
Design, Synthesis, and Biological Evaluation of N14-Amino Acid-Substituted Tetrandrine Derivatives as Potential Antitumor Agents against Human Colorectal Cancer

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
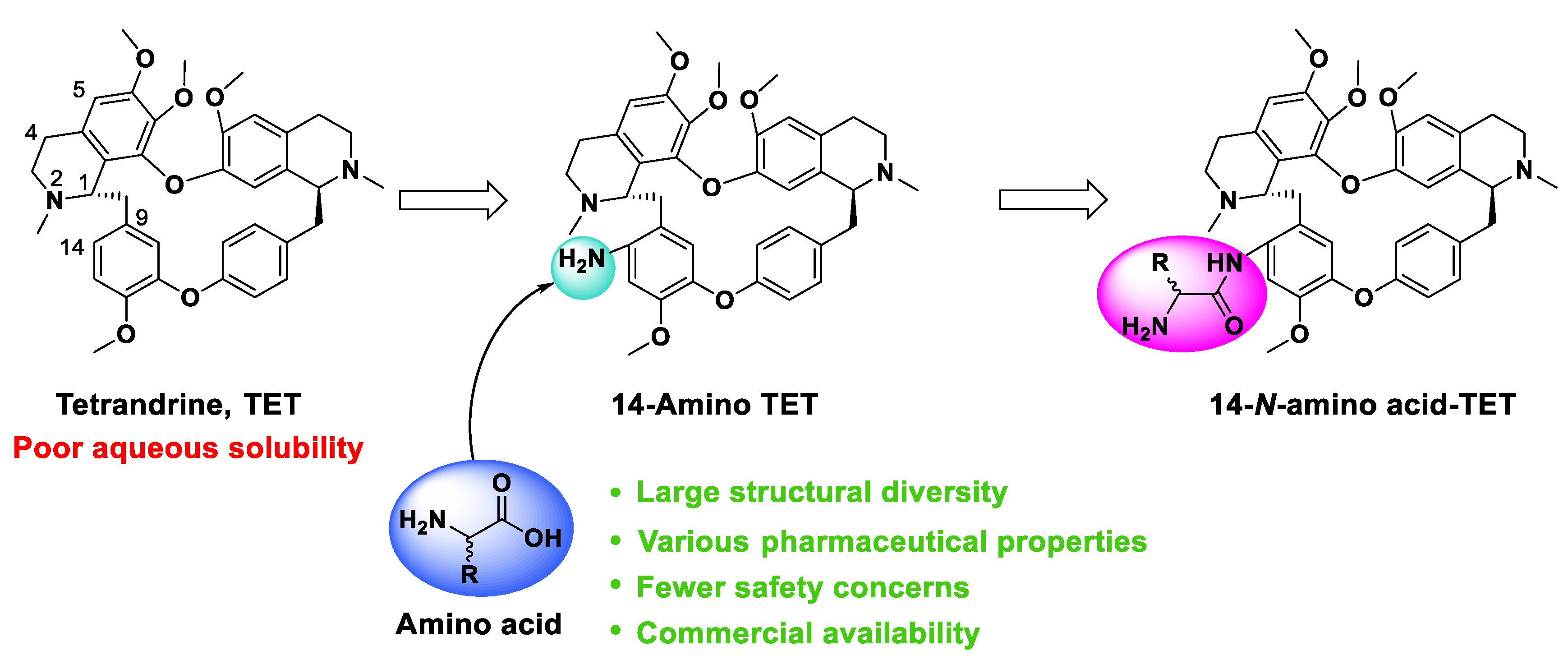
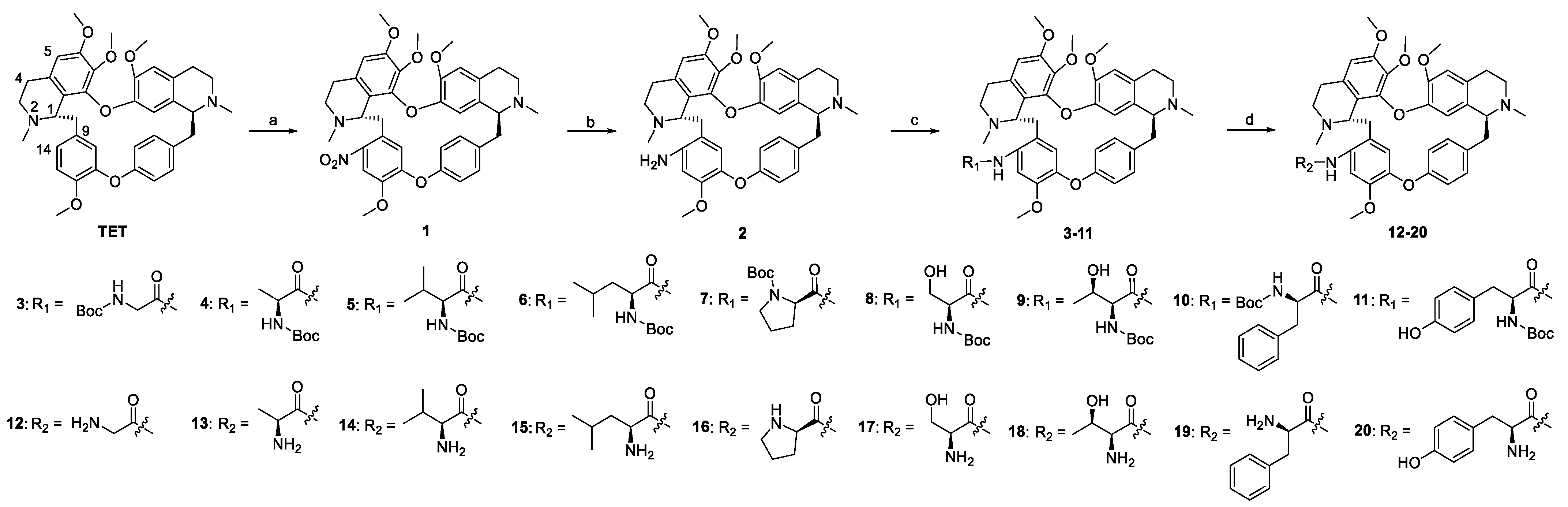
2.1. Chemistry
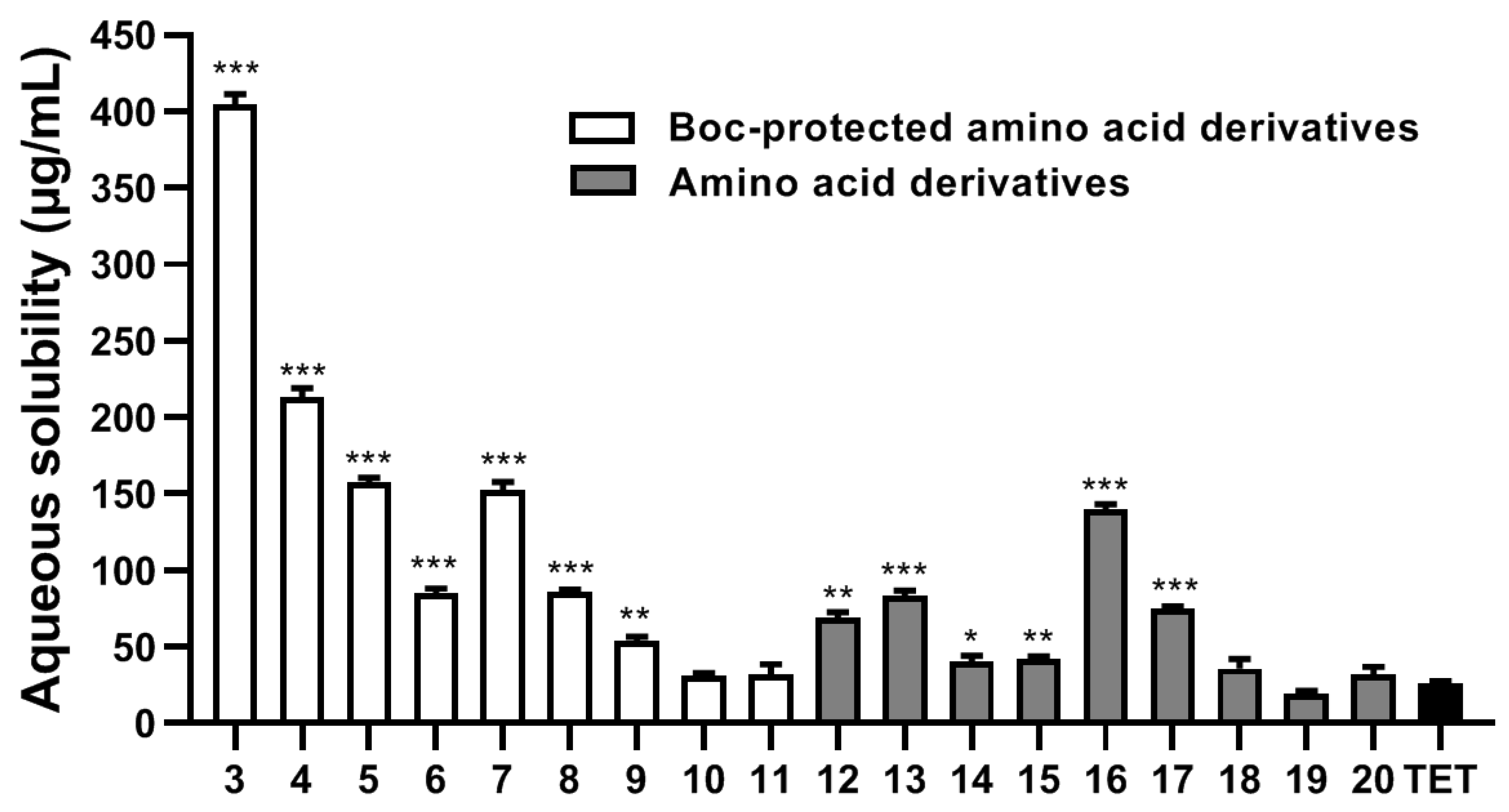
2.2. Aqueous Solubility Analysis
2.3. Antiproliferative Activity
2.3.1. Cytotoxicity Analysis
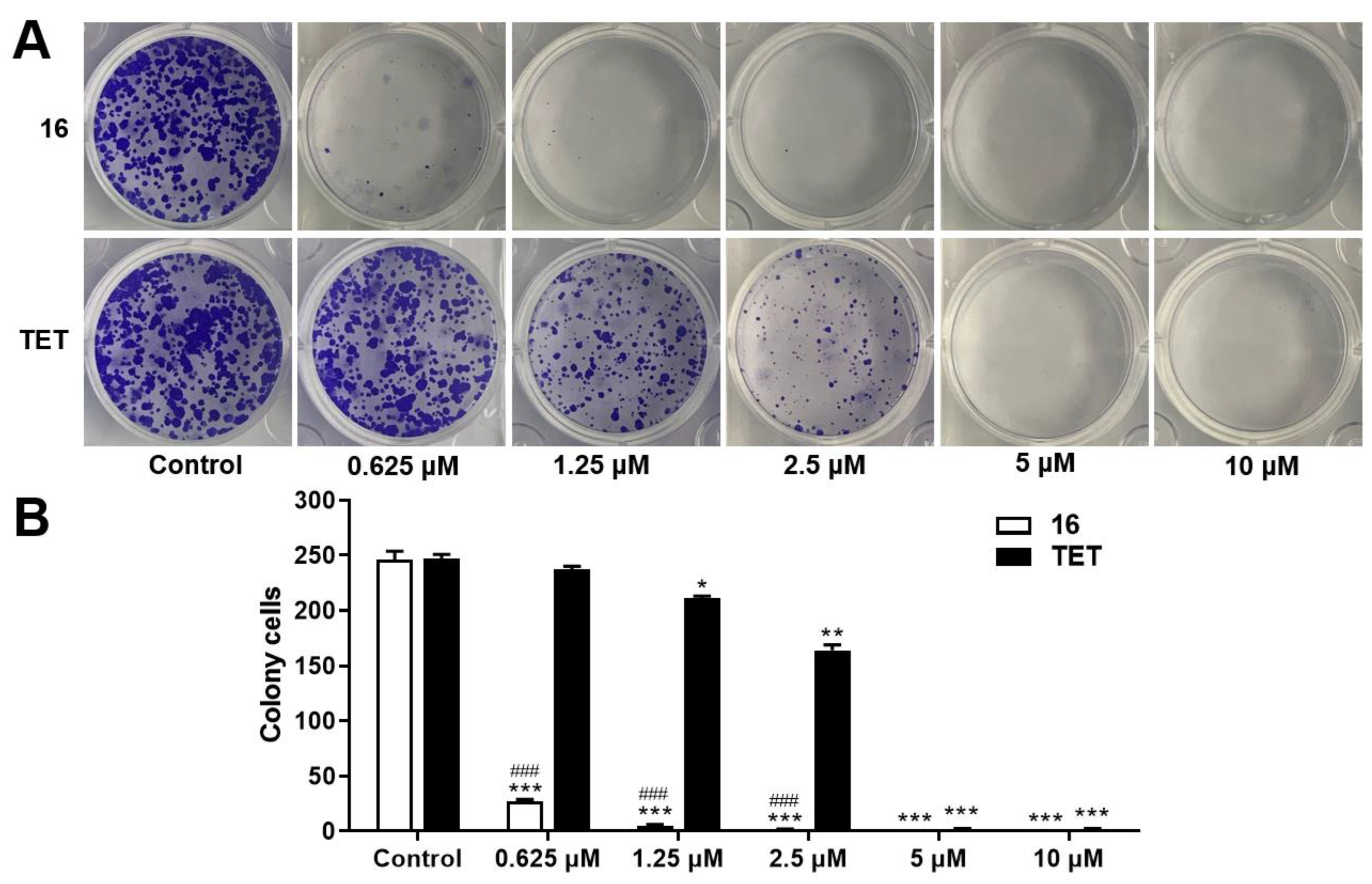
2.3.2. Colony Formation Assay
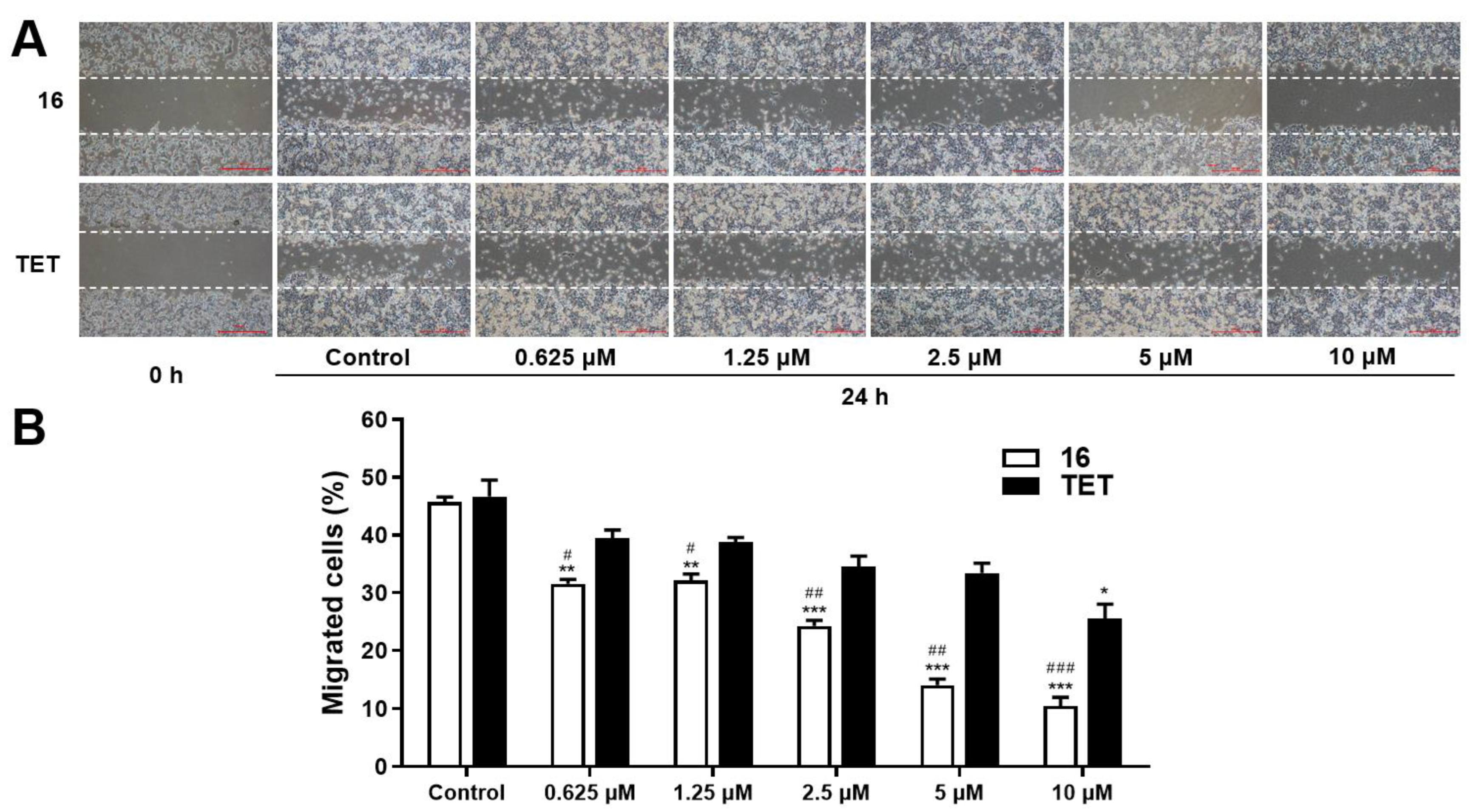
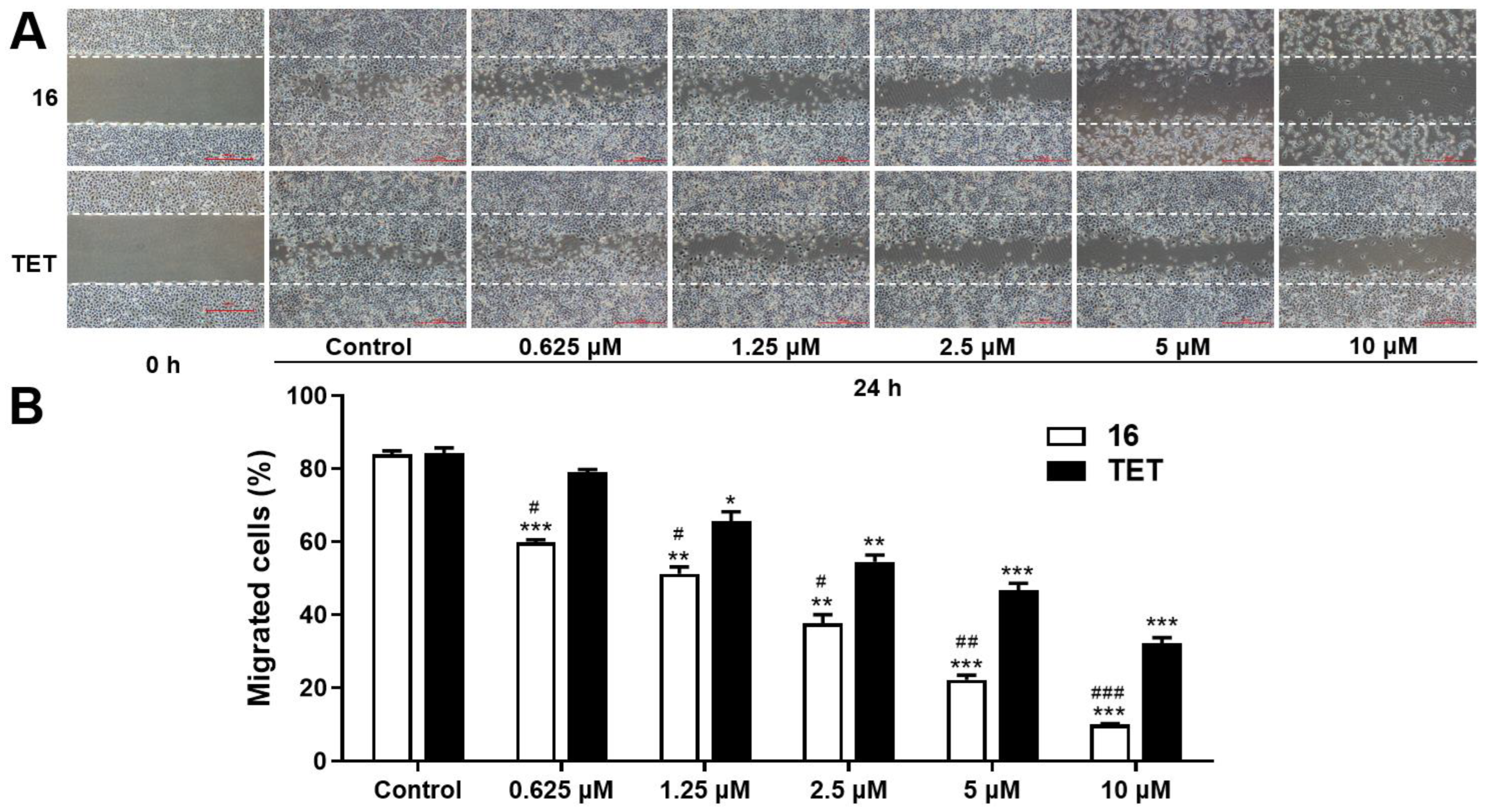
2.3.3. Migration Assay
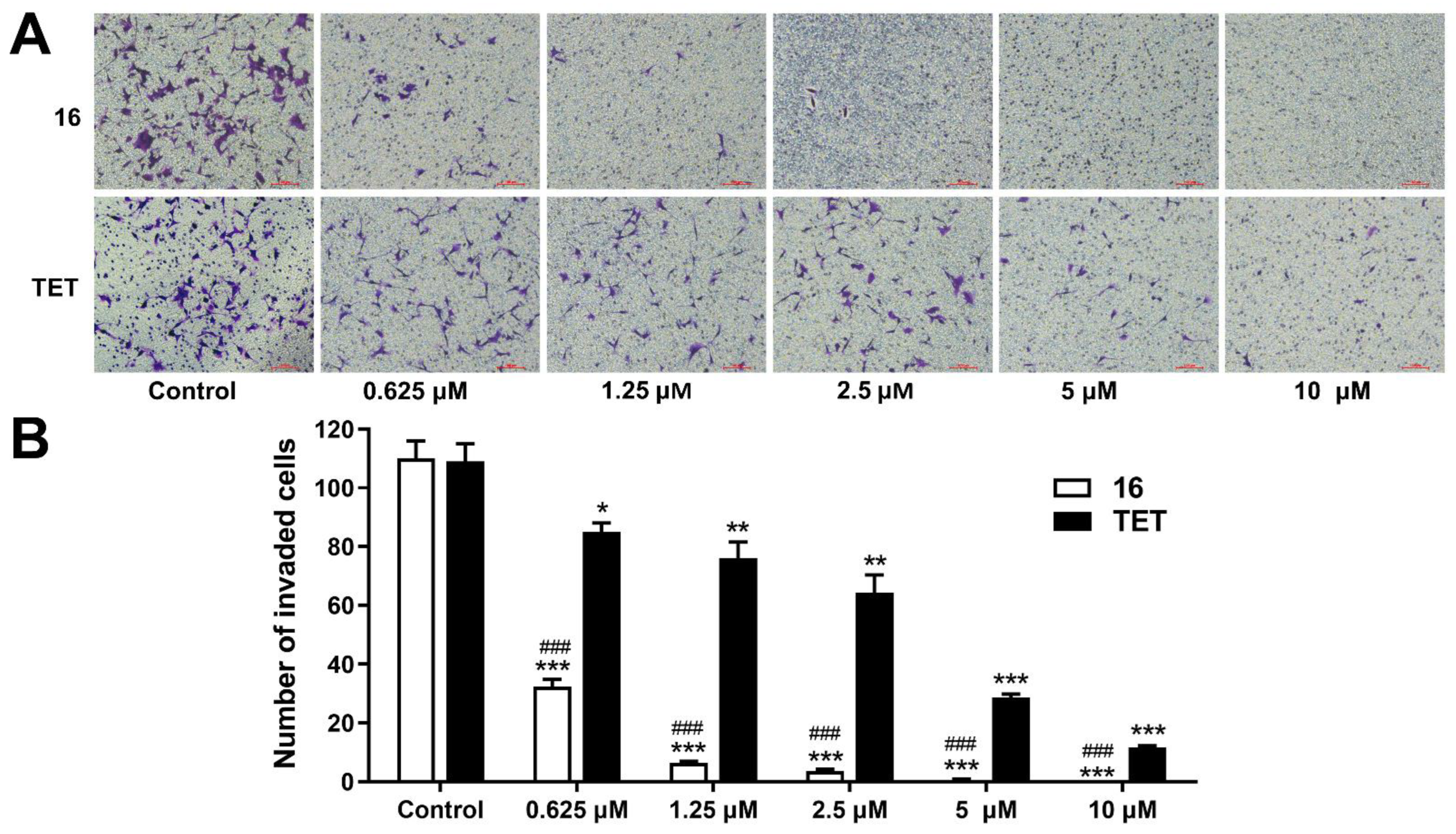
2.3.4. Invasion Assay
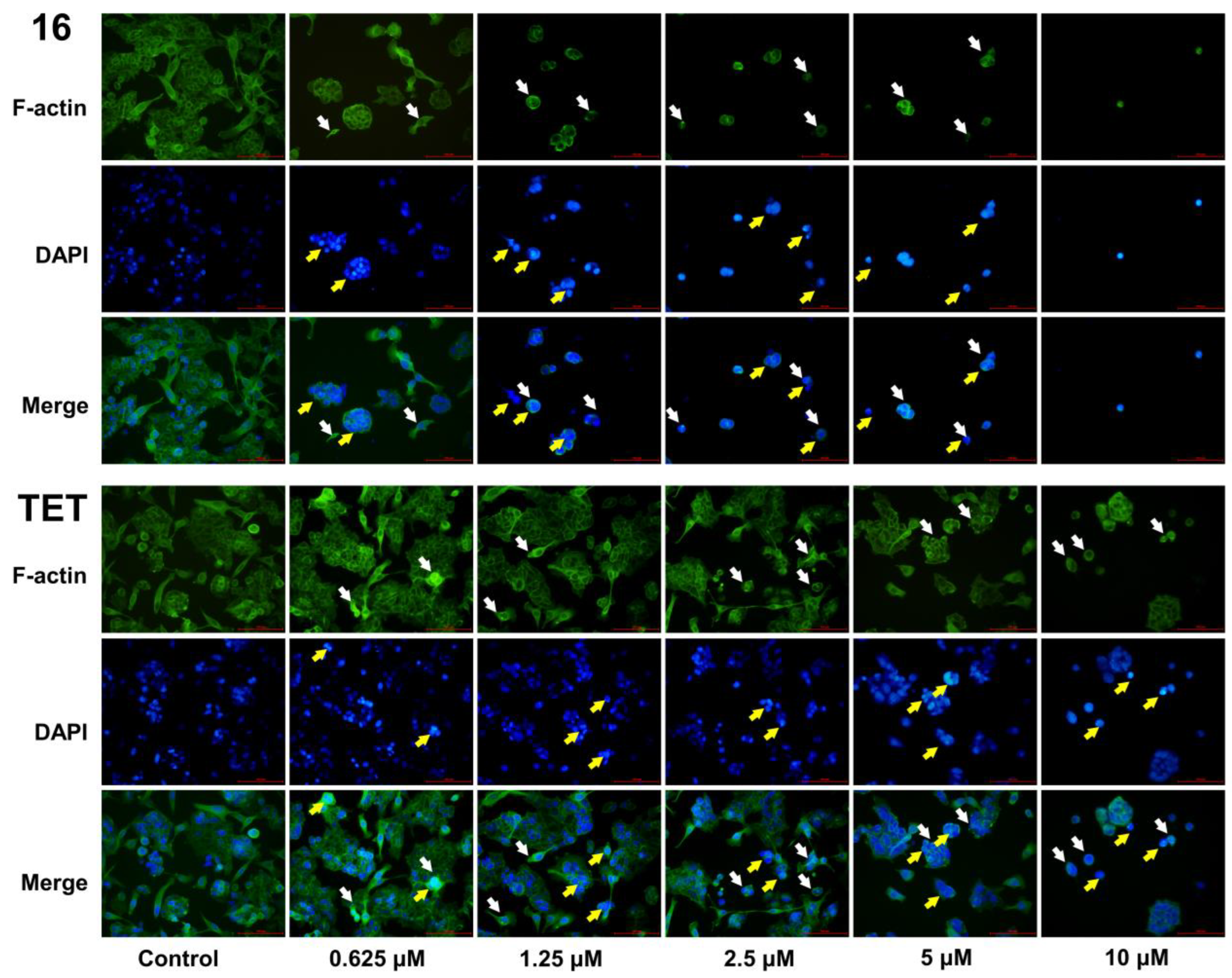
2.3.5. Morphological Analysis
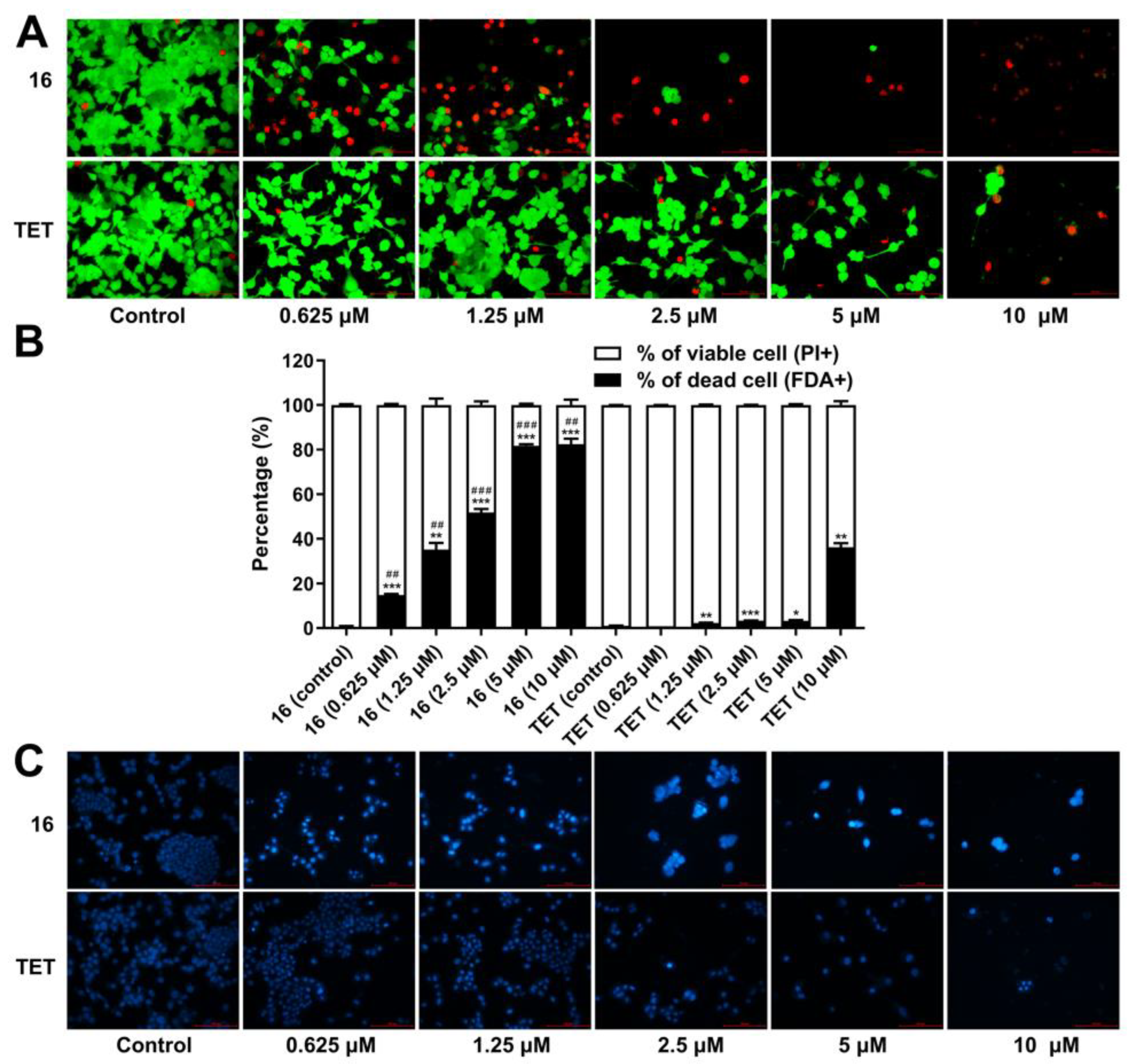
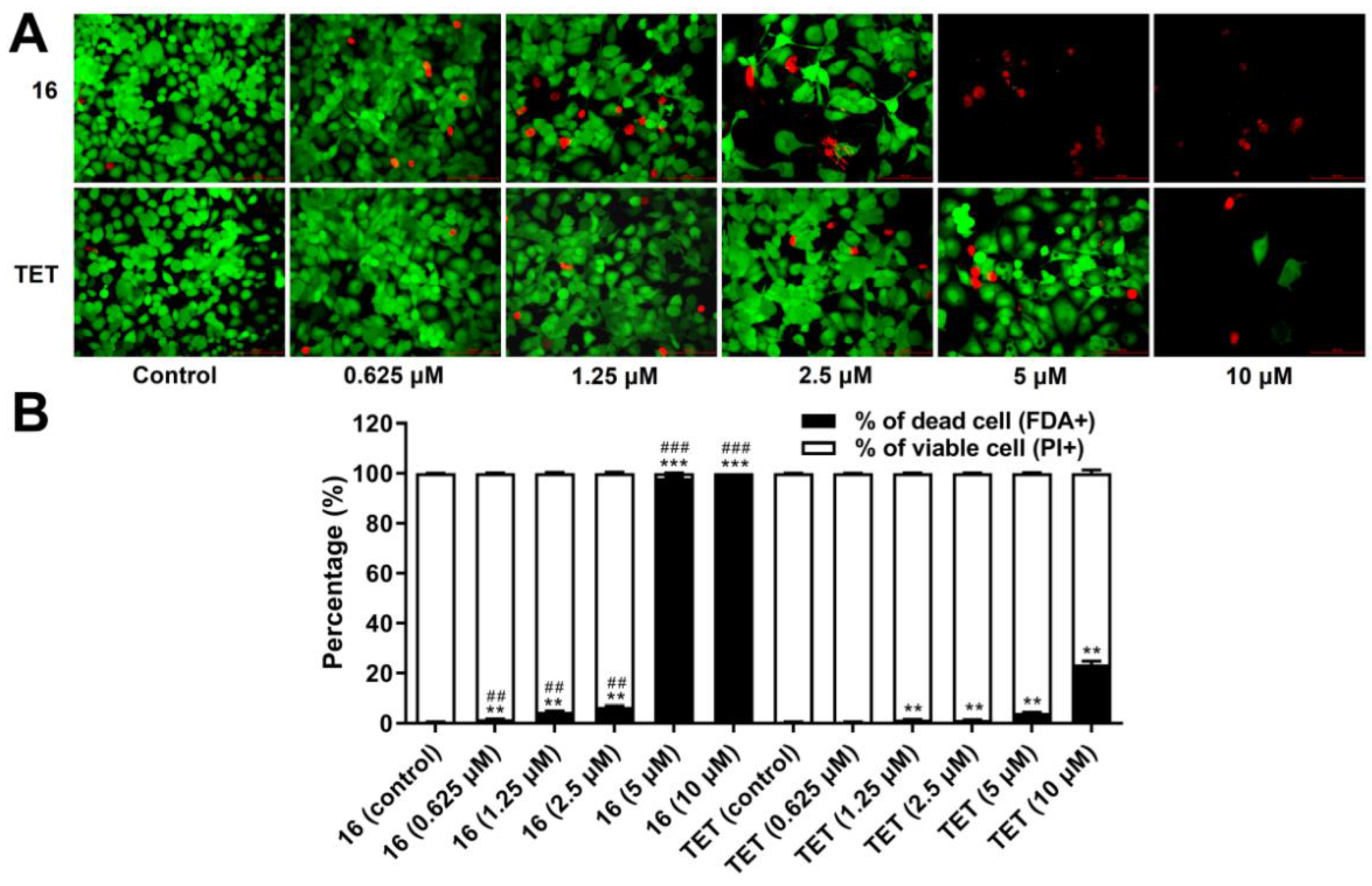
2.3.6. Cell Death Analysis
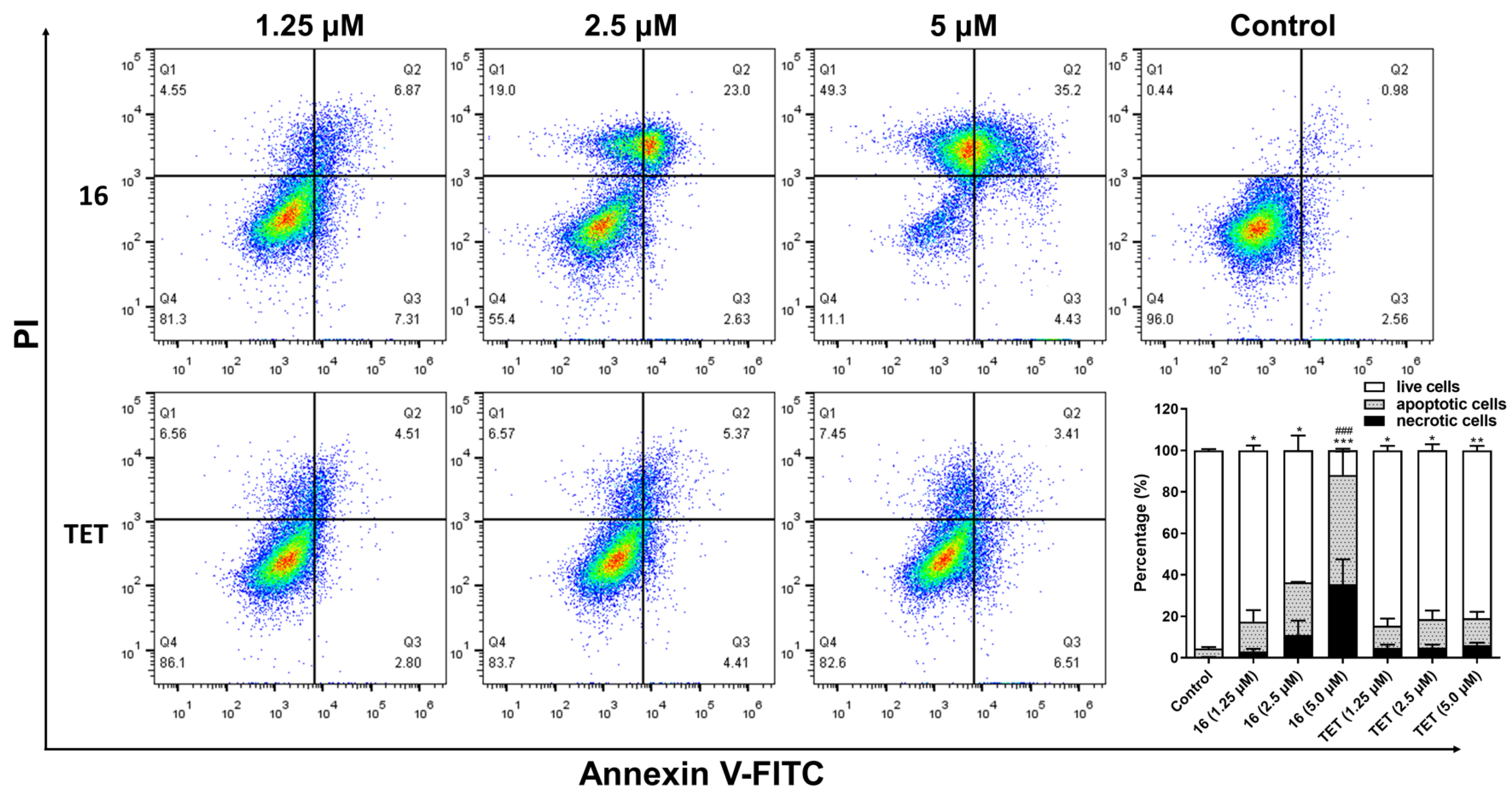
2.3.7. Flow Cytometry Assay
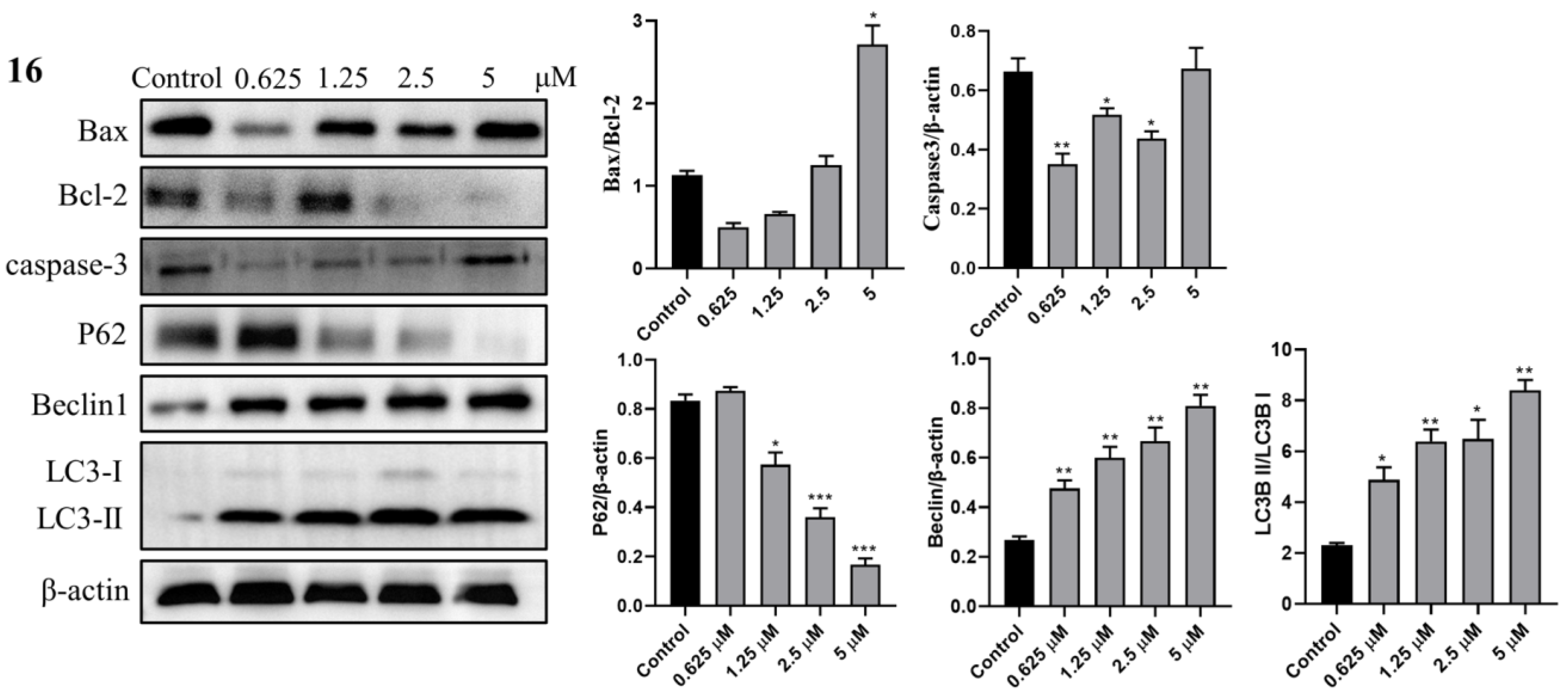
2.3.8. Western Blot Analysis
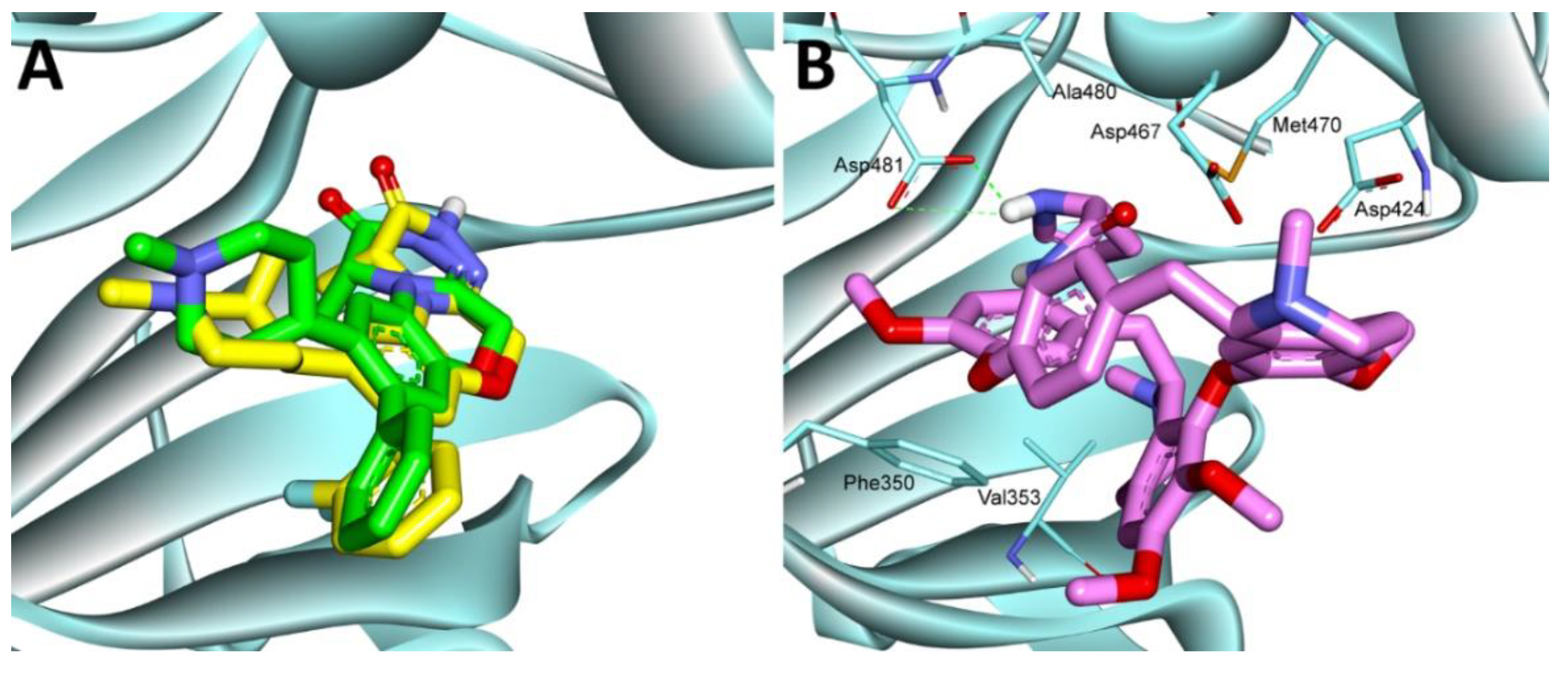
2.3.9. Molecular Docking Study
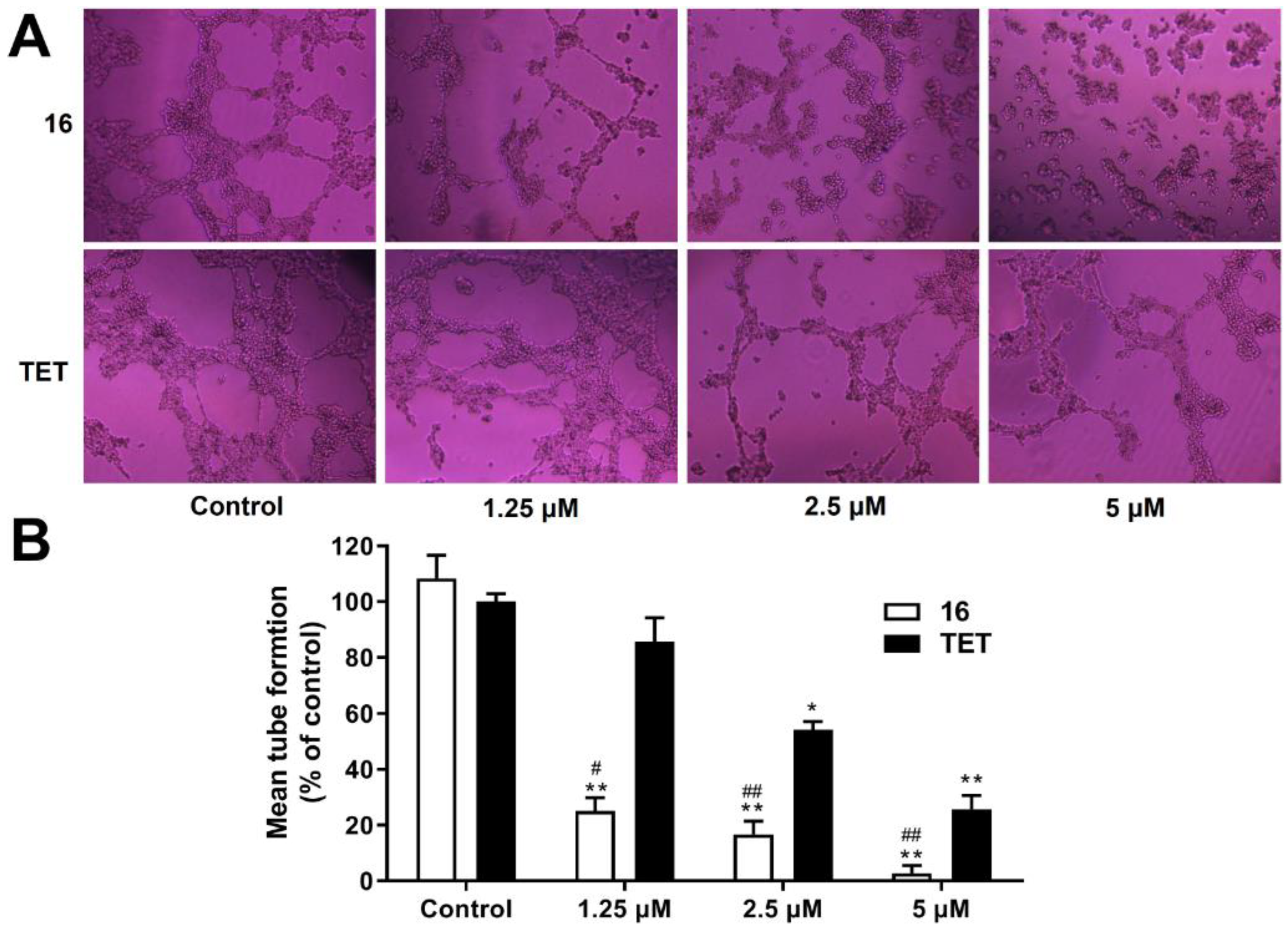
2.3.10. Anti-Angiogenesis Activity Analysis
3. Materials and Methods
3.1. Instruments and Materials
3.2. Methods of Synthesis
3.2.1. General Procedure for the Preparation of 14-Nitrotetrandrine (1)
3.2.2. General Procedure for the Preparation of 14-Aminotetrandrine (2)
3.2.3. General Procedure for the Synthesis of Compounds 3, 7, 8, and 11
3.2.4. General Procedure for the Synthesis of Compounds 12, 14–18, and 20
3.3. Cell Lines and Culture Conditions
3.4. MTT Assay
3.5. Aqueous Solubility Determination
3.6. Colony Formation Assay
3.7. Wound Healing Assays
3.8. Invasion Assay
3.9. F-Actin Phalloidin Staining
3.10. Live/Dead Cell Analysis
3.11. Hoechst 33258 Staining
3.12. Flow Cytometry Analysis
3.13. Cell Cycle Analysis
3.14. Western Blot
3.15. Molecular Docking
3.16. Tube Formation Assay
3.17. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sousa, E.M.F.; Wang, X.; Jansen, M.; Fessler, E.; Trinh, A.; de Rooij, L.P.; de Jong, J.H.; de Boer, O.J.; van Leersum, R.; Bijlsma, M.F.; et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat. Med. 2013, 19, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.P.; Wei, H.B.; Zheng, Z.H.; Guo, W.P.; Fang, J.F. Celecoxib increases retinoid sensitivity in human colon cancer cell lines. Cell. Mol. Biol. Lett. 2010, 15, 440–450. [Google Scholar] [CrossRef]
- Weitz, J.; Koch, M.; Debus, J.; Höhler, T.; Galle, P.R.; Büchler, M.W. Colorectal cancer. Lancet 2005, 365, 153–165. [Google Scholar] [CrossRef]
