
Assessing the Role of a Malonamide Linker in the Design of Potent Dual Inhibitors of Factor Xa and Cholinesterases

 ,
,  ,
,  ,
,  ,
,  , ,
, ,  and
and 
Abstract
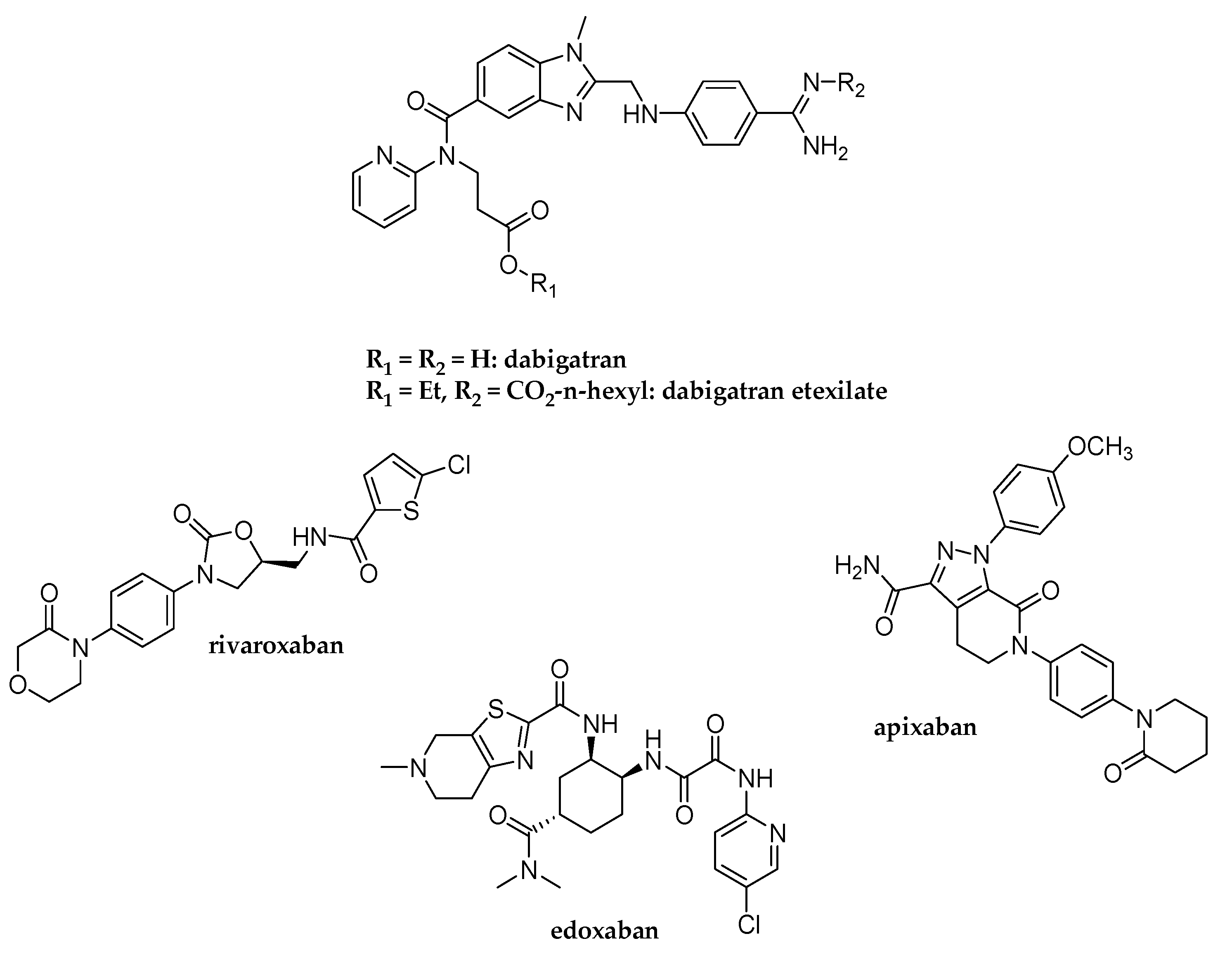
:1. Introduction
2. Chemistry
3. Results and Discussion
