
Going Retro, Going Viral: Experiences and Lessons in Drug Discovery from COVID-19

 and
and
Abstract
:
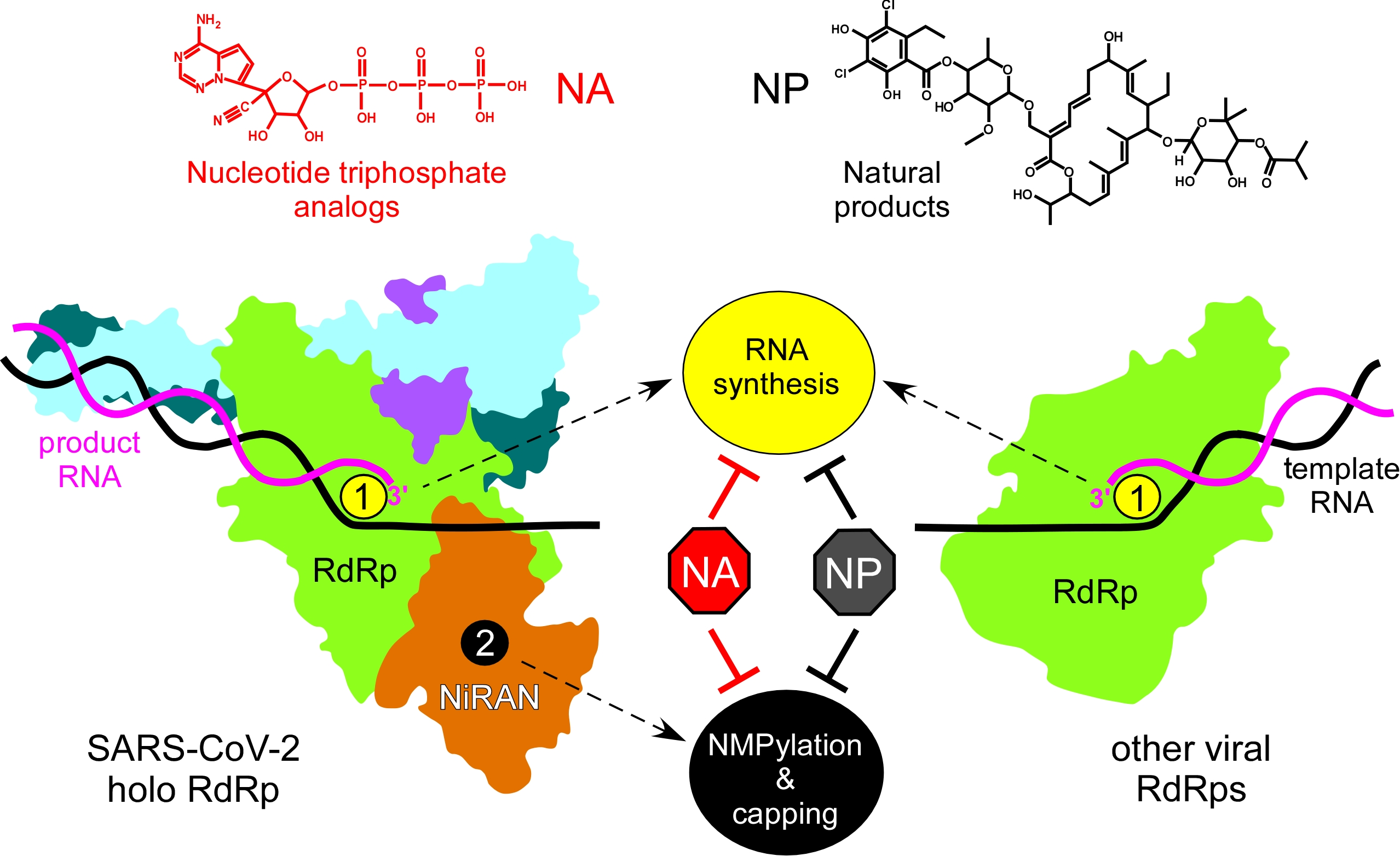{kind=link}
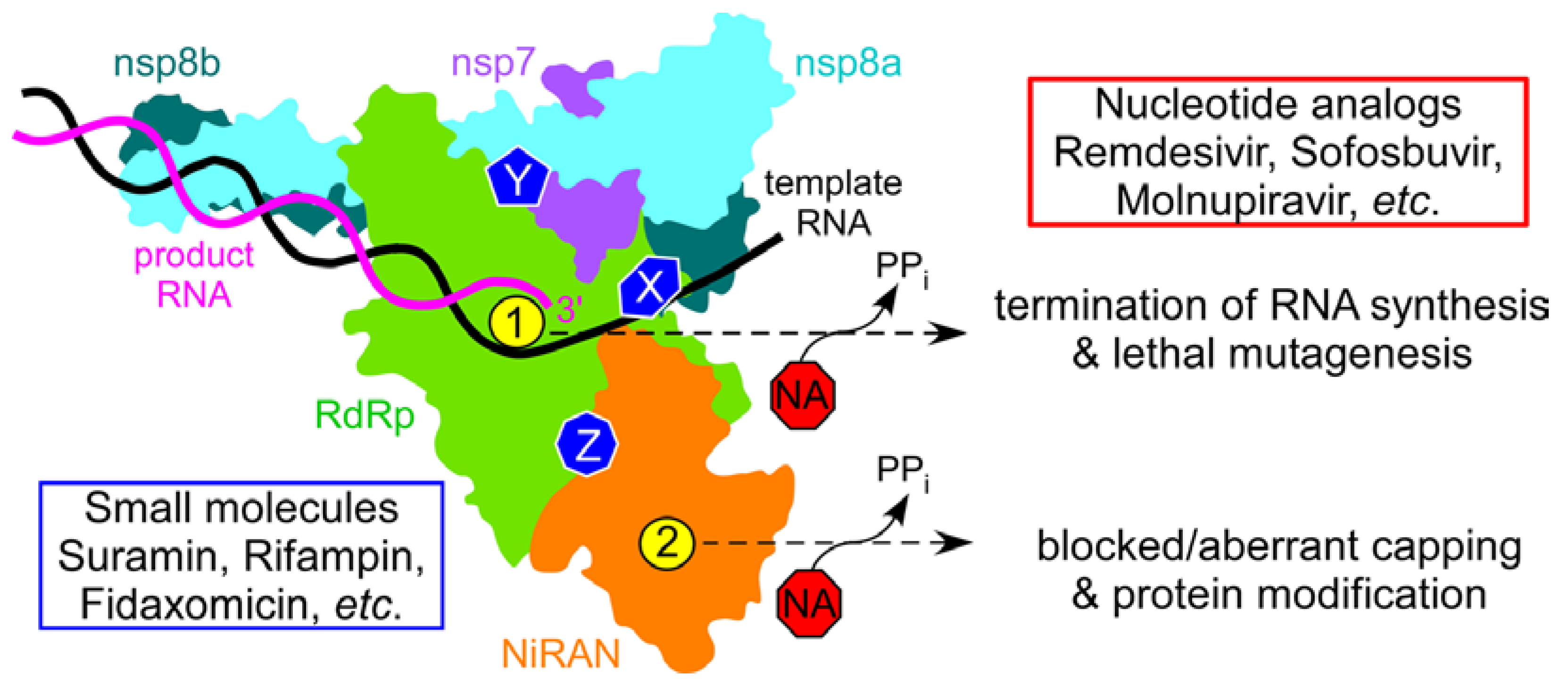{kind=link}
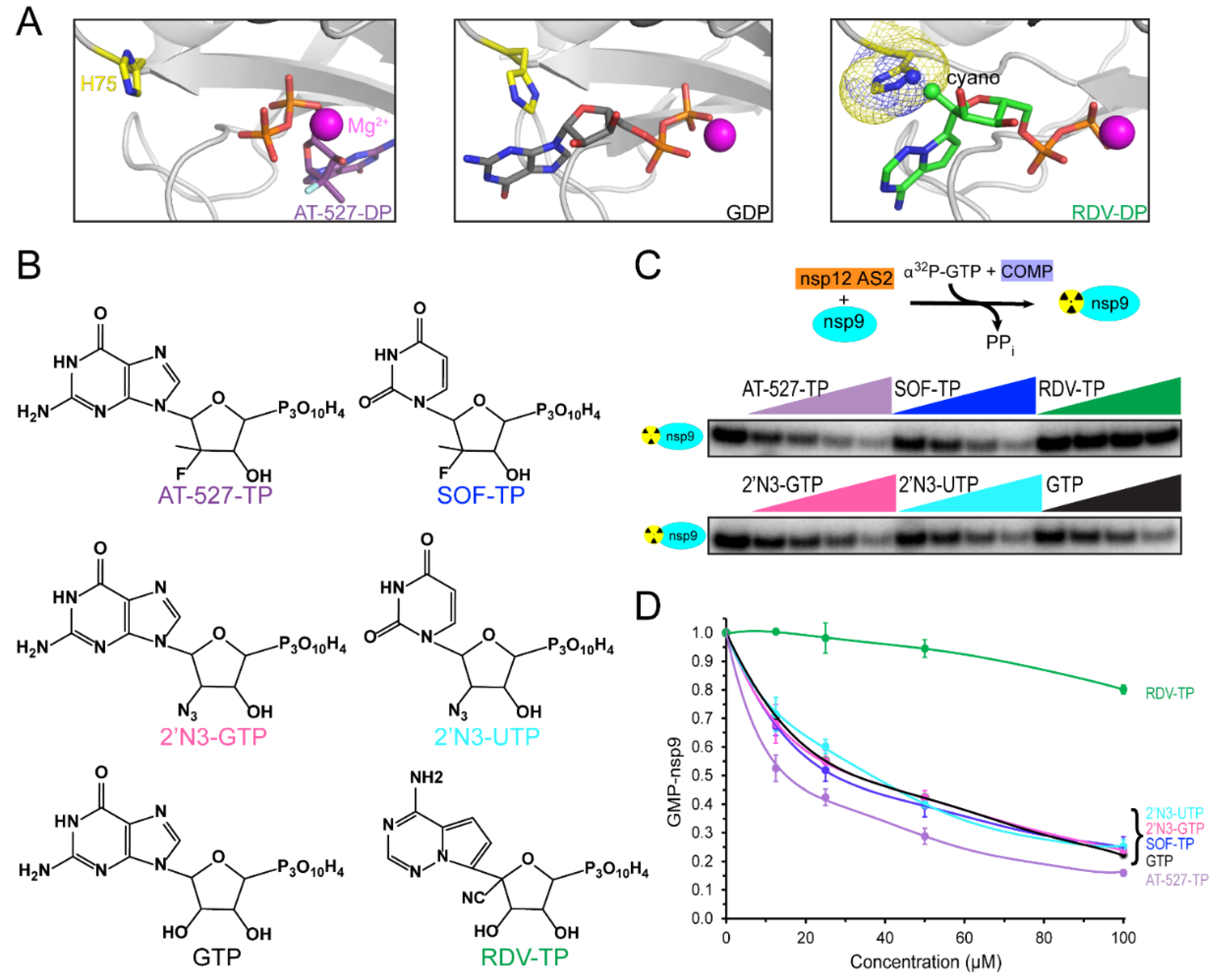{kind=link}
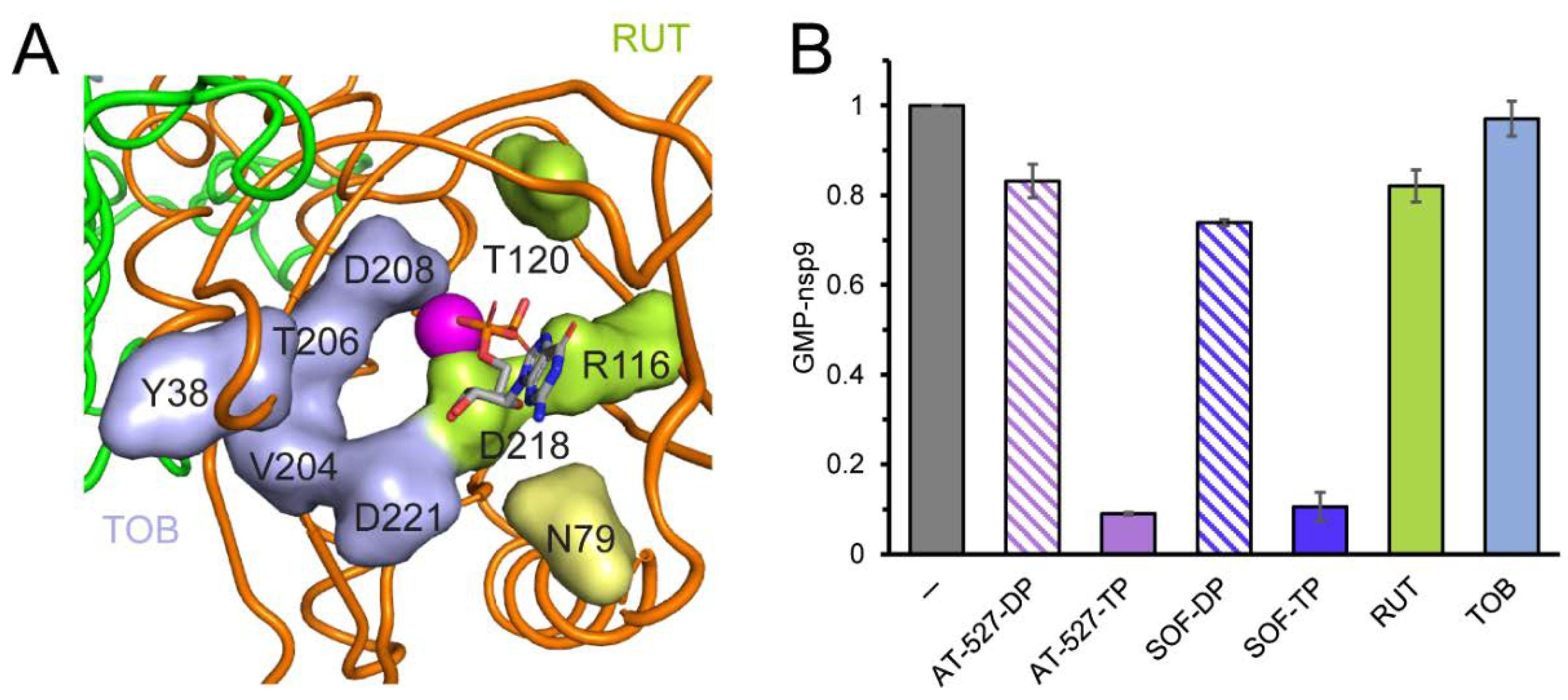{kind=link}
