov, R.A.; Talan, A.S.; Tarasov, A.V.; Garifzyanov, A.P. Synthesis of Novel Mono-and Bisaminophosphoryl Compounds and Their Membrane Transport Properties for Acidic Substrates. Russ. J. Gen. Chem. 2008, 78, 1330–1333. [Google Scholar] [CrossRef]
- Cherkasov, R.A.; Garifzyanov, A.R.; Talan, A.S.; Davletshin, R.R.; Kurnosova, N.V. Synthesis of New Liophilic Functionalized Aminomethylphosphine Oxides and Their Acid-Base and Membrane-Transport Properties toward Acidic Substrates. Russ. J. Gen. Chem. 2009, 79, 1835–1849. [Google Scholar] [CrossRef]
- Pudovik, A.N. Addition of Dialkyl Phosphites to Unsaturated Compounds. A New Method of Synthesis of β-Keto Phosphonic and Unsaturated α-Hydroxyphosphonic Esters. Dokl. Akad. Nauk SSSR 1950, 73, 499–502. [Google Scholar]
- Jagodić, V. Darstellung von Monoestern N-Substituierter Aminomethylphosphonsäuren Durch Teilweise Verseifung Entsprechender Diester. Chem. Ber. 1960, 93, 2308–2313. [Google Scholar] [CrossRef]
- Blicke, F.F. The Mannich Reaction. In Organic Reactions 1; Wiley: New York, NY, USA, 1942; pp. 303–341. [Google Scholar]
- Juribašić, M.; Bellotto, L.; Tušek-Božić, L. N–H···O=P Hydrogen-Bonded Dimers as the Main Structural Motif of Aminophosphonate Diesters. Struct. Chem. 2012, 23, 257–266. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds; Griffiths, P.R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; ISBN 9780470405840. [Google Scholar]
- Larkin, P.J. Infrared and Raman Spectroscopy: Principles and Spectral Interpretation, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 9780128042090. [Google Scholar]
- Lin-Vien, D.; Colthup, N.B.; Fateley, W.G.; Grasselli, J.G. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules; Academic Press: San Diego, CA, USA, 1991; ISBN 0-12-451160-0. [Google Scholar]
- Groves, J.A.; Wright, P.A.; Lightfoot, P. Two Closely Related Lanthanum Phosphonate Frameworks Formed by Anion-Directed Linking of Inorganic Chains. Inorg. Chem. 2005, 44, 1736–1739. [Google Scholar] [CrossRef]
- Silva, P.; Vieira, F.; Gomes, A.C.; Ananias, D.; Fernandes, J.A.; Bruno, S.M.; Soares, R.; Valente, A.A.; Rocha, J.; Paz, F.A.A. Thermal Transformation of a Layered Multifunctional Network into a Metal–Organic Framework Based on a Polymeric Organic Linker. J. Am. Chem. Soc. 2011, 133, 15120–15138. [Google Scholar] [CrossRef]
- Colodrero, R.M.P.; Olivera-Pastor, P.; Losilla, E.R.; Aranda, M.A.G.; Leon-Reina, L.; Papadaki, M.; McKinlay, A.C.; Morris, R.E.; Demadis, K.D.; Cabeza, A. Multifunctional Lanthanum Tetraphosphonates: Flexible, Ultramicroporous and Proton-Conducting Hybrid Frameworks. Dalt. Trans. 2012, 41, 4045. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Bao, S.-S.; Ferreira, R.A.S.; Zheng, L.-M.; Carlos, L.D. A Layered Erbium Phosphonate in Pseudo-D5h Symmetry Exhibiting Field-Tunable Magnetic Relaxation and Optical Correlation. Chem. Commun. 2014, 50, 7621. [Google Scholar] [CrossRef] [PubMed]