3.1. In Vitro Antithrombotic Activity
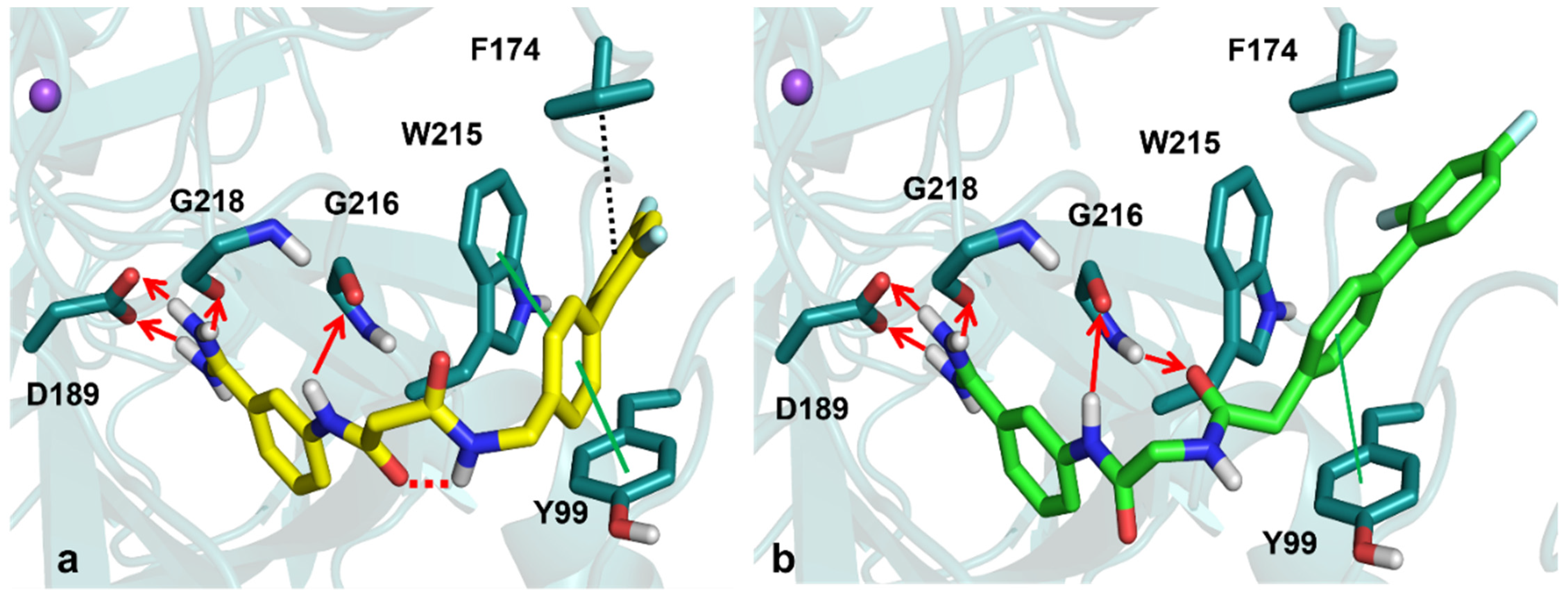
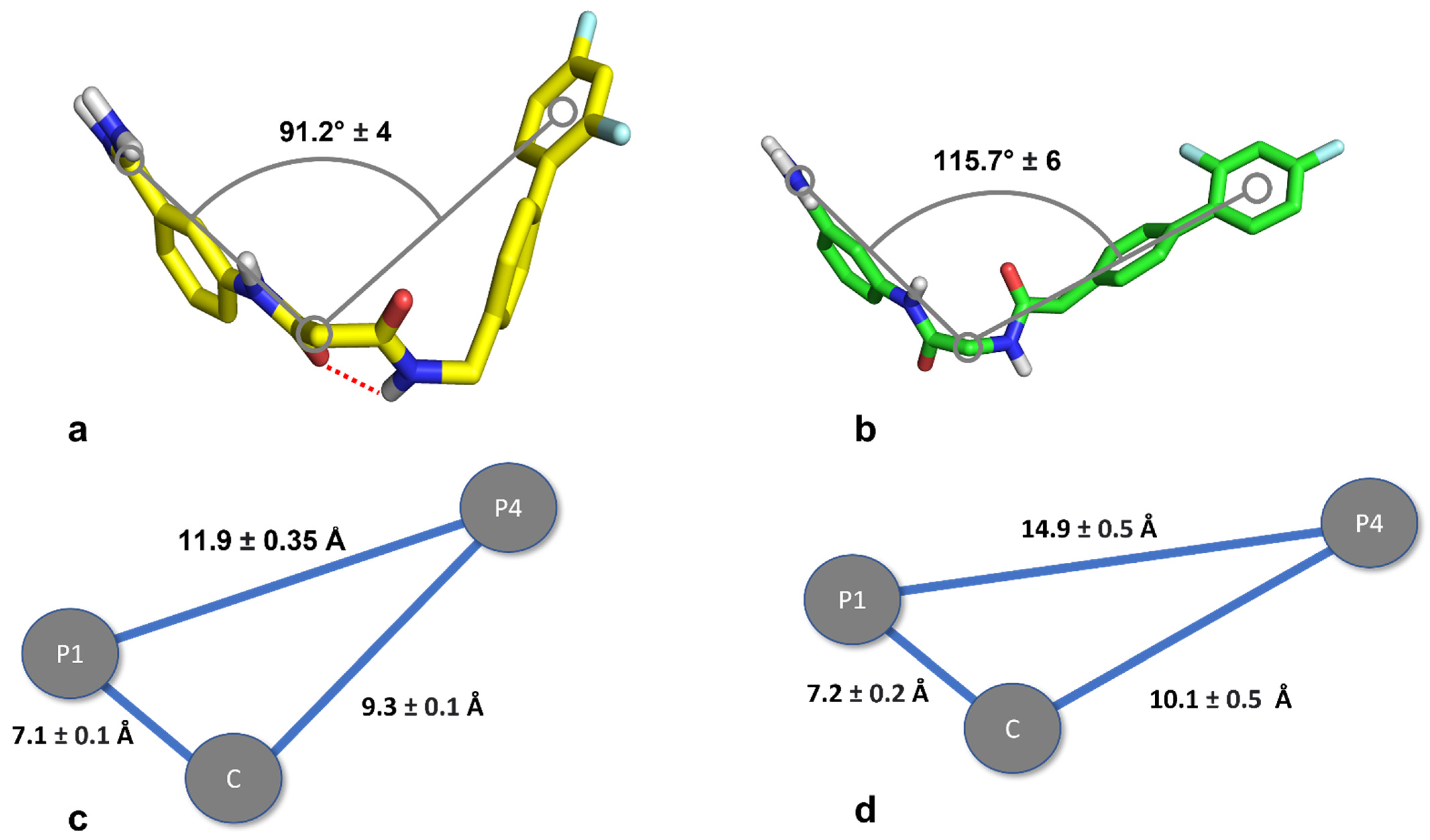
3.2. Molecular Modeling
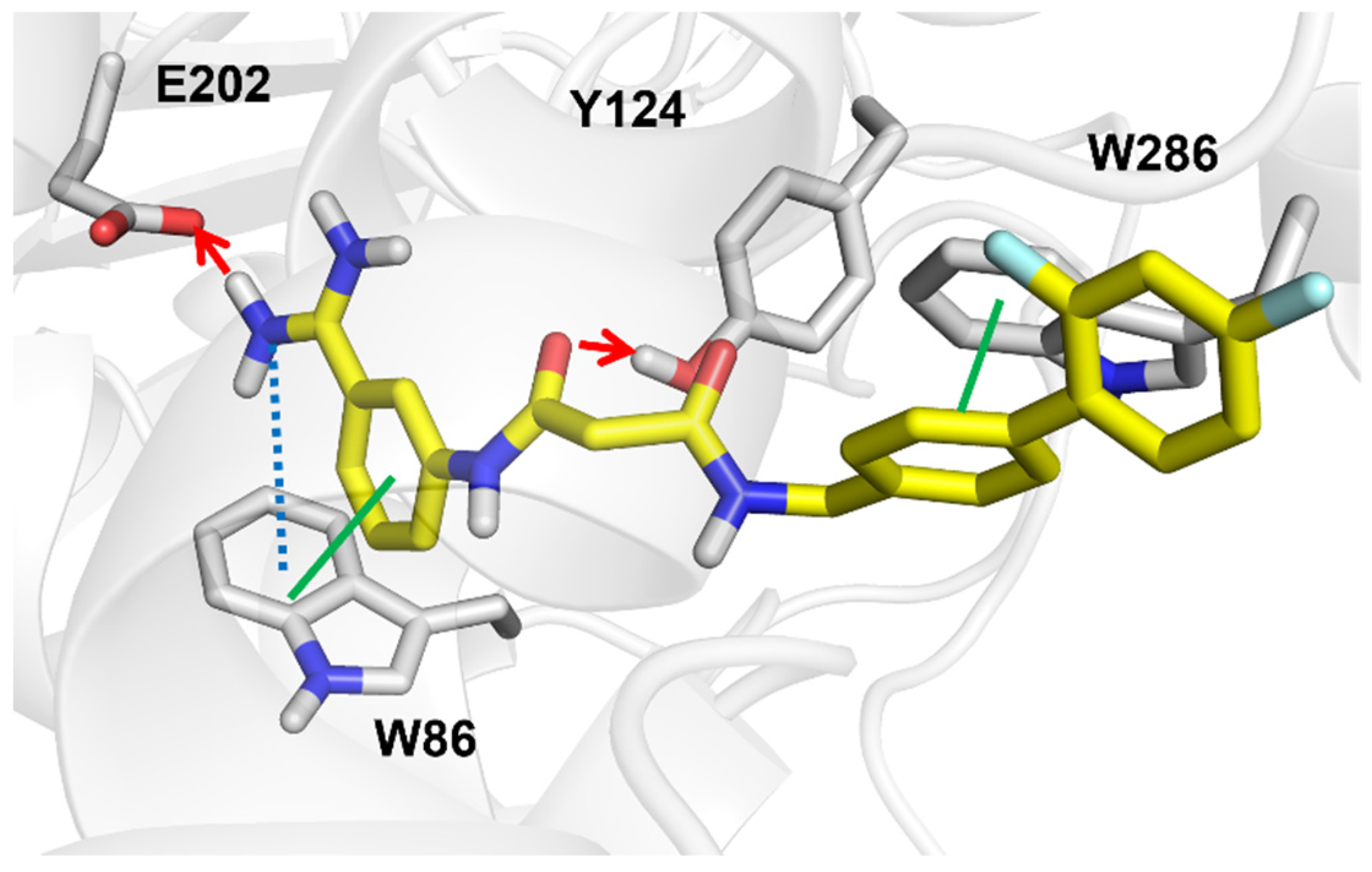
3.3. Anti-Cholinesterase Activity
4. Materials and Methods
4.1. Chemistry