- Zhang, R.H.; Wang, S.; Zhang, H.; Lan, J.J.; Xu, G.B.; Zhao, Y.L.; Wang, L.; Li, Y.J.; Wang, Y.L.; Zhou, Y.H.; et al. Discovery of tetrandrine derivatives as tumor migration, invasion and angiogenesis inhibitors. Bioorg. Chem. 2020, 101, 104025. [Google Scholar] [CrossRef]
- Pretzsch, E.; Bösch, F.; Neumann, J.; Ganschow, P.; Bazhin, A.; Guba, M.; Werner, J.; Angele, M. Mechanisms of metastasis in colorectal cancer and metastatic organotropism: Hematogenous versus peritoneal spread. J. Oncol. 2019, 2019, 7407190. [Google Scholar] [CrossRef]
- Na, H.; Liu, X.; Li, X.; Zhang, X.; Wang, Y.; Wang, Z.; Yuan, M.; Zhang, Y.; Ren, S.; Zuo, Y. Novel roles of DC-SIGNR in colon cancer cell adhesion, migration, invasion, and liver metastasis. J. Hematol. Oncol. 2017, 10, 28. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.T.; Wu, W.; Ng, Y.Z.; Zhang, X.; Yan, B.; Ong, C.W.; Tan, S.; Salto-Tellez, M.; Hooi, S.C.; Chung, M.C.M. Proteomic analysis of colorectal cancer metastasis: Stathmin-1 revealed as a player in cancer cell migration and prognostic marker. J. Proteome Res. 2012, 11, 1433–1445. [Google Scholar] [CrossRef]
- Rmali, K.A.; Puntis, M.C.; Jiang, W.G. Tumour-associated angiogenesis in human colorectal cancer. Colorectal Dis. 2007, 9, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Paulo, A.; Moreira, R.; Lavrado, J. Indoloquinolines as scaffolds for drug discovery. Curr. Med. Chem. 2010, 17, 2348–2370. [Google Scholar]
- Veeresham, C. Natural products derived from plants as a source of drugs. J. Adv. Pharm. Technol. Res. 2012, 3, 200–201. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.; Jacoby, R. Dimebon in Alzheimer’s disease: Old drug for new indication. Lancet 2008, 372, 179–180. [Google Scholar] [CrossRef]
- Chinembiri, T.N.; du Plessis, L.H.; Gerber, M.; Hamman, J.H.; du Plessis, J. Review of natural compounds for potential skin cancer treatment. Molecules 2014, 19, 11679–11721. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.R.; Islam, F.; Nafady, M.H.; Akter, M.; Mitra, S.; Das, R.; Urmee, H.; Shohag, S.; Akter, A.; Chidambaram, K.; et al. Natural small molecules in breast cancer treatment: Understandings from a therapeutic viewpoint. Molecules 2022, 27, 2165. [Google Scholar] [CrossRef]
- Huang, H.; Yao, Y.; He, Z.; Yang, T.; Ma, J.; Tian, X.; Li, Y.; Huang, C.; Chen, X.; Li, W. Antimalarial β-carboline and indolactam alkaloids from marinactinospora thermotolerans, a deep sea isolate. J. Nat. Prod. 2011, 74, 2122–2127. [Google Scholar] [CrossRef]
- Bhagya, N.; Chandrashekar, K.R. Tetrandrine-A molecule of wide bioactivity. Phytochemistry 2016, 125, 5–13. [Google Scholar] [CrossRef]
- Liu, T.; Liu, X.; Li, W. Tetrandrine, a Chinese plant-derived alkaloid, is a potential candidate for cancer chemotherapy. Oncotarget 2016, 7, 40800–40815. [Google Scholar] [CrossRef] [Green Version]