- Mendes, R.F.; Almeida Paz, F.A. Dynamic Breathing Effect in Metal-Organic Frameworks: Reversible 2D-3D-2D-3D Single-Crystal to Single-Crystal Transformation. Inorg. Chim. Acta 2017, 460, 99–107. [Google Scholar] [CrossRef]
- Bligh, S.W.A.; Choi, N.; Geraldes, C.F.G.C.; Knoke, S.; McPartlin, M.; Sanganee, M.J.; Woodroffe, T.M. A Novel Hexaaza Macrocycle with Methylenephosphonate Pendant Arms: A Potential Useful Chelate for Biomedical Applications. J. Chem. Soc. Dalton Trans. 1997, 3, 4119–4126. [Google Scholar] [CrossRef]
- Gałęzowska, J.; Janicki, R.; Kozłowski, H.; Mondry, A.; Młynarz, P.; Szyrwiel, Ł. Unusual Coordination Behaviour of a Phosphonate- and Pyridine-Containing Ligand in a Stable Lanthanide Complex. Eur. J. Inorg. Chem. 2010, 2010, 1696–1702. [Google Scholar] [CrossRef]
- Le Fur, M.; Beyler, M.; Lepareur, N.; Fougère, O.; Platas-Iglesias, C.; Rousseaux, O.; Tripier, R. Pyclen Tri- n -Butylphosphonate Ester as Potential Chelator for Targeted Radiotherapy: From Yttrium(III) Complexation to 90 Y Radiolabeling. Inorg. Chem. 2016, 55, 8003–8012. [Google Scholar] [CrossRef]
- Tang, S.-F.; Song, J.-L.; Li, X.-L.; Mao, J.-G. Luminescent Lanthanide(III) Carboxylate−Phosphonates with Helical Tunnels. Cryst. Growth Des. 2006, 6, 2322–2326. [Google Scholar] [CrossRef]
- Ayi, A.A.; Kinnibrugh, T.L.; Clearfield, A. Hydrothermal Synthesis and Structural Characterization of Ammonium Ion-Templated Lanthanide(III) Carboxylate-Phosphonates. Front. Chem. 2014, 2, 94. [Google Scholar] [CrossRef] [Green Version]
- Danesi, P.R.; Chiarizia, R.; Scibona, G. The Meaning of Slope Analysis in Solvent Extraction Chemistry: The Case of Zinc Extraction by Trilaurylammonium Chloride. J. Inorg. Nucl. Chem. 1970, 32, 2349–2355. [Google Scholar] [CrossRef]
- Fang, Y.; Yuan, X.; Wu, L.; Peng, Z.; Feng, W.; Liu, N.; Xu, D.; Li, S.; Sengupta, A.; Mohapatra, P.K.; et al. Ditopic CMPO-Pillar[5]Arenes as Unique Receptors for Efficient Separation of Americium(III) and Europium(III). Chem. Commun. 2015, 51, 4263–4266. [Google Scholar] [CrossRef]
- Gupta, C.K.; Krishnamurthy, N. Extractive Metallurgy of Rare Earths; CRC Press: Boca Raton, FL, USA, 2005; ISBN 978-0415333405. [Google Scholar]
- Xu, G.X.; Wang, W.Q.; Wu, J.G.; Li, B.L.; Wu, G.B.; Shi, N. Chemistry of Nuclear Fuels Extraction. At. Energy Sci. Technol. 1963, 7, 487–508. [Google Scholar]
- Zhang, Z.; Jia, Q.; Liao, W. Progress in the Separation Processes for Rare Earth Resources. In Handbook on the Physics and Chemistry of Rare Earths; Elsevier: Amsterdam, The Netherlands, 2015; Volume 48, pp. 287–376. [Google Scholar]