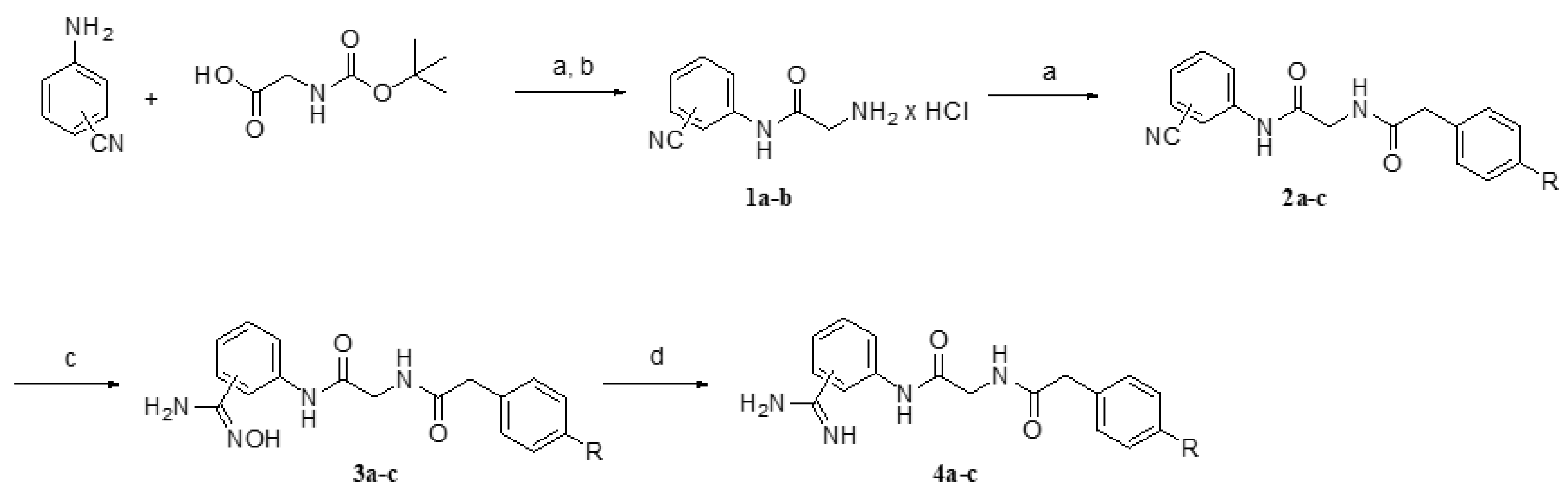
4.1.1. General Procedure for the Synthesis of Compounds 2a–c
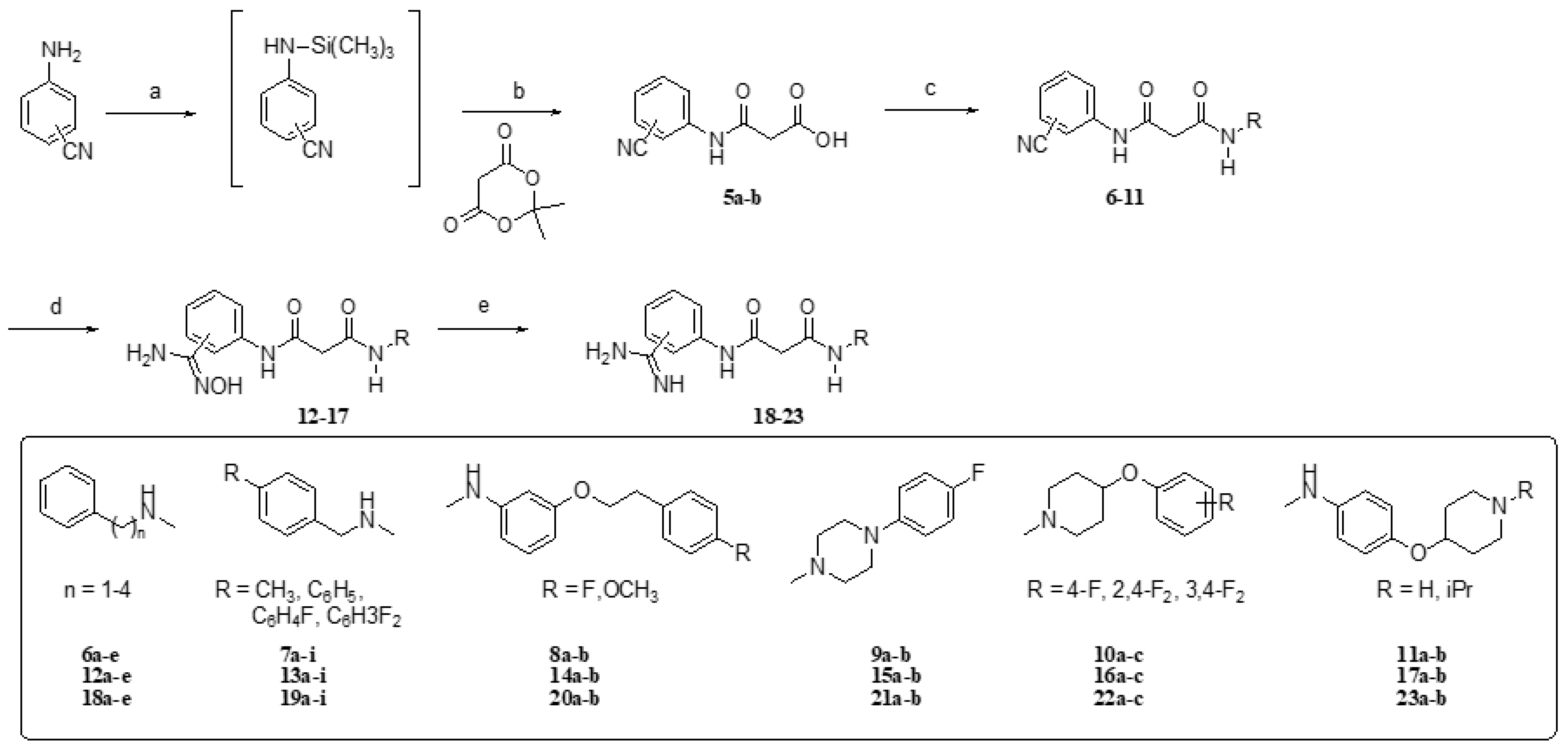
4.1.2. General Procedure for the Synthesis of Compounds 5a–c
4.1.3. General Procedure for the Synthesis of Malonamide Derivatives 6–11
4.1.4. General Procedure for the Synthesis of N-hydroxyamidine Derivatives
4.1.5. General Procedure for the Synthesis of Benzamidine Derivatives
4.2. Enzymatic Assays
4.2.1. Coagulative Serine Proteases Thrombin and fXa Inhibition Assay
4.2.2. Cholinesterases Inhibition Assay
4.3. Molecular Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Moran, A.E.; Roth, G.A.; Narula, J.; Mensah, G.A. 1990–2010 global cardiovascular disease atlas. Glob. Heart 2014, 9, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 4 July 2021).