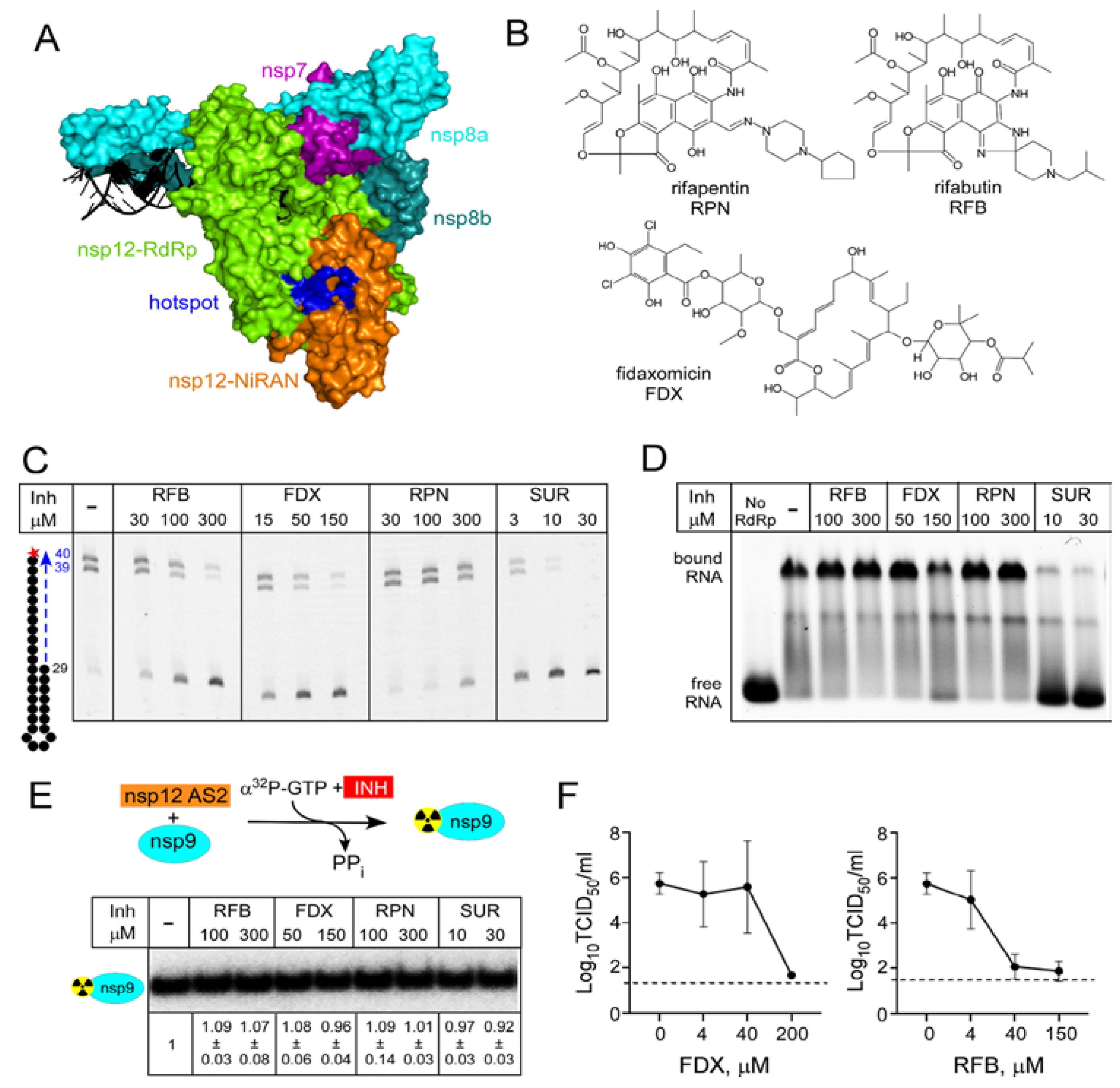{kind=link}
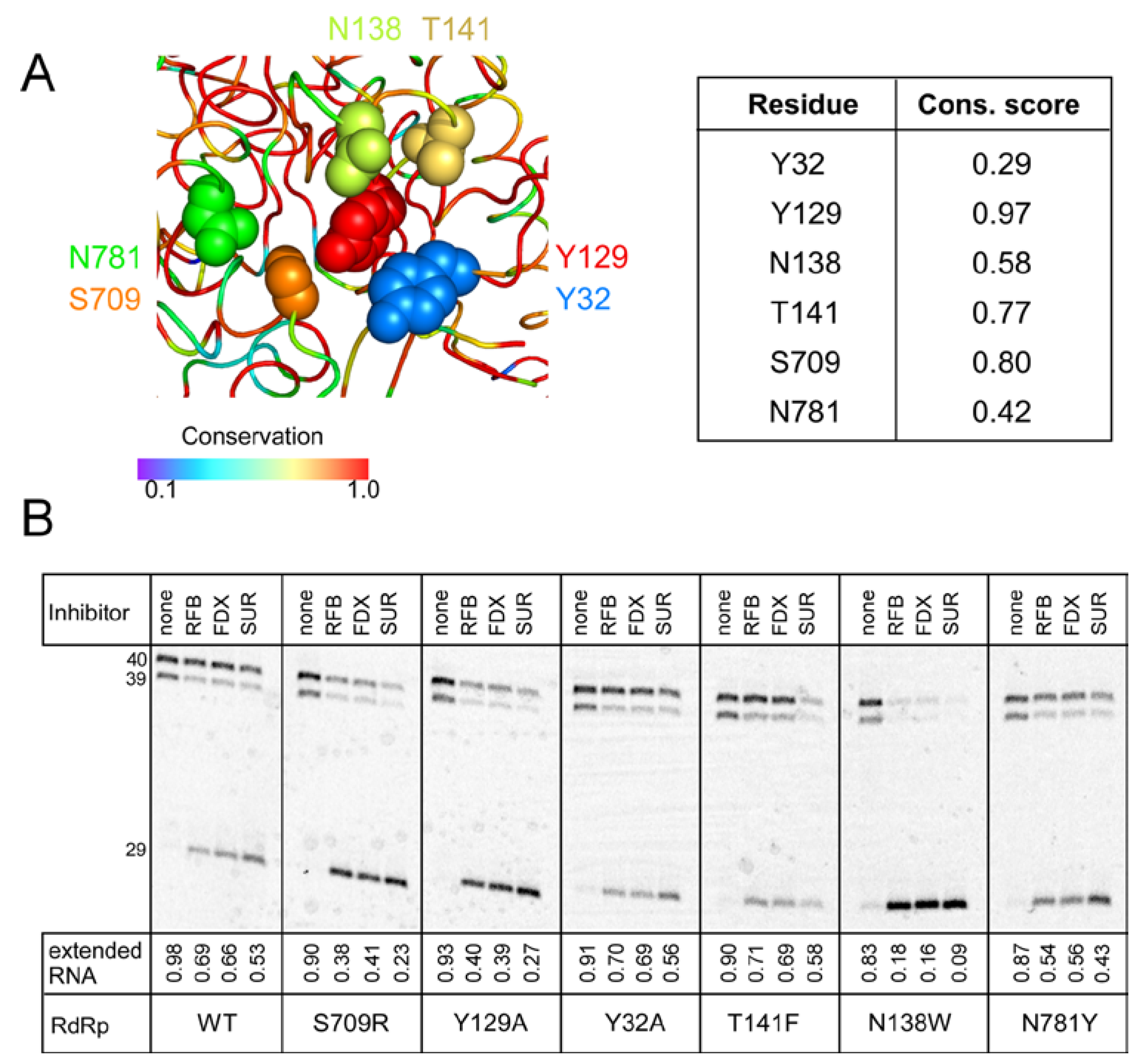{kind=link}
1. Introduction
2. Results
2.1. SARS-CoV-2 NiRAN Domain as a Target for Small-Molecule Ligands
2.2. Antibiotics Targeting Bacterial RNA Polymerase Also Inhibit SARS-CoV-2 RdRp
3. Discussion
3.1. Identification of Druggable Targets
3.2. Identification of Target-Specific Inhibitors
3.3. Conclusions and Perspectives
4. Materials and Methods
4.1. Expression Vectors and Protein Purification
4.2. NMPylation Assays
4.3. RNA Extension Assays
4.4. Electrophoretic Mobility Shift Assays
4.5. Cell-Based Viral Infectivity Assays
4.6. Sample Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UNAIDS. Available online: https://www.unaids.org (accessed on 2 June 2022).
- Johns Hopkins Coronavirus Resource Center. Available online: https://coronavirus.jhu.edu (accessed on 2 June 2022).