- Kuo, P.-L.; Lin, C.-C. Tetrandrine-induced cell cycle arrest and apoptosis in Hep G2 cells. Life Sci. 2003, 73, 243–252. [Google Scholar] [CrossRef]
- Guo, Y.; Pei, X. Tetrandrine-induced autophagy in MDA-MB-231 triple-negative breast cancer cell through the inhibition of PI3K/AKT/mTOR Signaling. Evid.-Based Complement. Altern. Med. 2019, 2019, 7517431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Mu, L.; Liu, X.L.; Li, Q.; Ding, L.X.; Chen, H.C.; Hu, Y.; Li, F.S.; Sun, W.J.; He, B.C.; et al. Tetrandrine inhibits proliferation of colon cancer cells by BMP9/PTEN/PI3K/AKT signaling. Genes Dis. 2021, 8, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Shishodia, G.; Koul, S.; Dong, Q.; Koul, H.K. Tetrandrine (TET) Induces death receptors apo trail R1 (DR4) and apo trail R2 (DR5) and sensitizes prostate cancer cells to TRAIL-induced apoptosis. Mol. Cancer Ther. 2018, 17, 1217–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.-M.; Chen, Y.; Chen, J.-C.; Lin, T.-Y.; Tseng, S.-H. Tetrandrine induces apoptosis and growth suppression of colon cancer cells in mice. Cancer Lett. 2010, 287, 187–195. [Google Scholar] [CrossRef]
- He, B.C.; Gao, J.L.; Zhang, B.Q.; Luo, Q.; Shi, Q.; Kim, S.H.; Huang, E.; Gao, Y.; Yang, K.; Wagner, E.R.; et al. Tetrandrine inhibits Wnt/β-catenin signaling and suppresses tumor growth of human colorectal cancer. Mol. Pharmacol. 2011, 79, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-S.; Kim, D.H.; Lee, I.-S.; Park, J.-H.; Martin, G.; Safe, S.; Kim, K.-J.; Kim, J.-H.; Jang, B.I.; Lee, S.-O. Plant alkaloid tetrandrine is a nuclear receptor 4A1 antagonist and inhibits Panc-1 cell growth in vitro and in vivo. Int. J. Mol. Sci. 2022, 23, 5280. [Google Scholar] [CrossRef]
- Shang, W.; Zhang, J.; Song, H.; Zhu, S.; Zhang, A.; Hua, Y.; Han, S.; Fu, Y. Mechanism of tetrandrine against endometrial cancer based on network pharmacology. Drug Des. Dev. Ther. 2021, 15, 2907–2919. [Google Scholar] [CrossRef]
- Wong, V.K.W.; Zeng, W.; Chen, J.; Yao, X.J.; Leung, E.L.H.; Wang, Q.Q.; Chiu, P.; Ko, B.C.B.; Law, B.Y.K. Tetrandrine, an activator of autophagy, induces autophagic cell death via PKC-α inhibition and mTOR-dependent mechanisms. Front. Pharmacol. 2017, 8, 351. [Google Scholar] [CrossRef] [Green Version]
- Liao, D.; Zhang, W.; Gupta, P.; Lei, Z.N.; Wang, J.Q.; Cai, C.Y.; Vera, A.A.; Zhang, L.; Chen, Z.S.; Yang, D.H. Tetrandrine interaction with ABCB1 Reverses multidrug resistance in cancer cells through competition with anti-cancer drugs followed by downregulation of ABCB1 expression. Molecules 2019, 24, 4383. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Yin, H.; Xu, H. Enhanced pro-apoptotic effect of tetrandrine loaded nanoparticles against osteosarcoma cells. Curr. Drug Deliv. 2016, 13, 946–952. [Google Scholar] [CrossRef]