- Sato, T. Liquid-Liquid Extraction of Rare-Earth Elements from Aqueous Acid Solutions by Acid Organophosphorus Compounds. Hydrometallurgy 1989, 22, 121–140. [Google Scholar] [CrossRef]
- Tasaki-Handa, Y.; Abe, Y.; Ooi, K.; Narita, H.; Tanaka, M.; Wakisaka, A. Selective Crystallization of Phosphoester Coordination Polymer for the Separation of Neodymium and Dysprosium: A Thermodynamic Approach. J. Phys. Chem. B 2016, 120, 12730–12735. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Guo, X.; Wang, Y.; Diefenbach, K.; Chen, L.; Wang, J.-Q.; Lin, J.; Wang, S. Size-Dependent Selective Crystallization Using an Inorganic Mixed-Oxoanion System for Lanthanide Separation. Dalton Trans. 2019, 48, 12808–12811. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Wang, Y.; Bai, X.; Wang, Y.; Chen, L.; Xiao, C.; Diwu, J.; Du, S.; Chai, Z.; Albrecht-Schmitt, T.E.; et al. Rare Earth Separations by Selective Borate Crystallization. Nat. Commun. 2017, 8, 14438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ya Gao, H.; Li Peng, W.; Pan Meng, P.; Feng Feng, X.; Qiang Li, J.; Qiong Wu, H.; Sheng Yan, C.; Yang Xiong, Y.; Luo, F. Lanthanide Separation Using Size-Selective Crystallization of Ln-MOFs. Chem. Commun. 2017, 53, 5737–5739. [Google Scholar] [CrossRef]
- Nelson, J.J.M.; Cheisson, T.; Rugh, H.J.; Gau, M.R.; Carroll, P.J.; Schelter, E.J. High-Throughput Screening for Discovery of Benchtop Separations Systems for Selected Rare Earth Elements. Commun. Chem. 2020, 3, 7. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extractant | Synthesis Strategy | Separation Method | Studied REs and Actinoids | Ref. |
|---|---|---|---|---|
| 1 MOABP | Pudovik | Solvent extraction | Y(iii), La(iii), Ce(iii), Eu(iii), Pr(iii), Tb(iii), Th(iv), U(iv), U(vi) | [31,32,33,52,53,54,55] |
| 2 MOCABP | Pudovik | Solvent extraction | La(iii), Ce(iii), Eu(iii), Pr(iii) | [30,31,32,33] |
| 3–5 | Mannich a | Solvent extraction | La(iii), Lu(iii) | [34,35] |
| 6 | Mannich a | Solvent extraction | La(iii) | [34] |
| 7 | Pudovik | Solvent extraction | La(iii) | [35] |
| 8 | Kabachnik–Fields | Solvent extraction | Sc(iii) | [11] |
| 9 DEHAMP | Kabachnik–Fields | Solvent extraction | Sc(iii), La(iii), Ce(iv), Gd(iii), Yb(iii), Th(iv) | [40] |
| 10 HEHHAP | Kabachnik–Fields | Solvent extraction, synergistic solvent extraction with Cyanex272 | La(iii), Ce(iii), Pr(iii), Nd(iii), Sm(iii), Eu(iii), Gd(iii), Tb(iii), Dy(iii), Ho(iii), Y(iii), Er(iii), Tm(iii), Yb(iii), Lu(iii) | [44,48] |
| 11 Cextrant 230 | Kabachnik–Fields | Solvent extraction | Sc(iii), La(iii), Ce(iv), Gd(iii), Yb(iii), Th(iv), U(vi) | [39,45,56] |