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, D.; Hui, H.C.; Doerffler, E.; Clarke, M.O.; Chun, K.; Zhang, L.; Neville, S.; Carra, E.; Lew, W.; Ross, B.; et al. Discovery and Synthesis of a Phosphoramidate Prodrug of a Pyrrolo[2,1-f][triazin-4-amino] Adenine C-Nucleoside (GS-5734) for the Treatment of Ebola and Emerging Viruses. J. Med. Chem. 2017, 60, 1648–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huchting, J. Targeting viral genome synthesis as broad-spectrum approach against RNA virus infections. Antivir. Chem. Chemother. 2020, 28, 2040206620976786. [Google Scholar] [CrossRef] [PubMed]
- Picarazzi, F.; Vicenti, I.; Saladini, F.; Zazzi, M.; Mori, M. Targeting the RdRp of Emerging RNA Viruses: The Structure-Based Drug Design Challenge. Molecules 2020, 25, 5695. [Google Scholar] [CrossRef]
- Peersen, O.B. A Comprehensive Superposition of Viral Polymerase Structures. Viruses 2019, 11, 745. [Google Scholar] [CrossRef] [Green Version]
- Parvathaneni, V.; Kulkarni, N.S.; Muth, A.; Gupta, V. Drug repurposing: A promising tool to accelerate the drug discovery process. Drug Discov. Today 2019, 24, 2076–2085. [Google Scholar] [CrossRef]
- Vicenti, I.; Zazzi, M.; Saladini, F. SARS-CoV-2 RNA-dependent RNA polymerase as a therapeutic target for COVID-19. Expert Opin. Ther. Pat. 2021, 31, 325–337. [Google Scholar] [CrossRef]
- Wahl, A.; Gralinski, L.E.; Johnson, C.E.; Yao, W.; Kovarova, M.; Dinnon, K.H., 3rd; Liu, H.; Madden, V.J.; Krzystek, H.M.; De, C.; et al. SARS-CoV-2 infection is effectively treated and prevented by EIDD-2801. Nature 2021, 591, 451–457. [Google Scholar] [CrossRef]
- Shannon, A.; Selisko, B.; Le, N.T.; Huchting, J.; Touret, F.; Piorkowski, G.; Fattorini, V.; Ferron, F.; Decroly, E.; Meier, C.; et al. Rapid incorporation of Favipiravir by the fast and permissive viral RNA polymerase complex results in SARS-CoV-2 lethal mutagenesis. Nat. Commun. 2020, 11, 4682. [Google Scholar] [CrossRef]
- Shannon, A.; Fattorini, V.; Sama, B.; Selisko, B.; Feracci, M.; Falcou, C.; Gauffre, P.; El Kazzi, P.; Delpal, A.; Decroly, E.; et al. A dual mechanism of action of AT-527 against SARS-CoV-2 polymerase. Nat. Commun. 2022, 13, 621. [Google Scholar] [CrossRef]
- Jockusch, S.; Tao, C.; Li, X.; Chien, M.; Kumar, S.; Morozova, I.; Kalachikov, S.; Russo, J.J.; Ju, J. Sofosbuvir terminated RNA is more resistant to SARS-CoV-2 proofreader than RNA terminated by Remdesivir. Sci. Rep. 2020, 10, 16577. [Google Scholar] [CrossRef] [PubMed]
- Mslati, H.; Gentile, F.; Perez, C.; Cherkasov, A. Comprehensive Consensus Analysis of SARS-CoV-2 Drug Repurposing Campaigns. J. Chem. Inf. Model. 2021, 61, 3771–3788. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H.S. Structure and function of SARS-CoV-2 polymerase. Curr. Opin. Virol. 2021, 48, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Malone, B.; Campbell, E.A.; Darst, S.A. CoV-er all the bases: Structural perspectives of SARS-CoV-2 RNA synthesis. Enzymes 2021, 49, 1–37. [Google Scholar] [CrossRef]
- Bera, S.C.; Seifert, M.; Kirchdoerfer, R.N.; van Nies, P.; Wubulikasimu, Y.; Quack, S.; Papini, F.S.; Arnold, J.J.; Canard, B.; Cameron, C.E.; et al. The nucleotide addition cycle of the SARS-CoV-2 polymerase. Cell Rep. 2021, 36, 109650. [Google Scholar] [CrossRef]
- Yan, L.; Ge, J.; Zheng, L.; Zhang, Y.; Gao, Y.; Wang, T.; Huang, Y.; Yang, Y.; Gao, S.; Li, M.; et al. Cryo-EM Structure of an Extended SARS-CoV-2 Replication and Transcription Complex Reveals an Intermediate State in Cap Synthesis. Cell 2021, 184, 184–193.e110. [Google Scholar] [CrossRef]
- Slanina, H.; Madhugiri, R.; Bylapudi, G.; Schultheiss, K.; Karl, N.; Gulyaeva, A.; Gorbalenya, A.E.; Linne, U.; Ziebuhr, J. Coronavirus replication-transcription complex: Vital and selective NMPylation of a conserved site in nsp9 by the NiRAN-RdRp subunit. Proc. Natl. Acad. Sci. USA 2021, 118, e2022310118. [Google Scholar] [CrossRef]
- Wang, B.; Svetlov, D.; Artsimovitch, I. NMPylation and de-NMPylation of SARS-CoV-2 nsp9 by the NiRAN domain. Nucleic. Acids Res. 2021, 49, 8822–8835. [Google Scholar] [CrossRef]
- Conti, B.J.; Leicht, A.S.; Kirchdoerfer, R.N.; Sussman, M.R. Mass spectrometric based detection of protein nucleotidylation in the RNA polymerase of SARS-CoV-2. Commun. Chem. 2021, 4, 41. [Google Scholar] [CrossRef]
- Park, G.J.; Osinski, A.; Hernandez, G.; Eitson, J.L.; Majumdar, A.; Tonelli, M.; Henzler-Wildman, K.; Pawłowski, K.; Chen, Z.; Li, Y.; et al. The mechanism of RNA capping by SARS-CoV-2. bioRxiv 2022. [Google Scholar] [CrossRef]
- Wang, B.; Svetlov, V.; Wolf, Y.I.; Koonin, E.V.; Nudler, E.; Artsimovitch, I. Allosteric activation of SARS-CoV-2 RdRp by remdesivir triphosphate and other phosphorylated nucleotides. mBio 2021, 12, e0142321. [Google Scholar] [CrossRef] [PubMed]
- Tee, W.V.; Tan, Z.W.; Lee, K.; Guarnera, E.; Berezovsky, I.N. Exploring the Allosteric Territory of Protein Function. J. Phys. Chem. B 2021, 125, 3763–3780. [Google Scholar] [CrossRef] [PubMed]
