
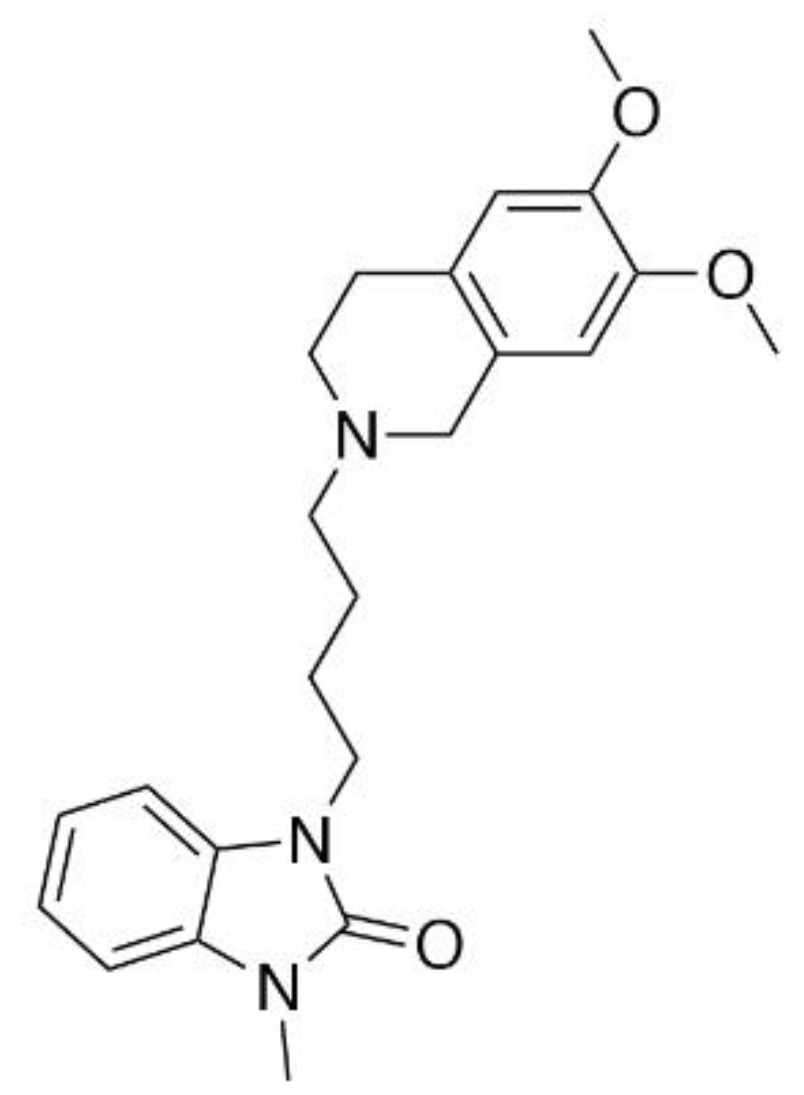
Characterization of CM-398, a Novel Selective Sigma-2 Receptor Ligand, as a Potential Therapeutic for Neuropathic Pain

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
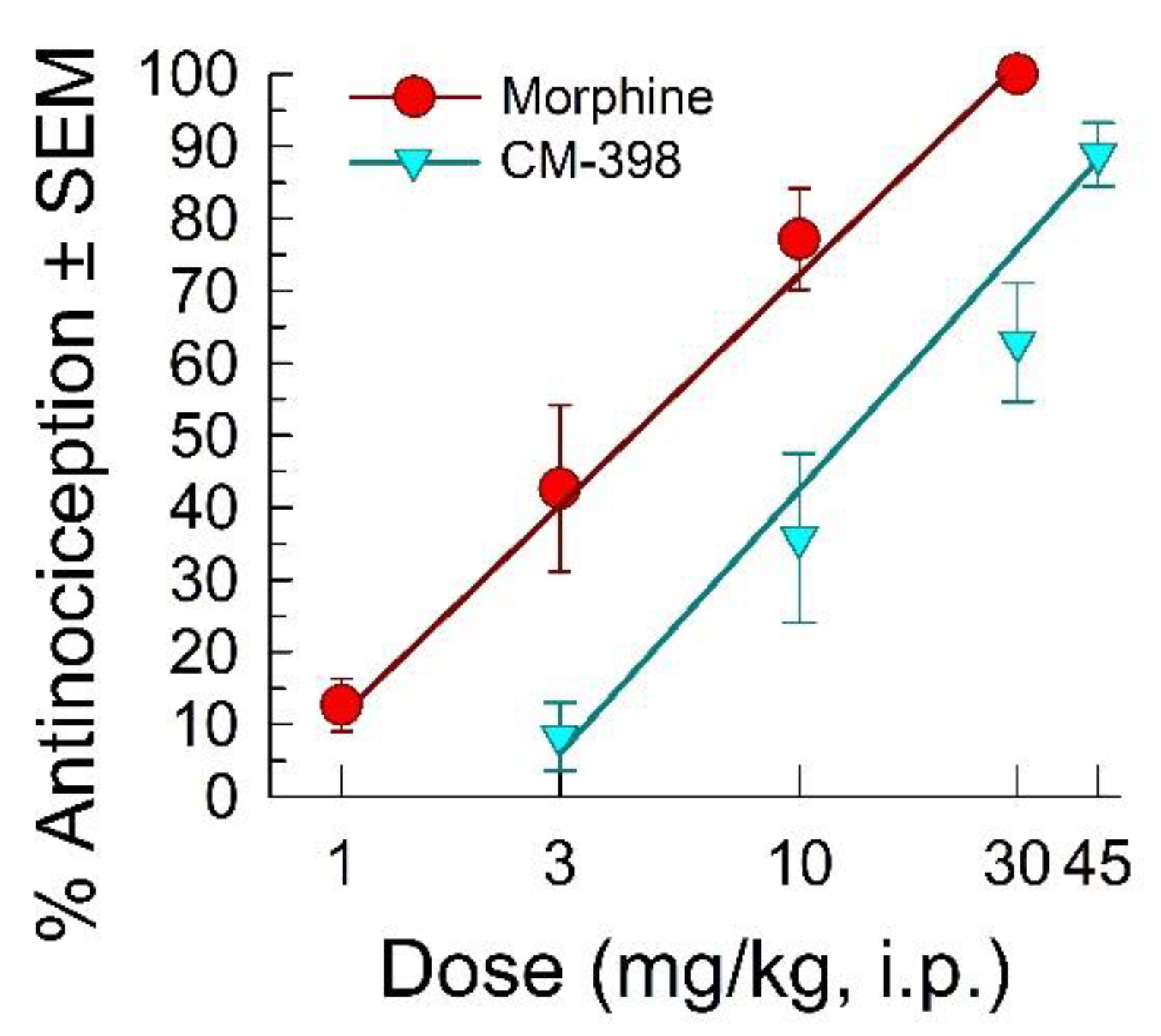
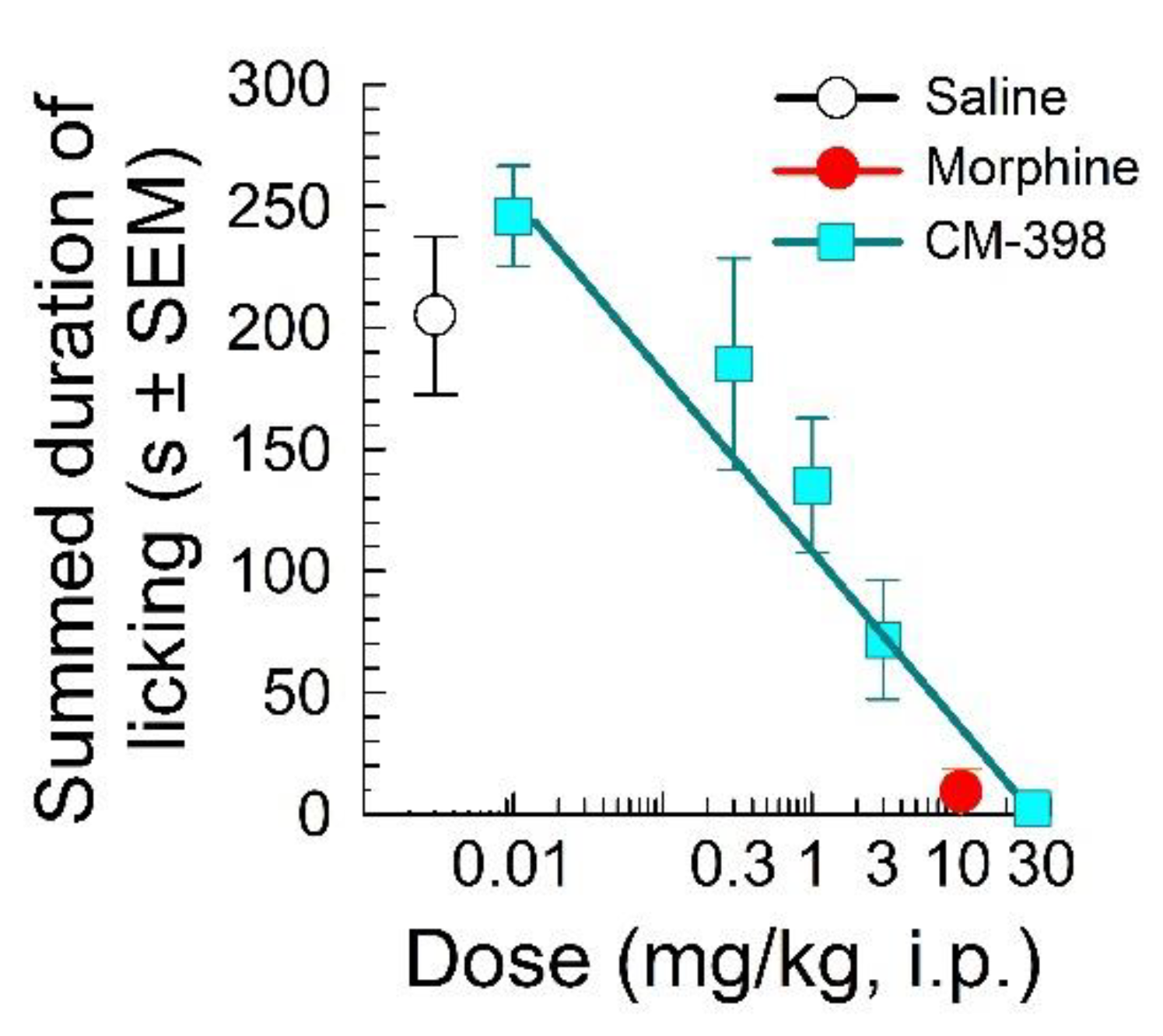
2.1. Assessment of CM-398 Antinociception in Visceral, Inflammatory, and Acute Thermal Models of Nociception
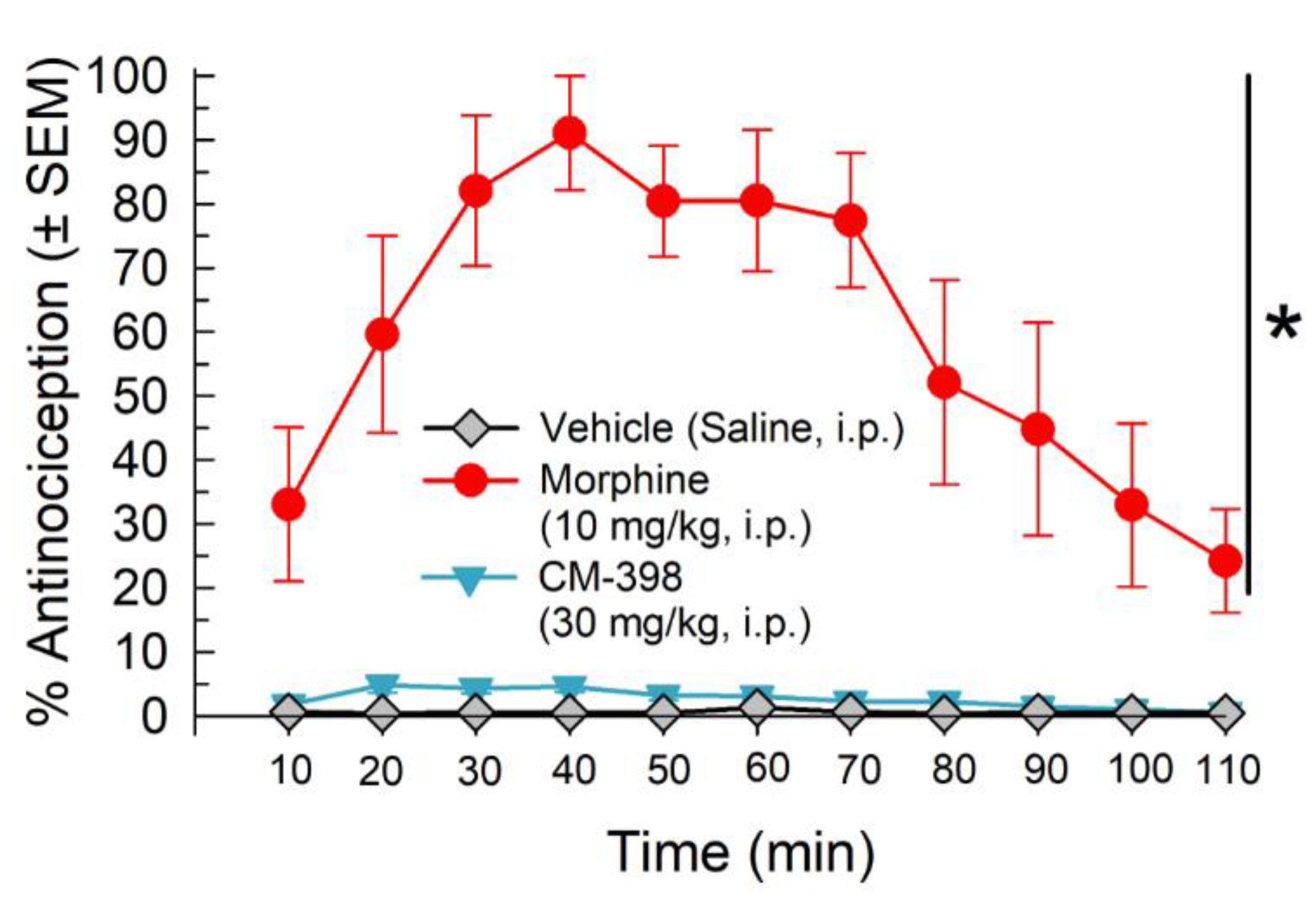
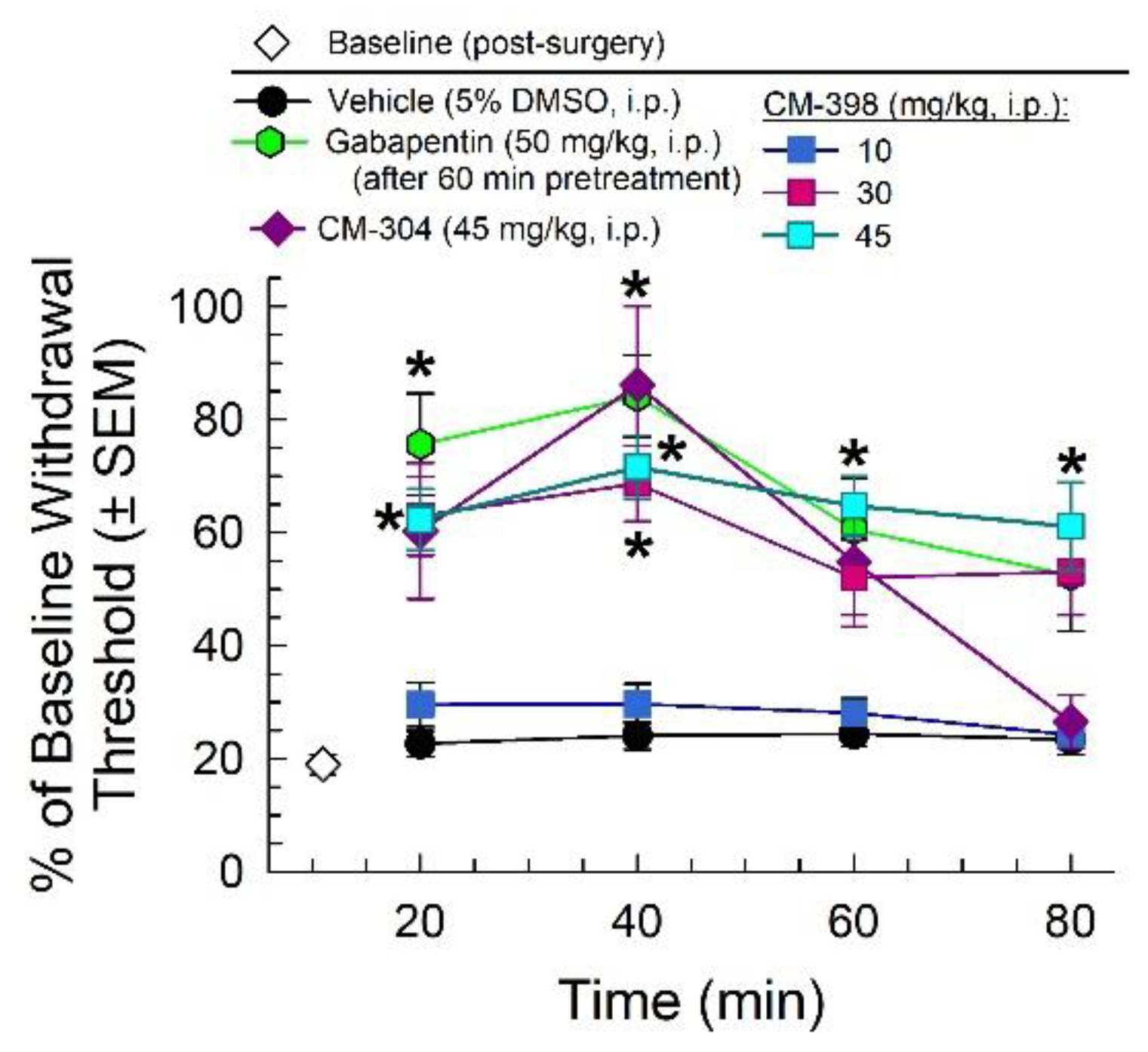
2.2. Anti-Allodynic Effects of CM-398 in the CCI Neuropathic Pain Model
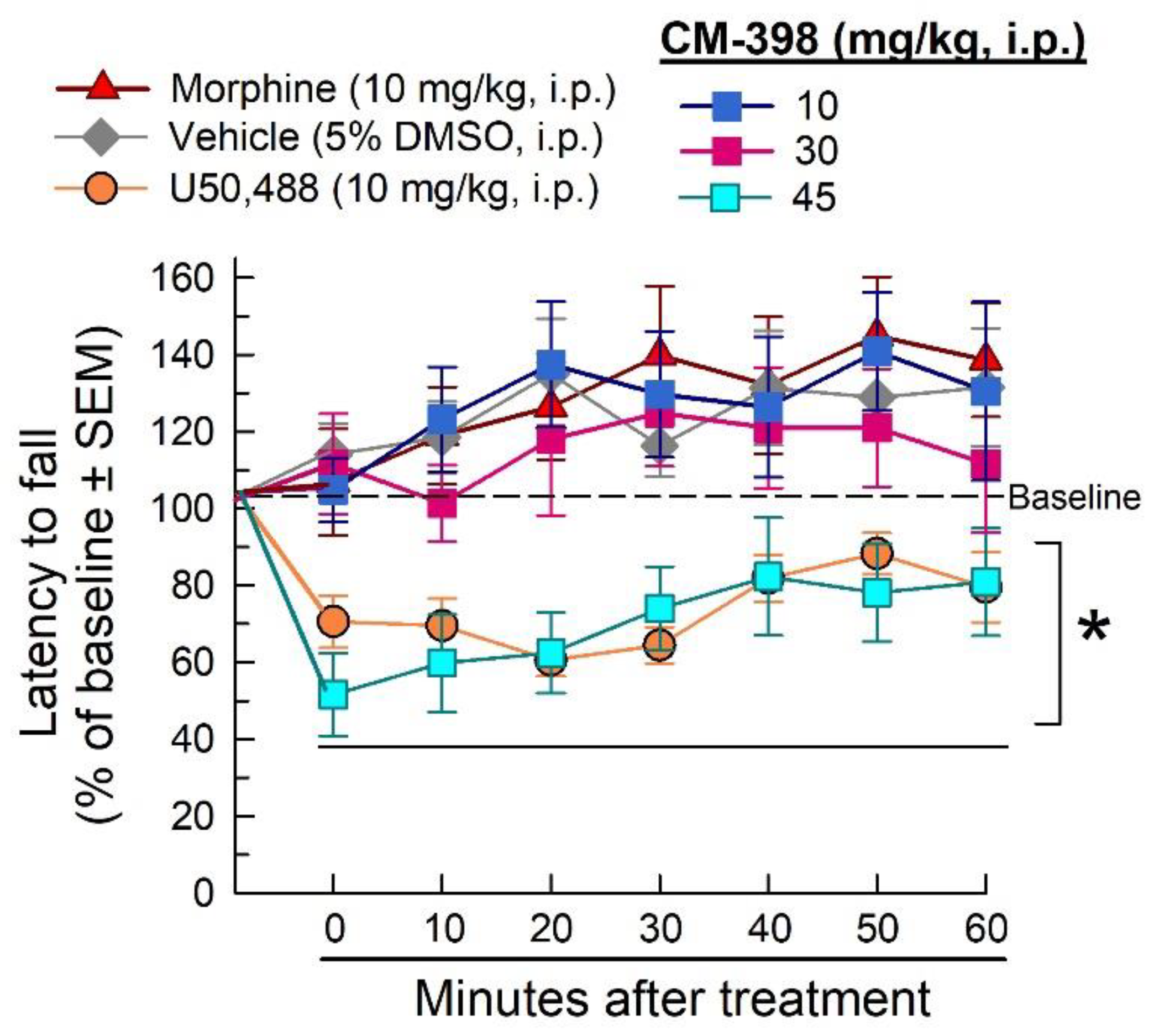
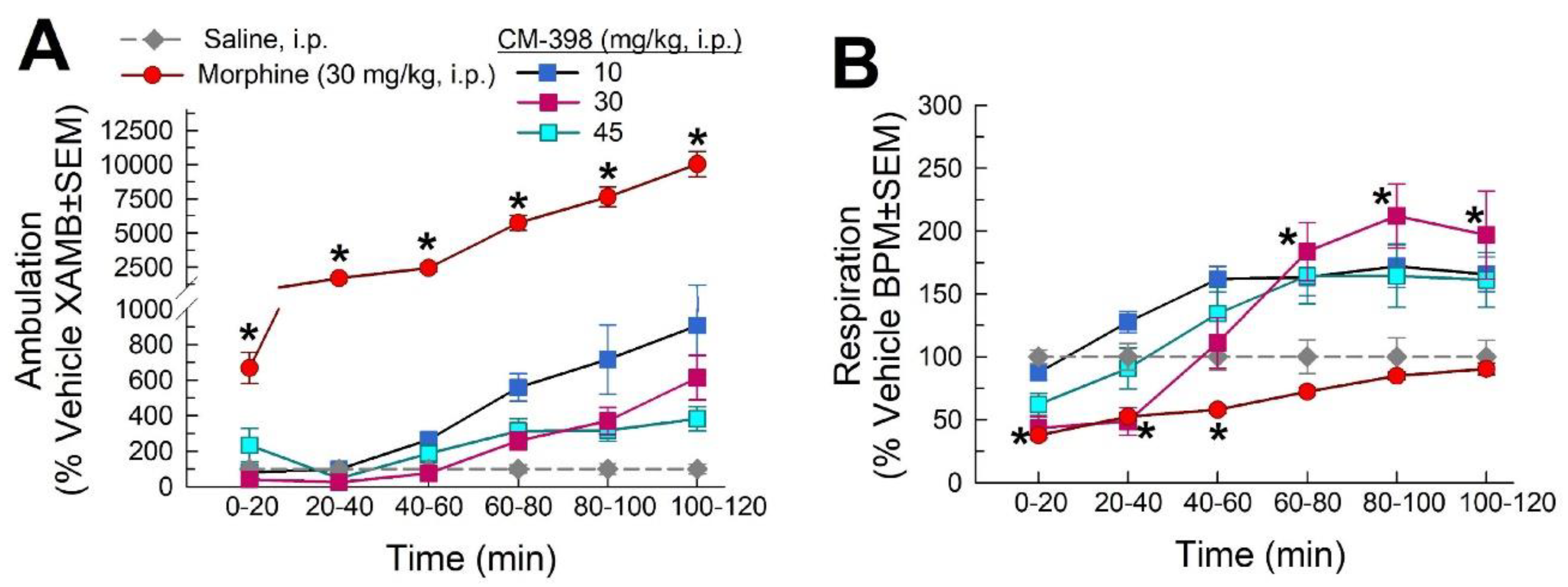
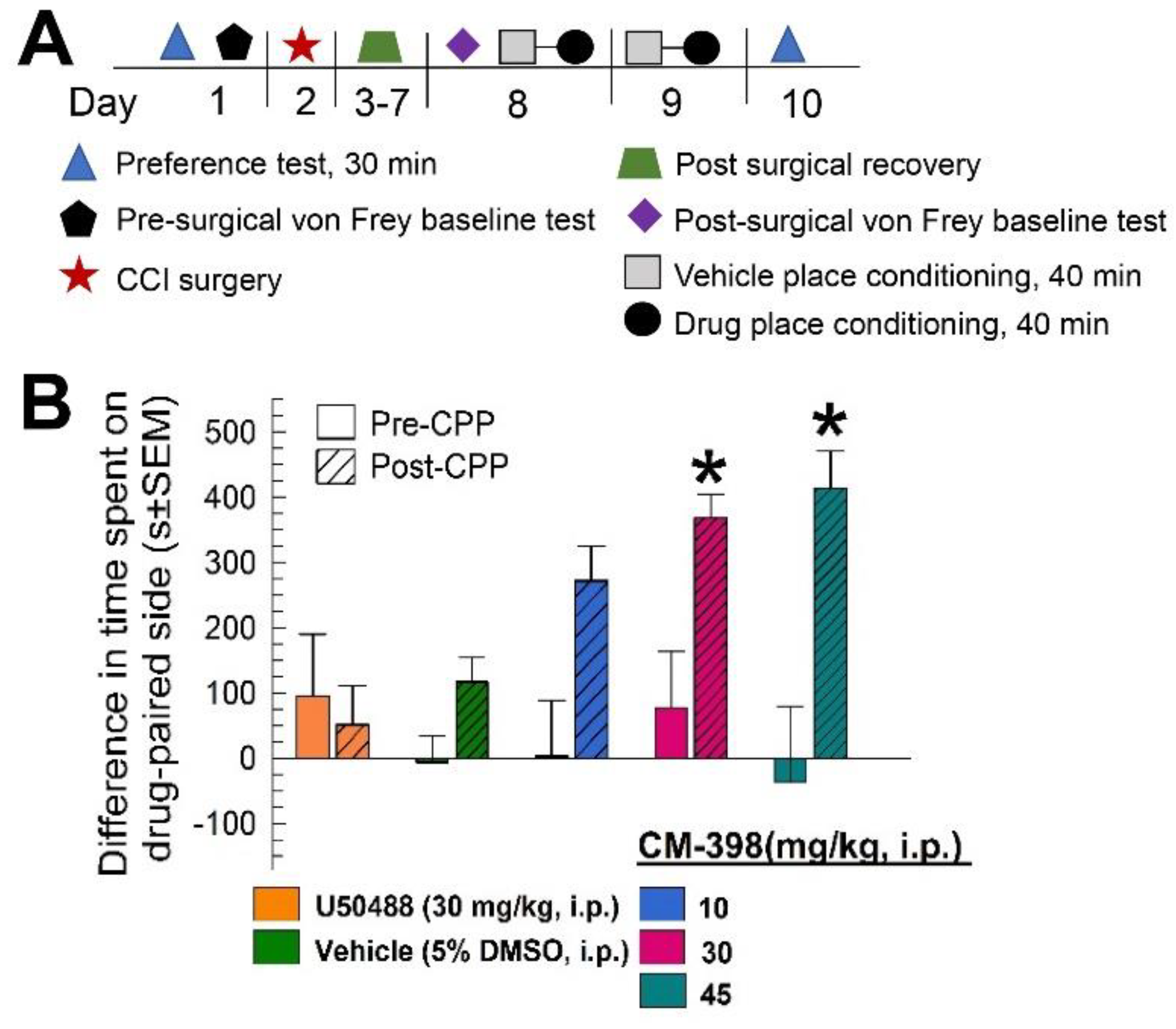
2.3. Assessment of CM-398-Induced Liabilities of Sedation, Respiratory Depression, and Drug-Seeking Behavior
2.4. Assessment of CM-398 Antinociception in an Operant Pain Model
3. Discussion
4. Materials and Methods
4.1. Materials
4.1.1. Subjects
4.1.2. Materials, Drug Preparation, and Administration
4.2. Behavioral Assays
4.2.1. Tail-Withdrawal Assay
4.2.2. Formalin Assay
4.2.3. Acetic Acid Writhing Test
mouse}]/[average stretches in vehicle group]) × 100.
4.2.4. Mechanical Allodynia von Frey Assessment
each mouse]/mean paw withdrawal force {g} in control group).
4.2.5. Chronic Constriction Injury
4.2.6. Rotarod Assay to Assess Motor Coordination
4.2.7. Respiratory Depression and Spontaneous Locomotor Testing with CLAMS
4.2.8. Conditioned Place Preference
4.2.9. Chronic Constrictive Nerve Injury/Conditioned Place Preference Operant Model of Pain (CCI/CPP)
4.3. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fornasari, D.; Coaccioli, S. Pharmacology of pain. Reumatismo 2014, 66, 14–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarzi-Puttini, P.; Vellucci, R.; Zuccaro, S.M.; Cherubino, P.; Labianca, R.; Fornasari, D. The appropriate treatment of chronic pain. Clin. Drug Investig. 2012, 32, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Russo, C.M.; Brose, W.G. Chronic pain. Annu. Rev. Med. 1998, 49, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Fitzgibbon, D.R.; Rathmell, J.P.; Michna, E.; Stephens, L.S.; Posner, K.L.; Domino, K.B. Malpractice claims associated with medication management for chronic pain. Anesthesiology 2010, 112, 948–956. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.M.; Bowen, W.D.; Walker, F.O.; Matsumoto, R.R.; DeCosta, B.; Rice, K.C. Sigma receptors: Biology and function. Pharmacol. Rev. 1990, 42, 355–402. [Google Scholar]