- Boothello, R.S.; Sankaranarayanan, N.V.; Afosah, D.K.; Karuturi, R.; Al-Horani, R.A.; Desai, U.R. Studies on fragment-based design of allosteric inhibitors of human factor XIa. Bioorg. Med. Chem. 2020, 28, 115762. [Google Scholar] [CrossRef] [PubMed]
- Choxi, R.; Kapoor, K.; Mackman, N.; Jovin, I.S. Direct Oral Anticoagulants and Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Grilusová, K.; Bolek, T.; Škorňová, I.; Staško, J.; Samoš, M.; Mokáň, M. Cancer-associated thrombosis-treatment and prevention with direct oral factor Xa inhibitors. Klin. Onkol. Summer 2021, 34, 283–290. [Google Scholar] [CrossRef]
- Fahmi, A.M.; Elewa, H.; El Jilany, I. Warfarin dosing strategies evolution and its progress in the era of precision medicine. A narrative review. Int. J. Clin. Pharm. 2022, 44, 599–607. [Google Scholar] [CrossRef]
- Burmeister, C.; Ghazaleh, S.; Beran, A.; Patel, N.J.; Khokher, W.; Bhuta, S.; Mhanna, M.; Eltahawy, E.; Assaly, R. Head-to-head Comparison Between Direct Oral Anticoagulants and Vitamin K Antagonists for Chronic Thromboembolic Pulmonary Hypertension: A Systematic Review and Meta-Analysis. Curr. Probl. Cardiol. 2022, 2022, 101232. [Google Scholar] [CrossRef]
- Haas, S. New oral Xa and IIa inhibitors: Updates on clinical trial results. J. Thromb. Thrombolysis 2008, 25, 52–60. [Google Scholar] [CrossRef]
- Alban, S. Pharmacological strategies for inhibition of thrombin activity. Curr. Pharm. Des. 2008, 14, 1152–1175. [Google Scholar] [CrossRef]
- Cheung, K.-S.; Leung, W.K. Gastrointestinal bleeding in patients on novel oral anticoagulants: Risk, prevention and management. World J. Gastroenterol. 2017, 23, 1954–1963. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, B.; Zhang, Y.; Dai, X.; Duan, Y.; Mao, Q.; Gao, J.; Yang, Y.; Bao, Z.; Fu, X.; et al. Design, synthesis and biological evaluation of novel FXIa inhibitors with 2-phenyl-1H-imidazole-5-carboxamide moiety as P1 fragment. Eur. J. Med. Chem. 2021, 220, 113437. [Google Scholar] [CrossRef]
- de Candia, M.; Fiorella, F.; Lopopolo, G.; Carotti, A.; Romano, M.R.; Lograno, M.D.; Martel, S.; Carrupt, P.A.; Belviso, B.D.; Caliandro, R.; et al. Synthesis and biological evaluation of direct thrombin inhibitors bearing 4-(piperidin-1-yl)pyridine at the P1 position with potent anticoagulant activity. J. Med. Chem. 2013, 56, 8696–8711. [Google Scholar] [CrossRef] [PubMed]
- de Candia, M.; Lopopolo, G.; Altomare, C. Novel factor Xa inhibitors: A patent review. Expert. Opin. Therm. Pat. 2009, 19, 1535–1580. [Google Scholar] [CrossRef] [PubMed]
- Lopopolo, G.; de Candia, M.; Panza, L.; Romano, M.R.; Lograno, M.D.; Campagna, F.; Altomare, C. β-D-glucosyl conjugates of highly potent inhibitors of blood coagulation factor Xa bearing 2-chorothiophene as a P1 motif. ChemMedChem 2012, 7, 1669–1677. [Google Scholar] [CrossRef]
- Aderibigbe, A.D.; Day, D.P. Syntheses and Applications of Malonamide Derivatives—A Minireview. Chem. Select. 2020, 5, 15222–15232. [Google Scholar]
- Islam, M.S.; Barakat, A.; Al-Majid, A.M.; Ghabbour, H.A.; Motiur Rahman, A.F.M.; Javaid, K.; Imad, R.; Yousuf, S.; Choudhary, M.I. A concise synthesis and evaluation of new malonamide derivatives as potential a-glucosidase inhibitors. Bioorg. Med. Chem. 2016, 24, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Islam, M.S.; Al-Majid, A.M.; Soliman, S.M.; Ghabbour, H.A.; Yousuf, S.; Choudhary, M.I.; Ul-Haq, Z. Synthesis, molecular structure, spectral analysis, and biological activity of new malonamide derivatives as a-glucosidase inhibitors. J. Mol. Struct. 2017, 1134, 253–264. [Google Scholar] [CrossRef]
- Chorev, M. The Partial Retro–Inverso Modification: A Road Traveled Together. Biopolymers 2005, 80, 67–84. [Google Scholar] [CrossRef]