| 12 DEHAPP | Kabachnik–Fields | Solvent extraction | Sc(III), La(III), Ce(III), Ce(iv), Gd(III), Y(III), Yb(III), Th(iv) | [41] |
| 13 HEHAPP | Kabachnik–Fields | Solvent extraction, synergistic solvent extraction with D2EHPA | La(iii), Ce(iii), Pr(iii), Nd(iii), Sm(iii), Eu(iii), Gd(iii), Tb(iii), Dy(iii), Ho(iii), Y(iii), Er(iii), Tm(iii), Yb(iii), Lu(iii) | [42,46] |
| 14 HEHAMP | Kabachnik–Fields | Solvent extraction, synergistic solvent extraction with HEHEHP | Sc(iii), La(iii), Pr(iii), Nd(iii), Sm(iii), Eu(iii), Gd(iii), Tb(iii), Dy(iii), Ho(iii), Y(iii), Er(iii), Tm(iii), Yb(iii), Lu(iii) | [43,47] |
| 15–20 | Kabachnik–Fields | Precipitation | Sc(iii), La(iii), Ce(iii), Pr(iii), Nd(iii), Sm(iii), Eu(iii), Gd(iii), Tb(iii), Dy(iii), Ho(iii), Y(iii), Er(iii), Tm(iii), Yb(iii), Lu(iii), Th(iv), U(vi) | [9] |
| 21 EEAMPA | Kabachnik–Fields | Solvent extraction | La(iii), Ce(iii), Pr(iii), Nd(iii), Sm(iii), Eu(iii), Gd(iii), Tb(iii), Dy(iii), Ho(iii), Y(iii), Er(iii), Tm(iii), Yb(iii), Lu(iii) | [50] |
| 22 | Kabachnik–Fields | Solvent extraction | Sc(iii), Nd(iii), Sm(iii), Dy(iii), Yb(iii), Lu(iii) | [11,36] |
| 23–28 | Kabachnik–Fields | Solvent extraction | Sc(iii) | [11] |
| 29, 31 | Kabachnik–Fields | Solvent extraction | Lu(iii) | [36] |
| 30 | Kabachnik–Fields | Solvent extraction | Sc(iii), La(iii), Ce(iii), Nd(iii), Sm(iii), Gd(iii), Y(iii), Lu(iii), U(vi) | [37] |
| 32 DEHAPO | Kabachnik–Fields | Solvent extraction | La(iii), Ce(iv), Gd(iii), Yb(iii), Th(iv) | [51] |
| Extractant | Metal | P-OH (cm−1) | νP=O (cm−1) | P-O-C (cm−1) | N-H (cm−1) | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ligand | Complex | Ligand | Complex | Ligand | Complex | Ligand | Complex | |||
| 1 | Ce(iii) Pr(iii) | 1050–1000 | weak | 1208 | 1225–1155 | 1050–1000 | weak | ν3330 | ν3330 | [30,33] |
| 2 | Ce(iii) Pr(iii) | 1050–1000 | weak | 1240 | 1215–1160 | 1050–1000 | weak | ν3300 | ν3300 | [30,33] |
| 9 | Ce(iv) Th(iv) | n/a | n/a | 1250 | 1244Ce | νas1014 | νas1014 | - | - | [40] |
| 1247Th | ||||||||||
| 10 | Yb(iii) Lu(iii) | νas2438 | / | 1216 | 1206Yb | νas1040 νs973 | νas1040 νs975 | - | - | [44] |
| 1207Lu | ||||||||||
| 10 + Cyanex272 | Yb(iii) | νas2319 | / | 1200 | 1203 | ν1041 ν957 | ν1041 ν954 | ν3373 δ1624 | ν3381 δ1615 | [48] |
| 11 | Ce(iv) Th(iv) | n/a | n/a | 1250 | 1200Ce | νas1014 | νas1014 | ν3451 | ν3451 | [39] |
| 1238Th | ||||||||||
| Sc(iii) | n/a | n/a | 1230 | 1250 | νas1046 νs1014 | νas1046 νs1014 | δ1650 | δ1612 | [45] | |
| U(vi) | n/a | n/a | 1235 | 1256 | νas1016 | νas1016 | ν3446 | ν3446 | [56] | |
| 12 | Ce(iv) | n/a | n/a | 1239 | 1126 | νas1043 νs1010 | νas1043 νs1010 | δ1650 | δ1600 | [41] |
| 13 | Yb(iii) Lu(iii) | νas2398 1643 | / | 1225 | 1175 | νas1050 νs998 | νas1050 νs998 | ν3300 a | ν3300 a | [42,46] |