- Matsumoto, R.R. Targeting sigma receptors: Novel medication development for drug abuse and addiction. Expert Rev. Clin. Pharmacol. 2009, 2, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.R.; Kruse, A.C. The molecular function of σ receptors: Past, present, and future. Trends Pharmacol. Sci. 2019, 40, 636–654. [Google Scholar] [CrossRef]
- Hanner, M.; Moebius, F.F.; Flandorfer, A.; Knaus, H.G.; Striessnig, J.; Kempner, E.; Glossmann, H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc. Natl. Acad. Sci. USA 1996, 93, 8072–8077. [Google Scholar] [CrossRef] [Green Version]
- Cirino, T.J.; Eans, S.O.; Medina, J.M.; Wilson, L.L.; Mottinelli, M.; Intagliata, S.; McCurdy, C.R.; McLaughlin, J.P. Characterization of Sigma 1 receptor Antagonist CM-304 and its analog, AZ-66: Novel therapeutics against allodynia and induced pain. Front. Pharmacol. 2019, 10, 678. [Google Scholar] [CrossRef] [Green Version]
- Romero, L.; Zamanillo, D.; Nadal, X.; Sánchez-Arroyos, R.; Rivera-Arconada, I.; Dordal, A.; Montero, A.; Muro, A.; Bura, A.; Segalés, C.; et al. Pharmacological properties of s1ra, a new sigma-1 receptor antagonist that inhibits neuropathic pain and activity-induced spinal sensitization. Br. J. Pharmacol. 2012, 166, 2289–2306. [Google Scholar] [CrossRef] [Green Version]
- Alon, A.; Schmidt, H.; Zheng, S.; Kruse, A.C. Structural perspectives on sigma-1 receptor function. Advances in Experimental Medicine and Biology. In Sigma Receptors: Their Role in Disease and as Therapeutic Targets. Advances in Experimental Medicine and Biology; Smith, S., Su, T.P., Eds.; Springer: Cham, Switzerland, 2017; Volume 964, pp. 5–13. [Google Scholar] [CrossRef]
- Bartz, F.; Kern, L.; Erz, D.; Zhu, M.; Gilbert, D.; Meinhof, T.; Wirkner, U.; Erfle, H.; Muckenthaler, M.; Pepperkok, R.; et al. Identification of cholesterol-regulating genes by targeted rnai screening. Cell Metab. 2009, 10, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.-Y.; Li, Y.-Y.; Ma, W.-H.; Chen, A.-F.; Sun, Y.-T.; Lee, J.Y.; Riad, A.; Xu, D.-H.; Mach, R.H.; Huang, Y.-S. Synthesis, binding, and functional properties of tetrahydroisoquinolino-2-alkyl phenones as selective σ2r/tmem97 ligands. Eur. J. Med. Chem. 2021, 209, 112906. [Google Scholar] [CrossRef] [PubMed]
- Bowen, W.D. Sigma receptors: Recent advances and new clinical potentials. Pharm. Acta Helv. 2000, 74, 211–218. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Lu, H.-L.; Zhang, L.-J.; Wu, Z. Sigma-2 receptor ligands and their perspectives in cancer diagnosis and therapy. Med. Res. Rev. 2013, 34, 532–566. [Google Scholar] [CrossRef]
