
A Study on the Direct Esterification of Monoalkylphosphates and Dialkylphosphates; The Conversion of the Latter Species to Trialkylphosphates by Alkylating Esterification †




Abstract
:1. Introduction
2. Results and Discussion
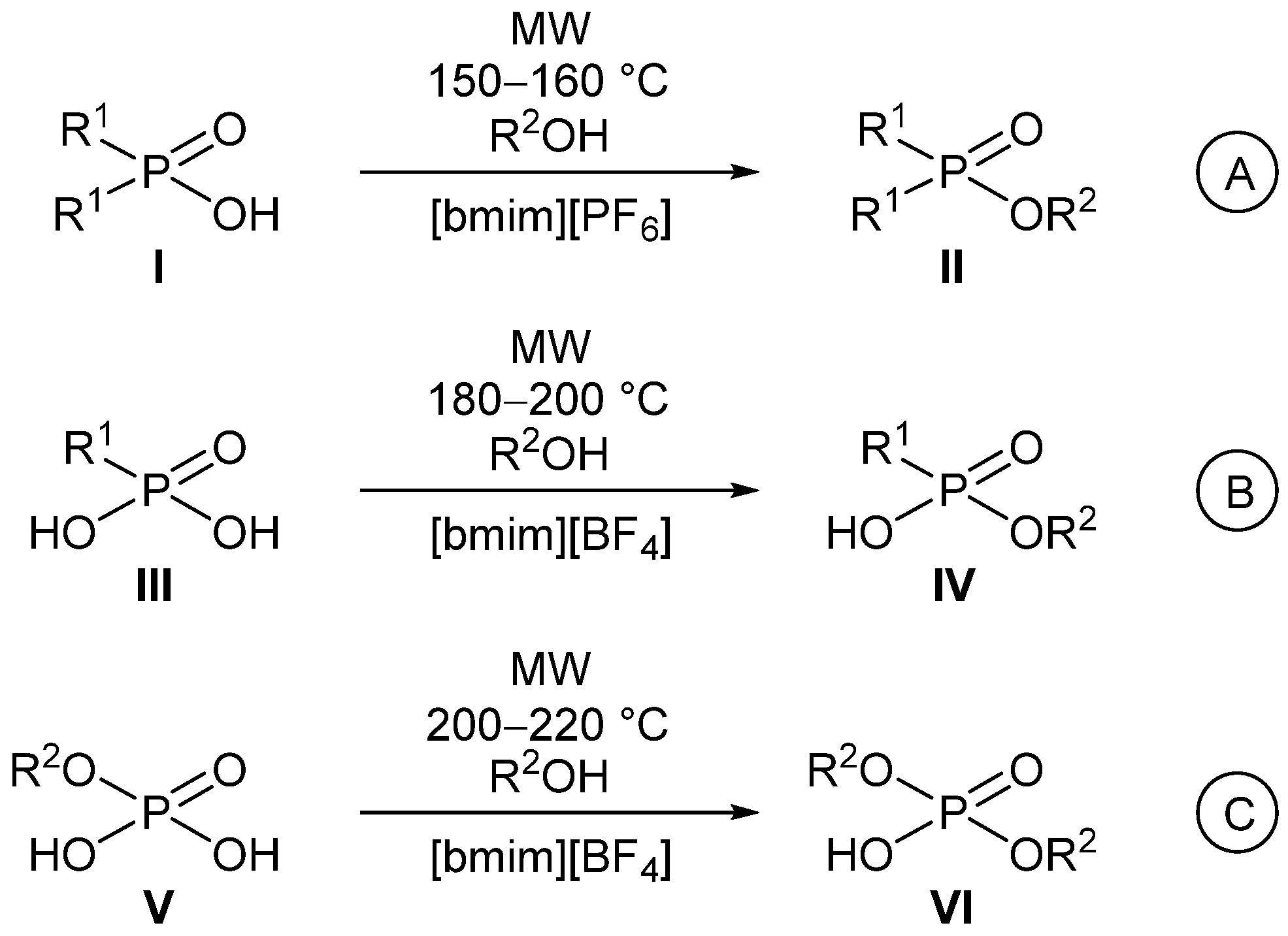
2.1. MW-Assisted Direct Esterification of Monoalkylphosphates
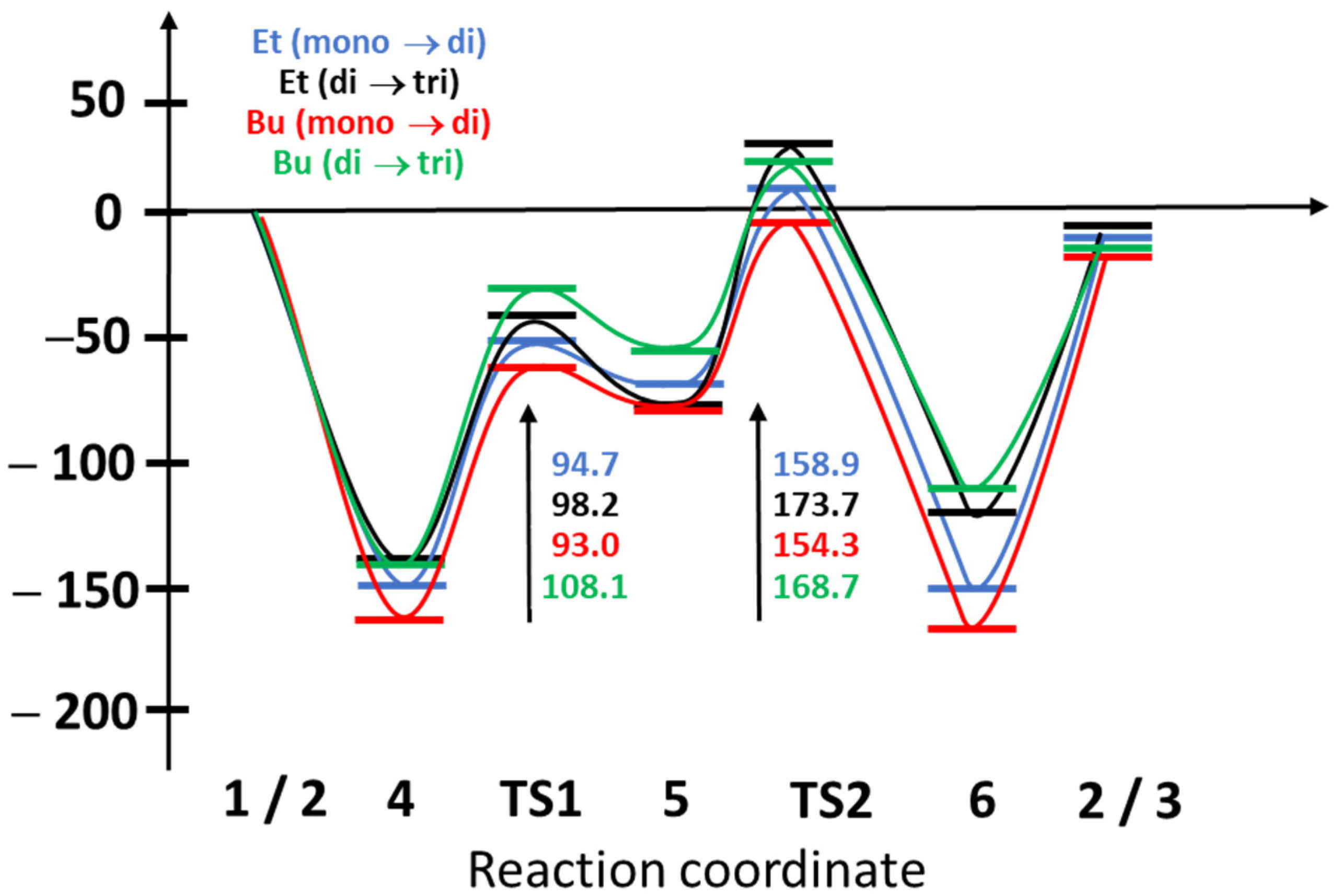
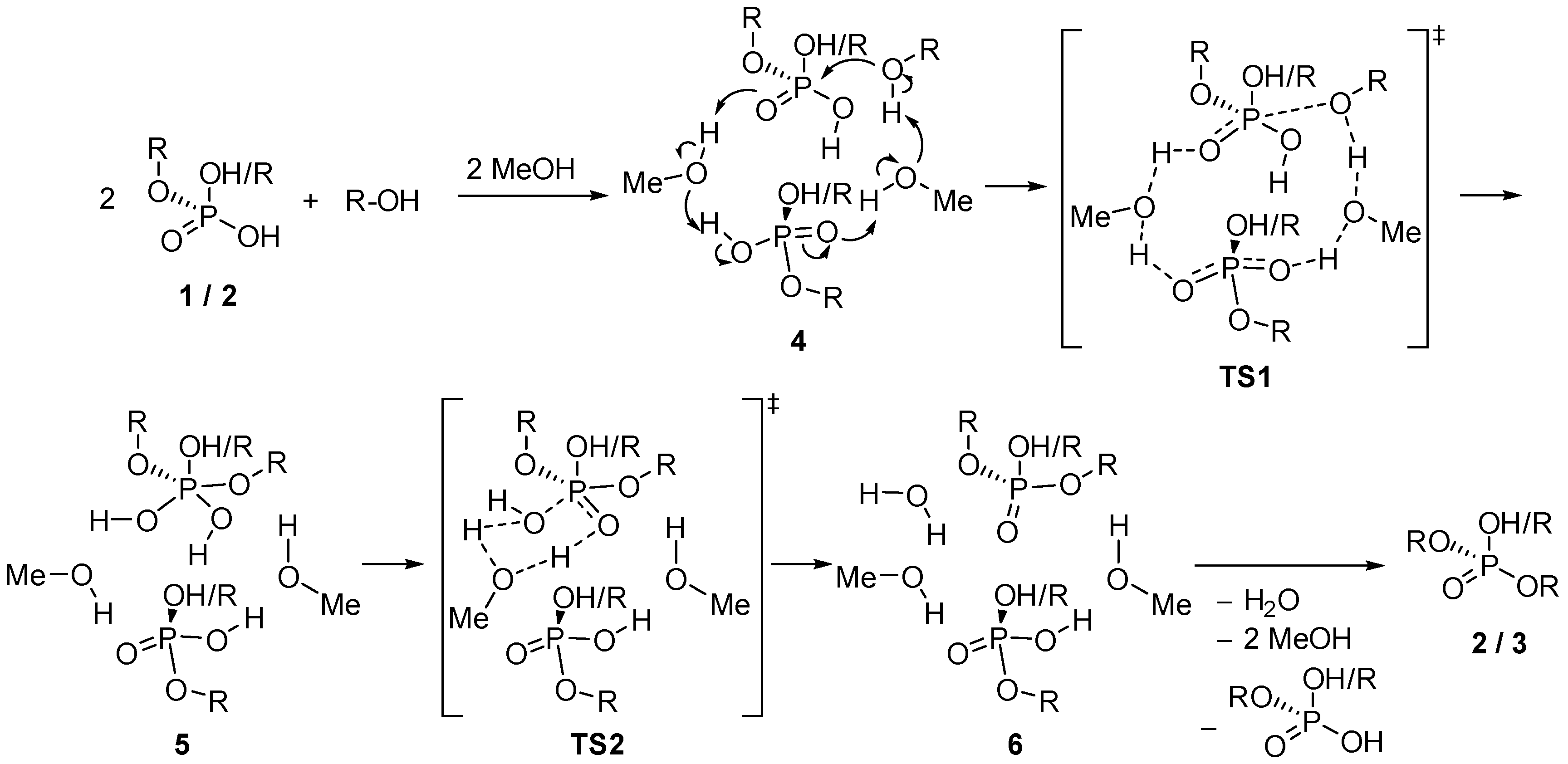
2.2. Theoretical Calculations on the Energetics and Mechanism of the Monoalkylphosphate → Dialkylphosphate → Trialkylphosphate Transformation
2.3. MW-Assisted Alkylation of Dialkylphosphates
3. Materials and Methods
3.1. General Information
3.2. The Direct Esterification of Monoalkylphosphates (1a–d)