- Yang, Q.H.; Jiang, C.S.; Jin, T.; Xu, J.F.; Qu, T.L.; Guo, Y.W.; Zhao, Z.B. Synthesis of novel tetrandrine derivatives and their inhibition against NSCLC A549 cells. Asian Nat. Prod. Res. 2018, 20, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lan, J.; Chen, C.; Hu, S.; Song, J.; Liu, W.; Zeng, X.; Lou, H.; Ben-David, Y.; Pan, W. Design, synthesis and bioactivity investigation of tetrandrine derivatives as potential anti-cancer agents. MedChemComm 2018, 9, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Wang, N.; Huang, L.; Liu, Y.; Ma, X.; Lou, H.; Chen, C.; Feng, Y.; Pan, W. Design and synthesis of novel tetrandrine derivatives as potential anti-tumor agents against human hepatocellular carcinoma. Eur. J. Med. Chem. 2017, 127, 554–566. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huang, L.; Sun, Q.; Zhang, M.; Li, T.; Liang, G.; Pan, W. Syntheses and anti-cancer activities of derivatives of tetrandrine and fangchinoline. Chem. Res. Chin. Univ. 2014, 30, 937–940. [Google Scholar] [CrossRef]
- Li, D.; Liu, H.; Liu, Y.; Zhang, Q.; Liu, C.; Zhao, S.; Jiao, B. Design, synthesis and biological activities of tetrandrine and fangchinoline derivatives as antitumer agents. Bioorg. Med. Chem. Lett. 2017, 27, 533–536. [Google Scholar] [CrossRef]
- Li, X.; Yu, N.; Li, J.; Bai, J.; Ding, D.; Tang, Q.; Xu, H. Novel “Carrier-Free” nanofiber codelivery systems with the synergistic antitumor effect of paclitaxel and tetrandrine through the enhancement of mitochondrial apoptosis. ACS Appl. Mater. Interfaces 2020, 12, 10096–10106. [Google Scholar] [CrossRef]
- Liu, R.; Wang, S.; Fang, S.; Wang, J.; Chen, J.; Huang, X.; He, X.; Liu, C. Liquid crystalline nanoparticles as an ophthalmic delivery system for tetrandrine: Development, characterization, and in vitro and in vivo evaluation. Nanoscale Res. Lett. 2016, 11, 254. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Ji, Z.; Zou, M.; Nie, X.; Shi, Y.; Cheng, G. Preparation, Characterization, pharmacokinetics and tissue distribution of solid lipid nanoparticles loaded with tetrandrine. AAPS PharmSciTech. 2011, 12, 1011. [Google Scholar] [CrossRef]
- Blaskovich, M.A. Unusual amino acids in medicinal chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef]
- Vig, B.S.; Huttunen, K.M.; Laine, K.; Rautio, J. Amino acids as promoieties in prodrug design and development. Adv. Drug Deliv. Rev. 2013, 65, 1370–1385. [Google Scholar] [CrossRef]