- de Candia, M.; Liantonio, F.; Carotti, A.; De Cristofaro, R.; Altomare, C. Fluorinated benzyloxyphenyl piperidine-4-carboxamides with dual function against thrombosis: Inhibitors of factor Xa and platelet aggregation. J. Med. Chem. 2009, 52, 1018–1028. [Google Scholar] [CrossRef]
- Lopopolo, G.; Fiorella, F.; de Candia, M.; Nicolotti, O.; Martel, S.; Carrupt, P.A.; Altomare, C.D. Biarylmethoxy isonipecotanilides as potent and selective inhibitors of blood coagulation factor Xa. Eur. J. Pharm. Sci. 2011, 42, 180–191. [Google Scholar] [CrossRef]
- Belviso, B.D.; Caliandro, R.; de Candia, M.; Zaetta, G.; Lopopolo, G.; Incampo, F.; Colucci, M.; Altomare, C.D. How a β-D-glucoside side chain enhances binding affinity to thrombin of inhibitors bearing 2-chlorothiophene as P1 moiety: Crystallography, fragment deconstruction study, and evaluation of antithrombotic properties. J. Med. Chem. 2014, 57, 8563–8575. [Google Scholar] [CrossRef]
- Khadse, A.N.; Sharma, M.K.; Murumkar, P.R.; Rajput, S.J.; Yadav, M.R. Advances in the Development of Novel Factor Xa Inhibitors: A Patent Review. Mini Rev. Med. Chem. 2018, 18, 1332–1353. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Schiavoni, M.M.; Mack, H.-G.; Ulic, S.E.; Della Vedova, C.O. Tautomers and conformers of malonamide, NH2-C(O)-CH2-C(O)-NH2: Vibrational analysis, NMR spectra and ab initio calculations. Spectrochimica Acta Part A 2000, 56, 1533–1541. [Google Scholar] [CrossRef]
- Sandrone, G.; Dixon, D.A.; Hay, B.P. Conformational analysis of malonamide, N,N-dimethylmalonamide, and N,N,N,N-tetramethylmalonamide. J. Phys. Chem. A 1999, 103, 3554–3561. [Google Scholar] [CrossRef]
- Purgatorio, R.; Gambacorta, N.; de Candia, M.; Catto, M.; Rullo, M.; Pisani, L.; Nicolotti, O.; Altomare, C.D. First-in-Class Isonipecotamide-Based Thrombin and Cholinesterase Dual Inhibitors with Potential for Alzheimer Disease. Molecules 2021, 26, 5208. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Prusoff, W.H. Relation between the inhibition constant (Ki) and the concentration of inhibitor which causes fifty per cent inhibition (IC50) of an enzymic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Purgatorio, R.; de Candia, M.; De Palma, A.; De Santis, F.; Pisani, L.; Campagna, F.; Cellamare, S.; Altomare, C.D.; Catto, M. Insights into Structure-Activity Relationships of 3-Arylhydrazonoindolin-2-One Derivatives for Their Multitarget Activity on β-Amyloid Aggregation and Neurotoxicity. Molecules 2018, 23, 1544. [Google Scholar] [CrossRef] [Green Version]
- Purgatorio, R.; de Candia, M.; Catto, M.; Rullo, M.; Pisani, L.; Denora, N.; Carrieri, A.; Nevskaya, A.A.; Voskressensky, L.G.; Altomare, C.D. Evaluation of Water-Soluble Mannich Base Prodrugs of 2,3,4,5-Tetrahydroazepino[4,3-b]indol-1(6H)-one as Multitarget-Directed Agents for Alzheimer’s Disease. ChemMedChem 2021, 16, 589–598. [Google Scholar] [CrossRef]
- Shi, Y.; Li, C.; O’Connor, S.P.; Zhang, J.; Shi, M.; Bisaha, S.N.; Wang, Y.; Sitkoff, D.; Pudzianowski, A.T.; Huang, C.; et al. Aroylguanidine-based factor Xa inhibitors: The discovery of BMS-344577. Bioorg. Med. Chem. Lett. 2009, 19, 6882–6889. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A High-Performance Quantum Chemistry Software Program with Strengths in Life and Materials Sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-4, Jaguar; Schrödinger, LLC: New York, NY, USA, 2021; Available online: https://www.schrodinger.com/products/jaguar (accessed on 6 June 2021).
- MacroModel; Schrödinger, LLC: New York, NY, USA, 2021; Available online: https://www.schrodinger.com/products/macromodel (accessed on 6 June 2021).