- Grundman, M.; Morgan, R.; Lickliter, J.D.; Schneider, L.S.; DeKosky, S.; Izzo, N.J.; Guttendorf, R.; Higgin, M.; Pribyl, J.; Mozzoni, K.; et al. A phase 1 clinical trial of the sigma-2 receptor complex allosteric antagonist CT1812, a novel therapeutic candidate for alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 5, 20–26. [Google Scholar] [CrossRef]
- Intagliata, S.; Sharma, A.; King, T.I.; Mesangeau, C.; Seminerio, M.; Chin, F.T.; Wilson, L.L.; Matsumoto, R.R.; McLaughlin, J.P.; Avery, B.A.; et al. Discovery of a highly selective sigma-2 receptor ligand, 1-(4-(6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1h)-yl)butyl)-3-methyl-1h-benzo[d]imidazol-2(3H)-one (CM398), with drug-like properties and antinociceptive effects in vivo. AAPS J. 2020, 22, 94. [Google Scholar] [CrossRef]
- Cobos, E.J.; Baeyens, J.M.; Del Pozo, E. Phenytoin differentially modulates the affinity of agonist and antagonist ligands for sigma-1 receptors of Guinea pig brain. Synapse 2005, 55, 192–195. [Google Scholar] [CrossRef]
- Guo, L.; Chen, Y.; Zhao, R.; Wang, G.; Friedman, E.; Zhang, A.; Zhen, X. Allosteric modulation of sigma-1 receptors elicits anti-seizure activities. Br. J. Pharmacol. 2015, 172, 4052–4065. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Lee, J.Y.; Hsieh, C.-J.; Riad, A.; Izzo, N.J.; Catalano, S.M.; Graham, T.J.A.; Mach, R.H. Screening of σ2 receptor ligands and in vivo evaluation of 11C-labeled 6,7-Dimethoxy-2-[4-(4-methoxyphenyl)butan-2-yl]-1,2,3,4-tetrahydroisoquinoline for potential use as a σ2 receptor brain PET tracer. J. Med. Chem. 2022, 65, 6261–6272. [Google Scholar] [CrossRef]
- Sahn, J.J.; Mejia, G.L.; Ray, P.R.; Martin, S.F.; Price, T.J. Sigma 2 receptor/TMEM97 agonists produce long lasting antineuropathic pain effects in mice. ACS Chem. Neurosci. 2017, 8, 1801–1811. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Blázquez, P.; Cortés-Montero, E.; Rodríguez-Muñoz, M.; Merlos, M.; Garzón-Niño, J. The sigma 2 receptor promotes and the sigma 1 receptor inhibits mu-opioid receptor-mediated antinociception. Mol. Brain 2020, 13, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Izzo, N.J.; Staniszewski, A.; To, L.; Fa, M.; Teich, A.F.; Saeed, F.; Wostein, H.; Walko, T.; Vaswani, A., III; Wardius, M.; et al. Alzheimer’s therapeutics targeting amyloid beta 1−42 oligomers I: Abeta 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits. PLoS ONE 2014, 9, e111898. [Google Scholar] [CrossRef] [PubMed]