- Barakat, K.; Ahmed, M.; Tabana, Y.; Ha, M. A ‘deep dive’ into the SARS-Cov-2 polymerase assembly: Identifying novel allosteric sites and analyzing the hydrogen bond networks and correlated dynamics. J. Biomol. Struct. Dyn. 2021, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Parvez, M.S.A.; Karim, M.A.; Hasan, M.; Jaman, J.; Karim, Z.; Tahsin, T.; Hasan, M.N.; Hosen, M.J. Prediction of potential inhibitors for RNA-dependent RNA polymerase of SARS-CoV-2 using comprehensive drug repurposing and molecular docking approach. Int. J. Biol. Macromol. 2020, 163, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Boyaci, H.; Campbell, E.A. Diverse and unified mechanisms of transcription initiation in bacteria. Nat. Rev. Microbiol. 2020, 19, 95–109. [Google Scholar] [CrossRef]
- Koch, U.; Narjes, F. Recent progress in the development of inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. Curr. Top Med. Chem. 2007, 7, 1302–1329. [Google Scholar] [CrossRef]
- Gioia, D.; Bertazzo, M.; Recanatini, M.; Masetti, M.; Cavalli, A. Dynamic Docking: A Paradigm Shift in Computational Drug Discovery. Molecules 2017, 22, 2029. [Google Scholar] [CrossRef] [Green Version]
- Pitsillou, E.; Liang, J.; Yu Meng Huang, H.; Hung, A.; Karagiannis, T.C. In silico investigation to identify potential small molecule inhibitors of the RNA-dependent RNA polymerase (RdRp) nidovirus RdRp-associated nucleotidyltransferase domain. Chem. Phys. Lett. 2021, 779, 138889. [Google Scholar] [CrossRef]
- Garriga, D.; Ferrer-Orta, C.; Querol-Audi, J.; Oliva, B.; Verdaguer, N. Role of motif B loop in allosteric regulation of RNA-dependent RNA polymerization activity. J. Mol. Biol. 2013, 425, 2279–2287. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Yu, J.; Huang, Y.; He, Z.; Luo, J.; Wu, Y.; Zheng, Y.; Wu, J.; Zhu, X.; Wang, H.; et al. Antibiotic fidaxomicin is an RdRp inhibitor as a potential new therapeutic agent against Zika virus. BMC Med. 2020, 18, 204. [Google Scholar] [CrossRef]
- Min, J.S.; Kwon, S.; Jin, Y.H. SARS-CoV-2 RdRp Inhibitors Selected from a Cell-Based SARS-CoV-2 RdRp Activity Assay System. Biomedicines 2021, 9, 996. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, K.C.; Gulyaeva, A.; Zevenhoven-Dobbe, J.C.; Janssen, G.M.; Ruben, M.; Overkleeft, H.S.; van Veelen, P.A.; Samborskiy, D.V.; Kravchenko, A.A.; Leontovich, A.M.; et al. Discovery of an essential nucleotidylating activity associated with a newly delineated conserved domain in the RNA polymerase-containing protein of all nidoviruses. Nucleic Acids Res. 2015, 43, 8416–8434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Malone, B.; Llewellyn, E.; Grasso, M.; Shelton, P.M.M.; Olinares, P.D.B.; Maruthi, K.; Eng, E.T.; Vatandaslar, H.; Chait, B.T.; et al. Structural Basis for Helicase-Polymerase Coupling in the SARS-CoV-2 Replication-Transcription Complex. Cell 2020, 182, 1560–1573.e1513. [Google Scholar] [CrossRef] [PubMed]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, A.K.; Orth, K. Enzymes Involved in AMPylation and deAMPylation. Chem. Rev. 2018, 118, 1199–1215. [Google Scholar] [CrossRef] [PubMed]
- Littler, D.R.; Mohanty, B.; Lowery, S.A.; Colson, R.N.; Gully, B.S.; Perlman, S.; Scanlon, M.J.; Rossjohn, J. Binding of a pyrimidine RNA base-mimic to SARS-CoV-2 nonstructural protein 9. J. Biol. Chem. 2021, 297, 101018. [Google Scholar] [CrossRef]
- Shuman, S.; Lima, C.D. The polynucleotide ligase and RNA capping enzyme superfamily of covalent nucleotidyltransferases. Curr. Opin. Struct. Biol. 2004, 14, 757–764. [Google Scholar] [CrossRef]
- Littler, D.R.; Liu, M.; McAuley, J.L.; Lowery, S.A.; Illing, P.T.; Gully, B.S.; Purcell, A.W.; Chandrashekaran, I.R.; Perlman, S.; Purcell, D.F.J.; et al. A natural product compound inhibits coronaviral replication in vitro by binding to the conserved Nsp9 SARS-CoV-2 protein. J. Biol. Chem. 2021, 297, 101362. [Google Scholar] [CrossRef]
- Naydenova, K.; Muir, K.W.; Wu, L.F.; Zhang, Z.; Coscia, F.; Peet, M.J.; Castro-Hartmann, P.; Qian, P.; Sader, K.; Dent, K.; et al. Structure of the SARS-CoV-2 RNA-dependent RNA polymerase in the presence of favipiravir-RTP. Proc. Natl. Acad. Sci. USA 2021, 118, e2021946118. [Google Scholar] [CrossRef]
- Malone, B.F.; Perry, J.K.; Olinares, P.D.B.; Chen, J.; Appelby, T.K.; Feng, J.Y.; Bilello, J.P.; Ng, H.; Sotiris, J.; Ebrahim, M.; et al. Structural basis for substrate selection by the SARS-CoV-2 replicase. bioRxiv 2022. [Google Scholar] [CrossRef]
- Tchesnokov, E.P.; Gordon, C.J.; Woolner, E.; Kocincova, D.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Gotte, M. Template-dependent inhibition of coronavirus RNA-dependent RNA polymerase by remdesivir reveals a second mechanism of action. J. Biol. Chem. 2020, 295, 16156–16165. [Google Scholar] [CrossRef] [PubMed]
- Dangerfield, T.L.; Huang, N.Z.; Johnson, K.A. Remdesivir Is Effective in Combating COVID-19 because It Is a Better Substrate than ATP for the Viral RNA-Dependent RNA Polymerase. iScience 2020, 23, 101849. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Zhang, X.; Zheng, W.; Sun, J.; Hua, L.; Xu, L.; Chu, X.J.; Ding, S.; Xiong, W. Development of a Simple In Vitro Assay To Identify and Evaluate Nucleotide Analogs against SARS-CoV-2 RNA-Dependent RNA Polymerase. Antimicrob. Agents Chemother. 2020, 65, e01508-20. [Google Scholar] [CrossRef] [PubMed]