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Exp. Opt. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  | ||
|---|---|---|---|
| Cmpd | Amidine Position | Ki (μM) a | |
| fXa | thr | ||
| 4a | p | >50 | 39.4 |
| 4b | m | 25.6 | 24.1 |
| 18a | p | 23.2 | >50 |
| 18b | m | 12.3 | >50 |
| Apixaban b | 0.00012 | ||
| Dabigatran b | 0.0042 | ||
 | ||||||
|---|---|---|---|---|---|---|
| Cmpd | Amidine Position | n | R | Ki (μM) a | ||
| fXa | thr | αCHT c | ||||
| 18c | p | 2 | H | >50 | >50 | |
| 18d | p | 3 | H | 39.8 | 25.5 | |
| 18e | p | 4 | H | 18.6 | 5.50 | |
| 19a | p | 1 | 4-CH3 | 4.40 | 36.5 | |
| 19b | m | 1 | 4-CH3 | 4.20 | 36.6 | |
| 19c | p | 1 | C6H5 | >50 | >50 | |
| 19d | m | 1 | C6H5 | 1.56 | >50 | 4.3 |
| 19e | m | 1 | 2-F(C6H4) | 0.932 | 14.6 | >50 |
| 19f | m | 1 | 3-F(C6H4) | 0.348 | 18.8 | 26.5 |
| 19g | m | 1 | 4-F(C6H4) | 0.907 | 6.19 | >50 |
| 19h | m | 1 | 2,4-F2(C6H3) | 0.057 | 16.1 | >50 |
| 19i | m | 1 | 3,5-F2(C6H3) | 0.264 | 26.7 | 30.2 |
 | |||||
|---|---|---|---|---|---|
| Cmpd | X-Y | R | Ki (μM) a | ||
| fXa | thr | αCHT | |||
| 4c | NHCO | H | 1.01 | 10.7 | 35.0 |
| 19h | CONH | H | 0.057 | 16.1 | >50 |
| 19l | CONH | CH3 | 0.214 | >50 | >50 |
 | |||||
|---|---|---|---|---|---|
| Cmpd | P4 | R | Ki (μM) a | ||
| fXa | thr | αCHT | |||
| 20a |  | 4-F | 0.413 | 12.0 | >50 |
| 20b | 4-OCH3 | 0.205 | 6.66 | >50 | |
| 21a |  | F | 0.423 | 31.4 | >50 |
| 22a |  | 4-F | 0.350 | >50 | >50 |
| 22b | 2,4-F2 | 0.089 | >50 | >50 | |
| 22c | 3,4-F2 | 0.120 | 27.0 | >50 | |
| 23a |  | H | 0.590 | 42.4 | >50 |
| 23b | CH(CH3)2 | 2.30 | >50 | >50 | |
 | ||||
|---|---|---|---|---|
| Cmpd | P4 | R | Ki (μM) a | |
| AchE b | BchE b | |||
| 19d |  | C6H5 | 9.33 | (28%) |
| 19e | 2-F(C6H4) | (35%) | 7.70 | |
| 19f | 3-F(C6H4) | 8.11 | 8.09 | |
| 19g | 4-F(C6H4) | 4.48 | (37%) | |
| 19h | 2,4-F2(C6H3) | 2.44 | 1.52 | |
| 19l c | 2,4-F2(C6H3) | 3.74 | 1.55 | |
| 19i | 3,5-F2(C6H3) | (5%) | 8.02 | |
| 20a |  | 4-F | 10.0 | 1.02 |
| 20b | 4-OCH3 | 1.56 | 5.84 | |
| 21a |  | 4-F | 4.68 | 10.0 |
| 22a |  | 4-F | 1.88 | (22%) |
| 22b | 2,4-F2 | 2.31 | (25%) | |
| 22c | 3,4-F2 | 2.68 | 10.9 | |
| 23a |  | H | (1%) | 10.0 |
| 23b | CH(CH3)2 | 1.30 | 0.650 | |
| Donepezil d | 0.021 | 2.25 | ||
| Galantamine d | 0.55 | 12.0 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Purgatorio, R.; Gambacorta, N.; Samarelli, F.; Lopopolo, G.; de Candia, M.; Catto, M.; Nicolotti, O.; Altomare, C.D. Assessing the Role of a Malonamide Linker in the Design of Potent Dual Inhibitors of Factor Xa and Cholinesterases. Molecules 2022, 27, 4269. https://doi.org/10.3390/molecules27134269
Purgatorio R, Gambacorta N, Samarelli F, Lopopolo G, de Candia M, Catto M, Nicolotti O, Altomare CD. Assessing the Role of a Malonamide Linker in the Design of Potent Dual Inhibitors of Factor Xa and Cholinesterases. Molecules. 2022; 27(13):4269. https://doi.org/10.3390/molecules27134269
Chicago/Turabian StylePurgatorio, Rosa, Nicola Gambacorta, Francesco Samarelli, Gianfranco Lopopolo, Modesto de Candia, Marco Catto, Orazio Nicolotti, and Cosimo D. Altomare. 2022. "Assessing the Role of a Malonamide Linker in the Design of Potent Dual Inhibitors of Factor Xa and Cholinesterases" Molecules 27, no. 13: 4269. https://doi.org/10.3390/molecules27134269
APA StylePurgatorio, R., Gambacorta, N., Samarelli, F., Lopopolo, G., de Candia, M., Catto, M., Nicolotti, O., & Altomare, C. D. (2022). Assessing the Role of a Malonamide Linker in the Design of Potent Dual Inhibitors of Factor Xa and Cholinesterases. Molecules, 27(13), 4269. https://doi.org/10.3390/molecules27134269