- Alon, A.; Lyu, J.; Braz, J.M.; Tummino, T.A.; Craik, V.; O’Meara, M.J.; Webb, C.M.; Radchenko, D.S.; Moroz, Y.S.; Huang, X.-P.; et al. Structures of the sigma-2 receptor enable docking for bioactive ligand discovery. Nature 2021, 600, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fernández, C.; Entrena, J.M.; Baeyens, J.M.; Cobos, E.J. Sigma-1 receptor antagonists: A new class of neuromodulatory analgesics. Advances in Experimental Medicine and Biology. In Sigma Receptors: Their Role in Disease and as Therapeutic Targets. Advances in Experimental Medicine and Biology; Smith, S., Su, T.P., Eds.; Springer: Cham, Switzerland, 2017; Volume 964, pp. 109–132. [Google Scholar] [CrossRef]
- Gris, G.; Portillo-Salido, E.; Aubel, B.; Darbaky, Y.; Deseure, K.; Vela, J.M.; Merlos, M.; Zamanillo, D. The selective sigma-1 receptor antagonist e-52862 attenuates neuropathic pain of different aetiology in rats. Sci. Rep. 2016, 6, 24591. [Google Scholar] [CrossRef] [Green Version]
- Vidal-Torres, A.; de la Puente, B.; Rocasalbas, M.; Touriño, C.; Andreea Bura, S.; Fernández-Pastor, B.; Romero, L.; Codony, X.; Zamanillo, D.; Buschmann, H.; et al. Sigma-1 receptor antagonism as opioid adjuvant strategy: Enhancement of opioid antinociception without increasing adverse effects. Eur. J. Pharmacol. 2013, 711, 63–72. [Google Scholar] [CrossRef]
- Yan, W.; Jiang, Z.; Fan, F. Analgesic action of rubimaillin in vitro and in vivo. Cell. Mol. Biol. 2020, 66, 171. [Google Scholar] [CrossRef]
- Millan, M.J. Descending control of pain. Prog. Neurobiol. 2002, 66, 355–474. [Google Scholar] [CrossRef]
- Tejada, M.A.; Montilla-García, A.; Sánchez-Fernández, C.; Entrena, J.M.; Perazzoli, G.; Baeyens, J.M.; Cobos, E.J. Sigma-1 receptor inhibition reverses acute inflammatory hyperalgesia in mice: Role of peripheral sigma-1 receptors. Psychopharmacology 2014, 231, 3855–3869. [Google Scholar] [CrossRef]
- de la Puente, B.; Nadal, X.; Portillo-Salido, E.; Sánchez-Arroyos, R.; Ovalle, S.; Palacios, G.; Muro, A.; Romero, L.; Entrena, J.M.; Baeyens, J.M.; et al. Sigma-1 receptors regulate activity-induced spinal sensitization and neuropathic pain after peripheral nerve injury. Pain 2009, 145, 294–303. [Google Scholar] [CrossRef]