| 13 + D2EHPA | Lu(iii) | ν2402 1643 | / | 1231 | 1176 | ν1031 | ν1031 | - | - | [46] |
| 14 | Yb(iii) | ν2314 δ981 | / | 1159 | 1204 | ν1026 | ν1041 | - | - | [43] |
| 14 + HEHEHP | Yb(iii) | ν2317 δ981 | / | 1206 | 1145 | νas1039 | νas1041 | δ1620 | δ1615 | [47] |
| 21 | Lu(iii) | νas2318 | / | 1146 | 1162 | n/a | n/a | δ1614 | δ1644 | [50] |
| 32 | Ce(iv) | n/a | n/a | 1054 | 1040 | n/a | n/a | 3311 1675 | 3396, 1666, 1614 | [51] |
| Complex | Acid | Diluent | Ref. | |
|---|---|---|---|---|
| 1 | U(vi)O2L2 U(iv)L4 | H2SO4 | Ligroin Recryst. from ethanol | [52] |
| 1 | ML3∙HL/ML2∙HL2 (M=Eu, Tb) | HCl, HNO3, and HClO4 | Ligroin | [53] |
| 1 | ML3HL (M=Ln, Eu) | HCl, HClO4 | Petroleum ether, CHCl3, CCl4 | [31] |
| 1 | Ce(iii)L3∙2HL | HCl | CHCl3, benzene | [33] |
| 1 | Ce(iii)L3∙HL | HCl | CCl4, cyclohexane | [33] |
| 1 | PrL3∙HL | HCl | CHCl3, benzene, CCl4 | [33] |
| 1 | PrL3 | HCl | cyclohexane | [33] |
| 2 | ML3∙HL (M=Ln, Eu) | HCl | CHCl3 | [31] |
| 2 | Ce(iii)L3∙2HL | HCl | CHCl3 | [33] |
| 2 | PrL3 | HCl | CHCl3 | [33] |
| 3 | LaLX3 | - | CHCl3 | [35] |
| 3 | LuL2X3 or LuLX3 * | - | CHCl3 | [35] |
| 4 | LaL2X3 or LaLX3 * | - | CHCl3 | [35] |
| 4 | LuL2X3 or LuLX3 * | - | CHCl3 | [35] |
| 5 | LaL2X3 or LaLX3 * | - | CHCl3 | [35] |
| 5 | LuL2X3 | - | CHCl3 | [35] |
| 6 | LaL2Pic3 | - | CHCl3 | [34] |
| 7 | LaLX3 | - | CHCl3 | [35] |
| 9 | Ce(iv)(SO4)2 ∙ 2L | H2SO4 | heptane | [40] |
| 9 | Th(HSO4)2SO4 ∙ L | H2SO4 | heptane | [40] |
| 10 | MClH2L4 (M=Lu, Yb) | HCl | heptane | [44] |
| 10 + Cyanex272 | MH2Cl2A2B (A=10, M=Yb, Lu) | HCl | heptane | [48] |
| 11 | Ce(iv)(HSO4)2SO4 ∙ 2L | H2SO4 | heptane | [39] |
| 11 | Th(HSO4)2SO4 ∙ L | H2SO4 | heptane | [39] |
| 11 | Sc(HSO4)SO4 ∙ 2L | H2SO4 | heptane | [45] |
| 11 | UO2SO4 ∙ 2L | H2SO4 | heptane | [56] |
| 12 | Ce(iv)(HSO4)2SO4 ∙ 2L | H2SO4 | heptane | [41] |
| 13 | ML3 (M=La, Gd, Y, Lu) | HCl | heptane | [42] |
| 13 + D2EHPA | LuCl2H4A3B2 (A=13) | HCl | heptane | [46] |
| 14 | MH2ClL4 (M=Tm, Yb, Lu) | HCl | heptane | [43] |
| 14 + HEHEHP | MA2B4 (A=14, M=Lu, Yb, Tm, Er, Y, Ho) | HCl | heptane | [47] |
| 15 | LuL(NO3)2 | HNO3 | water | [9] |
| 15 | LaL2(NO3) | HNO3 | water | [9] |
| 15 | YL3 | HNO3 | water | [9] |
| 21 | MHL3NO3 (M=La, Nd, Gd, Lu) | HNO3 | heptane | [50] |
| 22 | ScL2X3 | HClO4 | toluene | [11] |
| 32 | Ce(iv)(HSO4)2SO4 ∙ L | H2SO4 | heptane | [51] |
| Extractant | Ce(iv)/Th | U/Eu | Th/RE | U/RE | Th/Lu | U/Th | U/Lu | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | - | 26,000 * | - | - | - | - | - | [53] a |
| 11 | 14.7 | - | - | - | - | - | - | [39] b |
| 11 | - | - | >1000 | >1000 | - | - | - | [56] c |
| 12 | 754.2 | - | - | - | - | - | - | [41] d |