- Wu, Y.; Cao, D.; Wang, F.; Ma, L.; Gao, G.; Chen, L. Synthesis and evaluation of millepachine amino acid prodrugs with enhanced solubility as antitumor agents. Chem. Biol. Drug Des. 2015, 86, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, M.; Tanaka, K.; Tsuchida, Y.; Muto, Y.; Koketsu, M.; Watanabe, K. Increased bioavailability of tricin-amino acid derivatives via a prodrug approach. J. Med. Chem. 2011, 54, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Ran, L.; Li, X.; Liu, M.; Wang, Q. Synthesis and antiproliferative activities of polymethoxyflavones aminoalkyl and amino acid derivatives. Heterocycl. Commun. 2020, 26, 68–75. [Google Scholar] [CrossRef]
- Idzik, K.R.; Nodler, K.; Licha, T. Efficient synthesis of readily water-soluble sulfonic Acid carbamates. Molecules 2015, 20, 6856–6865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, A.K.; Gupta, M. Solubility and solution stability studies of different amino acid prodrugs of bromhexine. Drug Dev. Ind. Pharm. 2012, 38, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Balimane, P.V.; Tamai, I.; Guo, A.; Nakanishi, T.; Kitada, H.; Leibach, F.H.; Tsuji, A.; Sinko, P.J. Direct evidence for peptide transporter (PepT1)-mediated uptake of a nonpeptide prodrug, valacyclovir. Biochem. Biophys. Res. Commun. 1998, 250, 246–251. [Google Scholar] [CrossRef]
- Sugawara, M.; Huang, W.; Fei, Y.J.; Leibach, F.H.; Ganapathy, V.; Ganapathy, M.E. Transport of valganciclovir, a ganciclovir prodrug, via peptide transporters PEPT1 and PEPT2. J. Pharm. Sci. 2000, 89, 781–789. [Google Scholar] [CrossRef]
- Vale, N.; Ferreira, A.; Matos, J.; Fresco, P.; Gouveia, M.J. Amino acids in the development of prodrugs. Molecules 2018, 23, 2318. [Google Scholar] [CrossRef] [Green Version]
- Vig, B.; Rautio, J. Amino acid prodrugs for oral delivery: Challenges and opportunities. Ther. Deliv. 2011, 2, 959–962. [Google Scholar] [CrossRef] [Green Version]
- Bowden, N.A.; Sanders, J.P.M.; Bruins, M.E. Solubility of the proteinogenic α-amino acids in water, ethanol, and ethanol-water mixtures. J. Chem. Eng. Data. 2018, 63, 488–497. [Google Scholar] [CrossRef]
- Ali, S.M.; Hoemann, M.Z.; Aubé, J.; Mitscher, L.A.; Georg, G.I.; McCall, R.; Jayasinghe, L.R. Novel cytotoxic 3’-(tert-butyl) 3’-dephenyl analogs of paclitaxel and docetaxel. J. Med. Chem. 1995, 38, 3821–3828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, Q.; Zhang, H.; Jiang, X.H. Determination of apparent solubility, apparent partition coefficient and dissociation constant of tetrandrine. West China J. Pharmaceut. Sci. 2013, 28, 501–503. [Google Scholar]
- Mouradov, D.; Sloggett, C.; Jorissen, R.N.; Love, C.G.; Li, S.; Burgess, A.W.; Arango, D.; Strausberg, R.L.; Buchanan, D.; Wormald, S.; et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014, 74, 3238–3247. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, B. Arsenic trioxide (As(2)O(3)) inhibits peritoneal invasion of ovarian carcinoma cells in vitro and in vivo. Gynecol. Oncol. 2006, 103, 199–206. [Google Scholar]