- Merlos, M.; Burgueño, J.; Portillo-Salido, E.; Plata-Salamán, C.R.; Vela, J.M. Pharmacological modulation of the sigma 1 receptor and the treatment of pain. Adv. Exp. Med. Biol. 2017, 964, 85–107. [Google Scholar] [CrossRef]
- Kim, H.-W.; Roh, D.-H.; Yoon, S.-Y.; Seo, H.-S.; Kwon, Y.-B.; Han, H.-J.; Kim, K.-W.; Beitz, A.J.; Lee, J.-H. Activation of the spinal sigma-1 receptor enhances nmda-induced pain via pkc- and pka-dependent phosphorylation of the nr1 subunit in mice. Br. J. Pharmacol. 2008, 154, 1125–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.-R.; Roh, D.-H.; Yoon, S.-Y.; Kang, S.-Y.; Moon, J.-Y.; Kwon, S.-G.; Choi, H.-S.; Han, H.-J.; Beitz, A.J.; Oh, S.-B.; et al. Spinal sigma-1 receptors activate nadph oxidase 2 leading to the induction of pain hypersensitivity in mice and mechanical allodynia in neuropathic rats. Pharmacol. Res. 2013, 74, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fernández, C.; Montilla-García, Á.; González-Cano, R.; Nieto, F.R.; Romero, L.; Artacho-Cordón, A.; Montes, R.; Fernández-Pastor, B.; Merlos, M.; Baeyens, J.M.; et al. Modulation of peripheral μ-opioid analgesia by σ1 receptors. J. Pharmacol. Exp. Ther. 2013, 348, 32–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.N.; Meyer, R.A. Mechanisms of neuropathic pain. Neuron 2006, 52, 77–92. [Google Scholar] [CrossRef] [Green Version]
- Ong, K.S.; Keng, S.B. The biological, social, and psychological relationship between depression and chronic pain. J. Craniomandib. Sleep Pract. 2003, 21, 286–294. [Google Scholar] [CrossRef]
- Navratilova, E.; Xie, J.Y.; King, T.; Porreca, F. Evaluation of reward from pain relief. Ann. N. Y. Acad. Sci. 2013, 1282, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kochi, T.; Nakamura, Y.; Ma, S.; Hisaoka-Nakashima, K.; Wang, D.; Liu, K.; Wake, H.; Nishibori, M.; Irifune, M.; Morioka, N. Pretreatment with high mobility group box-1 monoclonal antibody prevents the onset of trigeminal neuropathy in mice with a distal infraorbital nerve chronic constriction injury. Molecules 2021, 26, 2035. [Google Scholar] [CrossRef]
- Evoy, K.E.; Morrison, M.D.; Saklad, S.R. Abuse and misuse of pregabalin and gabapentin. Drugs 2017, 77, 403–426. [Google Scholar] [CrossRef]
- Flórez, J.; Delgado, G.; Armijo, J.A. Adrenergic and serotonergic mechanisms in morphine-induced respiratory depression. Psychopharmacology 1972, 24, 258–274. [Google Scholar] [CrossRef]
- Seth, P.; Scholl, L.; Rudd, R.A.; Bacon, S. Overdose deaths involving opioids, cocaine, and psychostimulants—United states, 2015–2016. Am. J. Transplant. 2018, 18, 1556–1568. [Google Scholar] [CrossRef]