- Seifert, M.; Bera, S.C.; van Nies, P.; Kirchdoerfer, R.N.; Shannon, A.; Le, T.T.; Meng, X.; Xia, H.; Wood, J.M.; Harris, L.D.; et al. Inhibition of SARS-CoV-2 polymerase by nucleotide analogs from a single-molecule perspective. eLife 2021, 10, e70968. [Google Scholar] [CrossRef] [PubMed]
- Castro-Roa, D.; Garcia-Pino, A.; De Gieter, S.; van Nuland, N.A.J.; Loris, R.; Zenkin, N. The Fic protein Doc uses an inverted substrate to phosphorylate and inactivate EF-Tu. Nat. Chem. Biol. 2013, 9, 811–817. [Google Scholar] [CrossRef] [Green Version]
- Rothstein, D.M. Rifamycins, Alone and in Combination. Cold Spring Harb. Perspect Med. 2016, 6, a027011. [Google Scholar] [CrossRef]
- Dorst, A.; Jung, E.; Gademann, K. Recent Advances in Mode of Action and Biosynthesis Studies of the Clinically Used Antibiotic Fidaxomicin. Chimia 2020, 74, 270–273. [Google Scholar] [CrossRef]
- Moss, B.; Rosenblum, E.N.; Katz, E.; Grimley, P.M. Rifampicin: A specific inhibitor of vaccinia virus assembly. Nature 1969, 224, 1280–1284. [Google Scholar] [CrossRef]
- Wehrli, W.; Knusel, F.; Schmid, K.; Staehelin, M. Interaction of rifamycin with bacterial RNA polymerase. Proc. Natl. Acad. Sci. USA 1968, 61, 667–673. [Google Scholar] [CrossRef] [Green Version]
- Garriga, D.; Headey, S.; Accurso, C.; Gunzburg, M.; Scanlon, M.; Coulibaly, F. Structural basis for the inhibition of poxvirus assembly by the antibiotic rifampicin. Proc. Natl. Acad. Sci. USA 2018, 115, 8424–8429. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Luan, X.; Li, Z.; Zhou, Z.; Wang, Q.; Gao, M.; Wang, X.; Zhou, F.; Shi, J.; You, E.; et al. Structural basis for inhibition of the SARS-CoV-2 RNA polymerase by suramin. Nat. Struct. Mol. Biol. 2021, 28, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Elkarhat, Z.; Charoute, H.; Elkhattabi, L.; Barakat, A.; Rouba, H. Potential inhibitors of SARS-cov-2 RNA dependent RNA polymerase protein: Molecular docking, molecular dynamics simulations and MM-PBSA analyses. J. Biomol. Struct. Dyn. 2022, 40, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Dorst, A.; Berg, R.; Gertzen, C.G.W.; Schafle, D.; Zerbe, K.; Gwerder, M.; Schnell, S.D.; Sander, P.; Gohlke, H.; Gademann, K. Semisynthetic Analogs of the Antibiotic Fidaxomicin-Design, Synthesis, and Biological Evaluation. ACS Med. Chem. Lett. 2020, 11, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.K.; Xu, H.; Kirchhoff, P.D.; Cierpicki, T.; Turbiak, A.J.; Wan, B.; Zhang, N.; Peng, K.W.; Franzblau, S.G.; Garcia, G.A.; et al. Structure-based design of novel benzoxazinorifamycins with potent binding affinity to wild-type and rifampin-resistant mutant Mycobacterium tuberculosis RNA polymerases. J. Med. Chem. 2012, 55, 3814–3826. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Gill, S.K.; Kirchhoff, P.D.; Wan, B.; Franzblau, S.G.; Garcia, G.A.; Showalter, H.D. Synthesis and structure-activity relationships of novel substituted 8-amino, 8-thio, and 1,8-pyrazole congeners of antitubercular rifamycin S and rifampin. Bioorg. Med. Chem. Lett. 2011, 21, 6094–6099. [Google Scholar] [CrossRef]
- Malinen, A.M.; Nandymazumdar, M.; Turtola, M.; Malmi, H.; Grocholski, T.; Artsimovitch, I.; Belogurov, G.A. CBR antimicrobials alter coupling between the bridge helix and the beta subunit in RNA polymerase. Nat. Commun. 2014, 5, 3408. [Google Scholar] [CrossRef] [Green Version]
- Mullins, J.G.L. Drug repurposing in silico screening platforms. Biochem. Soc. Trans. 2022, 50, 747–758. [Google Scholar] [CrossRef]
- Geraghty, R.J.; Aliota, M.T.; Bonnac, L.F. Broad-Spectrum Antiviral Strategies and Nucleoside Analogues. Viruses 2021, 13, 667. [Google Scholar] [CrossRef]
- Steitz, T.A. A mechanism for all polymerases. Nature 1998, 391, 231–232. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 80, 1136–1138. [Google Scholar] [CrossRef]
- Waters, M.D.; Warren, S.; Hughes, C.; Lewis, P.; Zhang, F. Human genetic risk of treatment with antiviral nucleoside analog drugs that induce lethal mutagenesis: The special case of molnupiravir. Environ. Mol. Mutagen. 2022, 63, 37–63. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.J.; Sharma, S.D.; Feng, J.Y.; Ray, A.S.; Smidansky, E.D.; Kireeva, M.L.; Cho, A.; Perry, J.; Vela, J.E.; Park, Y.; et al. Sensitivity of mitochondrial transcription and resistance of RNA polymerase II dependent nuclear transcription to antiviral ribonucleosides. PLoS Pathog. 2012, 8, e1003030. [Google Scholar] [CrossRef] [PubMed]
- Kabinger, F.; Stiller, C.; Schmitzova, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Hobartner, C.; Cramer, P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Freese, E.; Bautz, E.; Freese, E.B. The chemical and mutagenic specificity of hydroxylamine. Proc. Natl. Acad. Sci. USA 1961, 47, 845–855. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Hill, C.S.; Sarkar, S.; Tse, L.V.; Woodburn, B.M.D.; Schinazi, R.F.; Sheahan, T.P.; Baric, R.S.; Heise, M.T.; Swanstrom, R. beta-d-N4-hydroxycytidine Inhibits SARS-CoV-2 Through Lethal Mutagenesis But Is Also Mutagenic To Mammalian Cells. J. Infect. Dis. 2021, 224, 415–419. [Google Scholar] [CrossRef]