- Choi, Y.J.; Hur, J.M.; Lim, S.; Jo, M.; Kim, D.H.; Choi, J.I. Induction of apoptosis by deinoxanthin in human cancer cells. Anticancer Res. 2014, 34, 1829–1835. [Google Scholar]
- Liu, Y.; Liu, C.; Tan, T.; Li, S.; Tang, S.; Chen, X. Sinomenine sensitizes human gastric cancer cells to cisplatin through negative regulation of PI3K/AKT/Wnt signaling pathway. Anticancer Drugs 2019, 30, 983–990. [Google Scholar] [CrossRef]
- Sato, A.; Hiramoto, A.; Kim, H.-S.; Wataya, Y. Anticancer strategy targeting cell death regulators: Switching the mechanism of anticancer floxuridine-induced cell death from necrosis to apoptosis. Int. J. Mol. Sci. 2020, 21, 5876. [Google Scholar] [CrossRef]
- Wu, Y.; Dong, G.; Sheng, C. Targeting necroptosis in anticancer therapy: Mechanisms and modulators. Acta Pharm. Sin. B 2020, 10, 1601–1618. [Google Scholar] [CrossRef]
- Wang, N.; Wang, X.; Tan, H.-Y.; Li, S.; Tsang, C.M.; Tsao, S.-W.; Feng, Y. Berberine suppresses cyclin D1 expression through proteasomal degradation in human hepatoma cells. Int. J. Mol. Sci. 2016, 17, 1899. [Google Scholar] [CrossRef] [Green Version]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, X.; Zhang, D.; Chen, J.; Liu, S.; Berk, M. The effects of apoptosis vulnerability markers on the myocardium in depression after myocardial infarction. BMC Med. 2013, 11, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.; Chen, C.; Niu, Z.; Gan, L.; Wang, Q.; Li, M.; Cai, X.; Gao, R.; Katakam, S.; Chen, H.; et al. Poly(I:C) preconditioning protects the heart against myocardial ischemia/reperfusion injury through TLR3/PI3K/Akt-dependent pathway. Signal Transduct. Target. Thery. 2020, 5, 216. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Men, Q.; Wu, G.; Yu, C.; Huang, Z.; Liu, X.; Li, W. Tetrandrine induces autophagy and differentiation by activating ROS and Notch1 signaling in leukemia cells. Oncotarget 2015, 6, 7992–8006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.-Y.; Liu, Y.-G.; Song, W.; Li, Y.-Y.; Hou, D.-S.; Luo, H.-M.; Liu, P. Anticancer activity of tetrandrine by inducing pro-death apoptosis and autophagy in human gastric cancer cells. J. Pharm. Pharmacol. 2018, 70, 1048–1058. [Google Scholar] [CrossRef]
- George, D.M.; Breinlinger, E.C.; Argiriadi, M.A.; Zhang, Y.; Wang, J.; Bansal-Pakala, P.; Duignan, D.B.; Honore, P.; Lang, Q.; Mittelstadt, S.; et al. Optimized protein kinase c θ (PKCθ) inhibitors reveal only modest anti-inflammatory efficacy in a rodent model of arthritis. J. Med. Chem. 2015, 58, 333–346. [Google Scholar] [CrossRef]