- Liu, Z.-H.; Shin, R.; Ikemoto, S. Dual role of medial A10 dopamine neurons in affective encoding. Neuropsychopharmacology 2008, 33, 3010–3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shippenberg, T.S.; Herz, A. Place preference conditioning reveals the involvement of D1-dopamine receptors in the motivational properties of mu-kappa-opioid agonists. Brain Res. 1987, 436, 169–172. [Google Scholar] [CrossRef]
- Chefer, V.I.; Bäckman, C.M.; Gigante, E.D.; Shippenberg, T.S. Kappa opioid receptors on dopaminergic neurons are necessary for kappa-mediated place aversion. Neuropsychopharmacology 2013, 38, 2623–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, A.D.; Reed, B.; Guariglia, C.; Dunn, A.M.; Hillman, J.M.; Kreek, M.J. Structurally related kappa opioid receptor agonists with substantial differential signaling bias: Neuroendocrine and behavioral effects in C57BL6 mice. Int. J. Neuropsychopharmacol. 2018, 21, 847–857. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, J.; Chen, J.; Tao, Y.; Wang, Y.; Xu, X.; Chen, J.; Xu, Y.; Xi, T.; Hu, X.; et al. Novel κ-opioid receptor agonist mb-1c-oh produces potent analgesia with less depression and sedation. Acta Pharmacol. Sin. 2015, 36, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Seminerio, M.J.; Robson, M.J.; Abdelazeem, A.H.; Mesangeau, C.; Jamalapuram, S.; Avery, B.A.; McCurdy, C.R.; Matsumoto, R.R. Synthesis and pharmacological characterization of a novel sigma receptor ligand with improved metabolic stability and antagonistic effects again methamphetamine. AAPS J. 2012, 14, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mei, J.; Pasternak, G.W. Modulation of brainstem opiate analgesia in the rat by σ1 receptors: A microinjection study. J. Pharmacol. Exp. Ther. 2007, 322, 1278–1285. [Google Scholar] [CrossRef] [Green Version]
- Mogil, J.S.; Smith, S.B.; O’Reilly, M.K.; Plourde, G. Influence of nociception and stress-induced antinociception on genetic variation in isoflurane anesthetic potency among mouse strains. Anesthesiology 2005, 103, 751–758. [Google Scholar] [CrossRef]
- Wilson, S.G.; Smith, S.B.; Chesler, E.J.; Melton, K.A.; Haas, J.J.; Mitton, B.; Strasburg, K.; Hubert, L.; Rodriguez-Zas, S.L.; Mogil, J.S. The heritability of antinociception: Common pharmacogenetic mediation of five neurochemically distinct analgesics. J. Pharmacol. Exp. Ther. 2003, 304, 547–559. [Google Scholar] [CrossRef] [Green Version]