- Tripathi, A.; Bankaitis, V.A. Molecular Docking: From Lock and Key to Combination Lock. J. Mol. Med. Clin. Appl. 2017, 2. [Google Scholar] [CrossRef]
- Zeng, X.; Song, X.; Ma, T.; Pan, X.; Zhou, Y.; Hou, Y.; Zhang, Z.; Li, K.; Karypis, G.; Cheng, F. Repurpose Open Data to Discover Therapeutics for COVID-19 Using Deep Learning. J. Proteome Res. 2020, 19, 4624–4636. [Google Scholar] [CrossRef]
- Group, R.C.; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Shytaj, I.L.; Fares, M.; Gallucci, L.; Lucic, B.; Tolba, M.M.; Zimmermann, L.; Adler, J.M.; Xing, N.; Bushe, J.; Gruber, A.D.; et al. The FDA-Approved Drug Cobicistat Synergizes with Remdesivir To Inhibit SARS-CoV-2 Replication In Vitro and Decreases Viral Titers and Disease Progression in Syrian Hamsters. mBio 2022, 13, e0370521. [Google Scholar] [CrossRef]
- Wiedemar, N.; Hauser, D.A.; Maser, P. 100 Years of Suramin. Antimicrob. Agents Chemother. 2020, 64, 3. [Google Scholar] [CrossRef]
- Salgado-Benvindo, C.; Thaler, M.; Tas, A.; Ogando, N.S.; Bredenbeek, P.J.; Ninaber, D.K.; Wang, Y.; Hiemstra, P.S.; Snijder, E.J.; van Hemert, M.J. Suramin Inhibits SARS-CoV-2 Infection in Cell Culture by Interfering with Early Steps of the Replication Cycle. Antimicrob. Agents Chemother. 2020, 64, e00900-20. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, E.; Pezzullo, M.; Tarantino, D.; Petazzi, R.; Germani, F.; Kramer, D.; Robel, I.; Rohayem, J.; Bolognesi, M.; Milani, M. Structure-based inhibition of Norovirus RNA-dependent RNA polymerases. J. Mol. Biol. 2012, 419, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Baddock, H.T.; Brolih, S.; Yosaatmadja, Y.; Ratnaweera, M.; Bielinski, M.; Swift, L.P.; Cruz-Migoni, A.; Fan, H.; Keown, J.R.; Walker, A.P.; et al. Characterization of the SARS-CoV-2 ExoN (nsp14ExoN-nsp10) complex: Implications for its role in viral genome stability and inhibitor identification. Nucleic Acids Res. 2022, 50, 1484–1500. [Google Scholar] [CrossRef]
- Artsimovitch, I.; Vassylyeva, M.N.; Svetlov, D.; Svetlov, V.; Perederina, A.; Igarashi, N.; Matsugaki, N.; Wakatsuki, S.; Tahirov, T.H.; Vassylyev, D.G. Allosteric modulation of the RNA polymerase catalytic reaction is an essential component of transcription control by rifamycins. Cell 2005, 122, 351–363. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Molodtsov, V.; Nawarathne, I.N.; Scharf, N.T.; Kirchhoff, P.D.; Showalter, H.D.; Garcia, G.A.; Murakami, K.S. X-ray crystal structures of the Escherichia coli RNA polymerase in complex with benzoxazinorifamycins. J. Med. Chem. 2013, 56, 4758–4763. [Google Scholar] [CrossRef] [Green Version]
- Boyaci, H.; Chen, J.; Lilic, M.; Palka, M.; Mooney, R.A.; Landick, R.; Darst, S.A.; Campbell, E.A. Fidaxomicin jams Mycobacterium tuberculosis RNA polymerase motions needed for initiation via RbpA contacts. eLife 2018, 7, e34823. [Google Scholar] [CrossRef]
- Adams, R.A.; Leon, G.; Miller, N.M.; Reyes, S.P.; Thantrong, C.H.; Thokkadam, A.M.; Lemma, A.S.; Sivaloganathan, D.M.; Wan, X.; Brynildsen, M.P. Rifamycin antibiotics and the mechanisms of their failure. J. Antibiot. 2021, 74, 786–798. [Google Scholar] [CrossRef]
- Guarnera, E.; Berezovsky, I.N. Allosteric drugs and mutations: Chances, challenges, and necessity. Curr. Opin. Struct. Biol. 2020, 62, 149–157. [Google Scholar] [CrossRef]
- Czarna, A.; Plewka, J.; Kresik, L.; Matsuda, A.; Karim, A.; Robinson, C.; O’Byrne, S.; Cunningham, F.; Georgiou, I.; Wilk, P.; et al. Refolding of lid subdomain of SARS-CoV-2 nsp14 upon nsp10 interaction releases exonuclease activity. Structure 2022. [Google Scholar] [CrossRef] [PubMed]
- Kalliokoski, T.; Kramer, C.; Vulpetti, A.; Gedeck, P. Comparability of mixed IC50 data—A statistical analysis. PLoS ONE 2013, 8, e61007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoint. Am. J. Hyg. 1938, 27, 493–497. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Svetlov, D.; Bartikofsky, D.; Wobus, C.E.; Artsimovitch, I. Going Retro, Going Viral: Experiences and Lessons in Drug Discovery from COVID-19. Molecules 2022, 27, 3815. https://doi.org/10.3390/molecules27123815
Wang B, Svetlov D, Bartikofsky D, Wobus CE, Artsimovitch I. Going Retro, Going Viral: Experiences and Lessons in Drug Discovery from COVID-19. Molecules. 2022; 27(12):3815. https://doi.org/10.3390/molecules27123815
Chicago/Turabian StyleWang, Bing, Dmitri Svetlov, Dylan Bartikofsky, Christiane E. Wobus, and Irina Artsimovitch. 2022. "Going Retro, Going Viral: Experiences and Lessons in Drug Discovery from COVID-19" Molecules 27, no. 12: 3815. https://doi.org/10.3390/molecules27123815
APA StyleWang, B., Svetlov, D., Bartikofsky, D., Wobus, C. E., & Artsimovitch, I. (2022). Going Retro, Going Viral: Experiences and Lessons in Drug Discovery from COVID-19. Molecules, 27(12), 3815. https://doi.org/10.3390/molecules27123815