- Park, H.J.; Zhang, Y.; Georgescu, S.P.; Johnson, K.L.; Kong, D.; Galper, J.B. Human umbilical vein endothelial cells and human dermal microvascular endothelial cells offer new insights into the relationship between lipid metabolism and angiogenesis. Stem Cell Rev. 2006, 2, 93–102. [Google Scholar] [CrossRef]
- Fan, H.; Wei, D.; Zheng, K.; Qin, X.; Yang, L.; Yang, Y.; Duan, Y.; Xu, Y.; Hu, L. Discovery of Dioxino[2,3-f]quinazoline derivative VEGFR-2 inhibitors exerting significant antipro-liferative activity in HUVECs and mice. Eur. J. Med. Chem. 2019, 175, 349–356. [Google Scholar] [CrossRef]
- Soeda, S.; Kozako, T.; Iwata, K.; Shimeno, H. Oversulfated fucoidan inhibits the basic fibroblast growth factor-induced tube formation by human umbilical vein endothelial cells: Its possible mechanism of action. Biochim. Biophys. Acta. 2000, 1497, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Huang, L.; Lou, H.; Chen, C.; Liu, T.; Hu, S.; Yao, Y.; Song, J.; Luo, J.; Liu, Y.; et al. Design and synthesis of novel C14-urea-tetrandrine derivatives with potent anti-cancer activity. Eur. J. Med. Chem. 2018, 143, 1968–1980. [Google Scholar] [CrossRef]
- Hu, S.C.; Yang, J.; Chen, C.; Song, J.R.; Pan, W.D. Design, Synthesis of novel tetrandrine-14-l-amino acid and tetrandrine-14-l-amino acid-urea derivatives as potential anti-cancer agents. Molecules 2020, 25, 1738. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, M.; Zhong, R.-F.; Liao, X.-M.; Deng, L.-L.; Xu, G.-B.; He, X.; Li, J.; Li, Y.-J.; Liu, T.; et al. Discovery and structure-activity relationship of auriculatone: A potent hepatoprotective agent against acetaminophen-induced liver injury. Bioorg. Med. Chem. Lett. 2017, 27, 3636–3642. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | Antiproliferative Activity (IC50/μM) a | ||||||
|---|---|---|---|---|---|---|---|
| A549 | HCT-15 | HepG2 | BXPC-3 | MCF-7 | HUVEC | L-02 | |
| 3 | 2.15 ± 0.17 | 2.42 ± 0.06 | 1.94 ± 0.27 | 2.55 ± 0.03 | 2.91 ± 0.17 | 1.68 ± 0.21 | 8.59 ± 0.13 |
| 4 | 7.44 ± 1.11 | 3.28 ± 0.32 | 3.90 ± 0.62 | 4.74 ± 0.37 | 5.78 ± 0.35 | 4.01 ± 0.41 | >20 |
| 5 | 7.24 ± 0.41 | 4.31 ± 0.09 | 3.54 ± 0.30 | 4.63 ± 0.09 | 7.40 ± 0.31 | 3.04 ± 0.34 | >20 |
| 6 | 5.54 ± 0.16 | 3.89 ± 0.47 | 3.11 ± 0.68 | 6.39 ± 0.20 | 4.99 ± 0.66 | 2.09 ± 0.19 | 13.58 ± 0.13 |
| 7 | 6.40 ± 0.36 | 1.15 ± 0.17 | 3.50 ± 1.37 | 3.10 ± 0.26 | 5.71 ± 0.29 | 3.49 ± 0.31 | 17.54 ± 0.21 |
| 8 | 7.45 ± 0.32 | 5.87 ± 0.30 | 3.76 ± 1.34 | 3.55 ± 0.32 | 5.84 ± 0.25 | 3.96 ± 0.36 | >20 |
| 9 | 12.28 ± 0.61 | 8.57 ± 0.57 | 6.97 ± 0.34 | 13.07 ± 0.37 | 11.29 ± 0.55 | 4.91 ± 1.05 | >20 |
| 10 | 1.96 ± 0.24 | 0.91 ± 0.05 | 1.54 ± 0.02 | 2.31 ± 0.08 | 2.59 ± 0.11 | 1.80 ± 0.02 | 15.54 ± 0.11 |