| 15 | - | - | - | - | 4.50 g | FP(U) i | FP(U) i | [9] |
| 16 | - | - | - | - | 6.02 f,^ | 2.01 e | 4.03 f,^ | [9] |
| 17 | - | - | - | - | 9.17 g | 2.40 e | 8.68 g | [9] |
| 18 | - | - | - | - | 44.41 g | FP(U) h | FP(U) h | [9] |
| 19 | - | - | - | - | FP(Lu) j | FP(U) h | FP(U) h | [9] |
| 20 | - | - | - | - | FP(Th) j | FP(U) e | FP(U) e | [9] |
| 32 | 100.3 | - | - | - | - | - | - | [51] k |
| Extractant | Dilution | Metal | Acid | Capacity | Ref. |
|---|---|---|---|---|---|
| 9 | 0.63 M in heptane | 0.23 M Ce(iv) (∑Ce 0.24 M) 0.02 M Th(iv) | H2SO4 | 30.0 g/L Ce(iv) 24.4 g/L Th(iv) | [40] |
| 10 | 30% (v/v) in heptane | 0.0985 M YbCl3 0.0986 M LuCl3 | HCl | 12.76 g/L Yb 15.43 g/L Lu | [44] |
| 11 | 30% (v/v) in heptane | Ce(iv) & Th(iv) | H2SO4 | >30 g/L Ce(iv) ~43 g/L Th(iv) | [39,83] |
| 11 | 30% (v/v) in heptane | 0.064 M Sc | H2SO4 | 3.85 g/L Sc | [45] |
| 11 | 5% (v/v) in heptane | 8.08 mM Th(iv) 21 mM U(vi) | H2SO4 | 4.08 g/L Th(iv) 6.16 g/L U(vi) | [56] |
| 12 | 30% (v/v) in heptane | 0.29 M Ce(iv) | H2SO4 | 31.43 g/L CeO2 | [41] |
| 13 | 30% (v/v) in heptane | 0.1 M RE | HCl | 0.201 M Ho 0.205 M Er 0.216 M Yb 0.229 M Lu | [42] |
| 14 | 30% (v/v) in heptane | 0.055 M YbCl3 * | HCl | 15.17 g/L Lu 14.46 g/L Yb 12.64 g/L Y | [43] |
| 14 + HEHEHP | 30% (v/v) in heptane (1:1 extractant ratio) | 96 mM Lu 92 mM Yb | HCl | 27.25 g/L Lu2O3 26.59 g/L Yb2O3 | [47] |
| 21 | 4 mM in heptane | 0.4 mM RE | HNO3 | 0.393 mM Ho 0.402 mM Er 0.422 mM Tm 0.435 mM Yb 0.450 mM Lu | [50] |
| 32 | 30% (v/v) in heptane | 0.143 M Ce(iv) | H2SO4 | 16.66 g/L CeO2 | [51] |
| Extractant | Ce/La | Pr/Ce | Nd/Pr | Sm/Nd | Eu/Sm | Gd/Eu | Tb/Gd | Dy/Tb | Ho/Dy | Er/Ho | Tm/Er | Yb/Tm | Lu/Yb | Y/Ho | Er/Y | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 1.47 | 1.23 | 0.85 | 1.93 | 1.14 | 0.62 | 1.76 | 1.39 | 1.39 | 2.28 | 4.29 | 1.59 | 1.63 | 1.04 | 2.18 | [44] a |
| 10 | - | - | - | - | - | - | - | - | 1.27 | 1.23 | 2.36 | 3.18 | 1.59 | 1.41 | 0.88 | [48] b |
| 10 + Cyanex272 | - | - | - | - | - | - | - | - | 2.57 | 3.33 | 3.07 | 3.58 | 1.60 | 1.60 | 2.08 | [48] c |
| 12 | 135.1 * | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [41] d |
| 13 | - | - | - | - | - | - | - | - | - | 2.83 | 3.87 | 5.64 | 4.89 | 2.24 | 2.35 | [42] e |
| 13 + D2EHPA | 0.72 | 1.31 | 0.93 | 0.92 | 1.03 | 0.97 | 1.01 | 1.13 | 1.03 | 1.45 | 2.58 | 2.77 | 1.77 | 1.35 | 0.93 ** | [46] f |
| 14 | - | - | 1.43 | 1.35 | 1.11 | 1.19 | 1.44 | 1.07 | 1.32 | 1.78 | 1.93 | 1.36 | 1.24 | 1.13 | 1.58 | [43] g |
| 14 + HEHEHP | - | - | 1.20 | 1.14 | 1.45 | 1.13 | 1.16 | 1.17 | 1.05 | 2.11 | 1.78 | 1.76 | 1.20 | 1.32 | 1.61 | [47] h |
| 15 | 2.56 m | 2.06 l | 1.16 l | 1.41 m | 1.28 m | 1.22 m | 1.01 n | 2.67 l | 1.28 m,^ | 2.00 l,^ | - | 2.88 l | 2.52 k | - | 3.02 l | [9] |