- Reilley, K.J.; Giulianotti, M.; Dooley, C.T.; Nefzi, A.; Mclaughlin, J.P.; Houghten, R.A. Identification of two novel, potent, low-liability antinociceptive compounds from the direct in vivo screening of a large mixture-based combinatorial library. AAPS J. 2010, 12, 318–329. [Google Scholar] [CrossRef] [Green Version]
- Brabant, C.; Quertemont, E.; Tirelli, E. Influence of the dose and the number of drug-context pairings on the magnitude and the long-lasting retention of cocaine-induced conditioned place preference in c57bl/6j mice. Psychopharmacology 2005, 180, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Orsini, C.; Bonito-Oliva, A.; Conversi, D.; Cabib, S. Susceptibility to conditioned place preference induced by addictive drugs in mice of the C57BL/6 and DBA/2 inbred strains. Psychopharmacology 2005, 181, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The arrive guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- James, M.L.; Shen, B.; Nielsen, C.H.; Behera, D.; Buckmaster, C.L.; Mesangeau, C.; Zavaleta, C.; Vuppala, P.K.; Jamalapuram, S.; Avery, B.A.; et al. Evaluation of σ-1 receptor Radioligand 18F-FTC-146 in rats and squirrel monkeys using pet. J. Nucl. Med. 2014, 55, 147–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCurdy, C.R.; Mesangeau, C.; Matsumoto, R.R.; Poupaert, J.H.; Avery, B.A.; Abdelazeem, A.H. Highly Selective Sigma Receptor Ligands. U.S. Patent No. 8,686,008, 1 April 2014. [Google Scholar]
- Cheng, H.-Y.; Pitcher, G.M.; Laviolette, S.R.; Whishaw, I.Q.; Tong, K.I.; Kockeritz, L.K.; Wada, T.; Joza, N.A.; Crackower, M.; Goncalves, J.; et al. Dream is a critical transcriptional repressor for pain modulation. Cell 2002, 108, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Bidlack, J.M.; Cohen, D.J.; McLaughlin, J.P.; Lou, R.; Ye, Y.; Wentland, M.P. 8-carboxamidocyclazocine: A long-acting, novel benzomorphan. J. Pharmacol. Exp. Ther. 2002, 302, 374–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eans, S.O.; Ganno, M.L.; Mizrachi, E.; Houghten, R.A.; Dooley, C.T.; McLaughlin, J.P.; Nefzi, A. Parallel synthesis of hexahydrodiimidazodiazepines heterocyclic peptidomimetics and their in vitro and in vivo activities at μ (MOR), δ (DOR), and κ (KOR) opioid receptors. J. Med. Chem. 2015, 58, 4905–4917. [Google Scholar] [CrossRef]