| 11 | 6.26 ± 0.41 | 3.56 ± 0.27 | 2.81 ± 0.35 | 3.58 ± 0.15 | 5.03 ± 0.04 | 2.16 ± 0.32 | 11.79 ± 0.61 |
| 12 | 4.94 ± 0.45 | 3.54 ± 0.36 | 3.19 ± 0.76 | 3.98 ± 0.16 | 6.91 ± 0.66 | 3.19 ± 0.61 | >20 |
| 13 | 8.12 ± 0.43 | 4.69 ± 0.47 | 3.27 ± 1.00 | 6.95 ± 0.57 | 11.06 ± 0.26 | 2.20 ± 0.37 | >20 |
| 14 | 6.11 ± 0.91 | 4.02 ± 0.15 | 3.26 ± 0.05 | 4.95 ± 0.29 | 7.96 ± 1.08 | 2.48 ± 0.15 | 14.49 ± 0.37 |
| 15 | 5.92 ± 0.73 | 2.71 ± 0.22 | 3.66 ± 0.82 | 4.55 ± 0.27 | 6.74 ± 0.16 | 3.09 ± 0.36 | 12.08 ± 0.04 |
| 16 | 1.19 ± 0.01 | 0.57 ± 0.03 | 1.05 ± 0.03 | 1.12 ± 0.10 | 1.46 ± 0.13 | 1.48 ± 0.01 | >20 |
| 17 | 7.44 ± 1.02 | 3.94 ± 0.37 | 3.13 ± 0.70 | 3.67 ± 0.19 | 7.53 ± 0.11 | 2.88 ± 0.26 | >20 |
| 18 | 6.81 ± 1.10 | 5.28 ± 0.37 | 3.57 ± 0.67 | 5.60 ± 0.38 | 10.24 ± 0.40 | 3.25 ± 0.69 | >20 |
| 19 | 2.67 ± 0.14 | 3.39 ± 0.07 | 1.71 ± 0.46 | 1.75 ± 0.13 | 3.31 ± 0.01 | 1.92 ± 0.17 | 9.02 ± 0.26 |
| 20 | 4.12 ± 0.27 | 3.33 ± 0.08 | 1.44 ± 0.18 | 2.36 ± 0.15 | 3.70 ± 0.91 | 1.91 ± 0.09 | 10.03 ± 1.28 |
| TET | 6.39 ± 0.83 | 6.12 ± 0.51 | 3.25 ± 0.11 | 7.60 ± 0.16 | 3.90 ± 0.47 | 3.62 ± 0.02 | 16.02 ± 1.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-C.; Zhang, R.-H.; Hu, S.-C.; Zhang, H.; Yang, D.; Zhang, W.-L.; Zhao, Y.-L.; Cui, D.-B.; Li, Y.-J.; Pan, W.-D.; et al. Design, Synthesis, and Biological Evaluation of N14-Amino Acid-Substituted Tetrandrine Derivatives as Potential Antitumor Agents against Human Colorectal Cancer. Molecules 2022, 27, 4040. https://doi.org/10.3390/molecules27134040
Wang Y-C, Zhang R-H, Hu S-C, Zhang H, Yang D, Zhang W-L, Zhao Y-L, Cui D-B, Li Y-J, Pan W-D, et al. Design, Synthesis, and Biological Evaluation of N14-Amino Acid-Substituted Tetrandrine Derivatives as Potential Antitumor Agents against Human Colorectal Cancer. Molecules. 2022; 27(13):4040. https://doi.org/10.3390/molecules27134040
Chicago/Turabian StyleWang, Yu-Chan, Rong-Hong Zhang, Sheng-Cao Hu, Hong Zhang, Dan Yang, Wen-Li Zhang, Yong-Long Zhao, Dong-Bing Cui, Yong-Jun Li, Wei-Dong Pan, and et al. 2022. "Design, Synthesis, and Biological Evaluation of N14-Amino Acid-Substituted Tetrandrine Derivatives as Potential Antitumor Agents against Human Colorectal Cancer" Molecules 27, no. 13: 4040. https://doi.org/10.3390/molecules27134040
APA StyleWang, Y. -C., Zhang, R. -H., Hu, S. -C., Zhang, H., Yang, D., Zhang, W. -L., Zhao, Y. -L., Cui, D. -B., Li, Y. -J., Pan, W. -D., Liao, S. -G., & Zhou, M. (2022). Design, Synthesis, and Biological Evaluation of N14-Amino Acid-Substituted Tetrandrine Derivatives as Potential Antitumor Agents against Human Colorectal Cancer. Molecules, 27(13), 4040. https://doi.org/10.3390/molecules27134040