| 16 | 1.33 m,^ | 1.23 i | 1.48 j | 1.50 k | 1.45 j,^ | 1.52 j,^ | 1.05 i | 1.23 m | 1.41 m | 1.09 i | - | 1.77 m,^ | 1.30 n | - | 2.75 i | [9] |
| 17 | 2.92 j | 1.36 k | 1.75 j | 1.44 l | 1.76 l | 1.41 l | 0.84 l | 1.51 l | 1.44 l | 1.18 l | - | 2.22 l,^ | 1.18 l | - | 3.33 m | [9] |
| 18 | 3.81 l | 1.26 m | 1.11 n | 1.70 l | 1.07 n,^ | 1.21 m | 1.49 l | 1.50 l | 1.47 l | 1.53 l | - | 3.60 k | 2.32 n | - | 2.21 l | [9] |
| 19 | 2.11 l | 2.18 k | 1.54 k | 2.04 k | 1.20 n | 1.42 k | 1.14 k | 1.06 k | 1.12 k | 1.57 n | - | 4.33 n | FP(Lu) n | - | 1.87 n | [9] |
| 20 | 1.88 m | 1.50 n | 1.15 m | 1.73 m | 1.29 l | 1.16 l | 1.77 l | 1.20 l | 1.08 l | 1.36 l | - | 1.94 l | 2.33 n | - | 2.03 l | [9] |
| 21 | 1.54 | 2.57 | 1.09 | 1.43 | 1.62 | 0.92 | 1.83 | 1.56 | 1.35 | 1.71 | 1.97 | 2.37 | 1.63 | 1.68 | 1.00 | [50] o |
| 32 | 167.0 * | - | - | - | - | - | - | - | - | - | - | - | - | - | - | [51] p |
| D2EHPA | 2.14 | 1.07 | 1.06 | 4.86 | 2.23 | 1.69 | 1.60 | 1.42 | 1.24 | 1.70 | 1.50 | 1.30 | 1.03 | - | - | [84] q |
| HEHEHP | 1.30 | 1.09 | 1.17 | 2.00 | 1.96 | 1.46 | 2.35 | 1.62 | 2.58 | 1.25 | 1.33 | 1.12 | 1.13 | - | - | [84] q |
| Extractant | Ho | Er | Tm | Yb | Lu | Y | Ref. |
|---|---|---|---|---|---|---|---|
| 13 + D2EHPA | - | - | - | - | 3.96 (0.5) | - | [46] a |
| 14 + HEHEHP | 2.18 (0.4) | 2.14 (0.5) | 2.54 (0.5) | 2.76 (0.5) | 2.89 (0.5) | 2.14 (0.5) | [47] b |
| 10 + Cyanex272 | 1.95 (0.4) | 2.71 (0.6) | 2.43 (0.6) | 3.67 (0.5) | 3.39 (0.5) | - | [48] c |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kukkonen, E.; Virtanen, E.J.; Moilanen, J.O. α-Aminophosphonates, -Phosphinates, and -Phosphine Oxides as Extraction and Precipitation Agents for Rare Earth Metals, Thorium, and Uranium: A Review. Molecules 2022, 27, 3465. https://doi.org/10.3390/molecules27113465
Kukkonen E, Virtanen EJ, Moilanen JO. α-Aminophosphonates, -Phosphinates, and -Phosphine Oxides as Extraction and Precipitation Agents for Rare Earth Metals, Thorium, and Uranium: A Review. Molecules. 2022; 27(11):3465. https://doi.org/10.3390/molecules27113465
Chicago/Turabian StyleKukkonen, Esa, Emilia Josefiina Virtanen, and Jani Olavi Moilanen. 2022. "α-Aminophosphonates, -Phosphinates, and -Phosphine Oxides as Extraction and Precipitation Agents for Rare Earth Metals, Thorium, and Uranium: A Review" Molecules 27, no. 11: 3465. https://doi.org/10.3390/molecules27113465
APA StyleKukkonen, E., Virtanen, E. J., & Moilanen, J. O. (2022). α-Aminophosphonates, -Phosphinates, and -Phosphine Oxides as Extraction and Precipitation Agents for Rare Earth Metals, Thorium, and Uranium: A Review. Molecules, 27(11), 3465. https://doi.org/10.3390/molecules27113465