- Bennett, G.J.; Xie, Y.-K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Pitcher, G.M.; Ritchie, J.; Henry, J.L. Nerve constriction in the rat: Model of neuropathic, surgical and central pain. Pain 1999, 83, 37–46. [Google Scholar] [CrossRef]
- Xu, M. Neuropathic pain activates the endogenous opioid system in mouse spinal cord and induces opioid receptor tolerance. J. Neurosci. 2004, 24, 4576–4584. [Google Scholar] [CrossRef] [Green Version]
- Hoot, M.R.; Sim-Selley, L.J.; Poklis, J.L.; Abdullah, R.A.; Scoggins, K.L.; Selley, D.E.; Dewey, W.L. Chronic constriction injury reduces cannabinoid receptor 1 activity in the rostral anterior cingulate cortex of mice. Brain Res. 2010, 1339, 18–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hummel, M.; Lu, P.; Cummons, T.A.; Whiteside, G.T. The persistence of a long-term negative affective state following the induction of either acute or chronic pain. Pain 2008, 140, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Salte, K.; Lea, G.; Franek, M.; Vaculin, S. Baclofen reversed thermal place preference in rats with chronic constriction injury. Physiol. Res. 2016, 65, 349–355. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, L.L.; Alleyne, A.R.; Eans, S.O.; Cirino, T.J.; Stacy, H.M.; Mottinelli, M.; Intagliata, S.; McCurdy, C.R.; McLaughlin, J.P. Characterization of CM-398, a Novel Selective Sigma-2 Receptor Ligand, as a Potential Therapeutic for Neuropathic Pain. Molecules 2022, 27, 3617. https://doi.org/10.3390/molecules27113617
Wilson LL, Alleyne AR, Eans SO, Cirino TJ, Stacy HM, Mottinelli M, Intagliata S, McCurdy CR, McLaughlin JP. Characterization of CM-398, a Novel Selective Sigma-2 Receptor Ligand, as a Potential Therapeutic for Neuropathic Pain. Molecules. 2022; 27(11):3617. https://doi.org/10.3390/molecules27113617
Chicago/Turabian StyleWilson, Lisa L., Amy R. Alleyne, Shainnel O. Eans, Thomas J. Cirino, Heather M. Stacy, Marco Mottinelli, Sebastiano Intagliata, Christopher R. McCurdy, and Jay P. McLaughlin. 2022. "Characterization of CM-398, a Novel Selective Sigma-2 Receptor Ligand, as a Potential Therapeutic for Neuropathic Pain" Molecules 27, no. 11: 3617. https://doi.org/10.3390/molecules27113617
APA StyleWilson, L. L., Alleyne, A. R., Eans, S. O., Cirino, T. J., Stacy, H. M., Mottinelli, M., Intagliata, S., McCurdy, C. R., & McLaughlin, J. P. (2022). Characterization of CM-398, a Novel Selective Sigma-2 Receptor Ligand, as a Potential Therapeutic for Neuropathic Pain. Molecules, 27(11), 3617. https://doi.org/10.3390/molecules27113617