3.3. The Alkylating Esterification of Dialkylphosphates (2a–2d)
3.4. Characterization of New Trialkylphosphates (7a–d and 8a–d)
3.4.1. Dibutyl-ethylphosphate (7a)
3.4.2. Dibutyl-propylphosphate (7b)
3.4.3. Dibutyl-isopropylphosphate (7c)
3.4.4. Dibutyl-pentylphosphate (7d)
3.4.5. Dipentyl-ethylphosphate (8a)
3.4.6. Dipentyl-propylphosphate (8b)
3.4.7. Dipentyl-isopropylphosphate (8c)
3.4.8. Dipentyl-butylphosphate (8d)
3.5. Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De la Hoz, A.; Diaz-Ortiz, A.; Prieto, P. Microwave-assisted green organic synthesis. In Alternative Energy Sources for Green Chemistry; Stefanidis, G., Stankiewicz, A., Eds.; Royal Society of Chemistry: London, UK, 2016; Volume 47, pp. 1–33. [Google Scholar] [CrossRef]
- Nasir Baig, R.B.; Varma, R.S. Alternative energy input: Mechanochemical, microwave and ultrasound-assisted organic synthesis. Chem. Soc. Rev. 2012, 41, 1559–1584. [Google Scholar] [CrossRef] [PubMed]
- Nain, S.; Singh, R.; Ravichandran, S. Importance of Microwave heating in organic synthesis. Adv. J. Chem. A 2019, 2, 94–104. [Google Scholar] [CrossRef]
- Sharma, N.; Sharma, U.K.; Van der Eycken, E.V. Microwave-assisted organic synthesis: Overview of recent applications. In Green Techniques for Organic Synthesis and Medicinal Chemistry, 2nd ed.; Zhang, W., Cue, B.W., Eds.; JohnWiley & Sons Ltd.: New York, NY, USA, 2018; pp. 441–468. [Google Scholar] [CrossRef]
- Romanova, N.N.; Gravis, A.G.; Zyk, N.V. Microwave irradiation in organic synthesis. Russ. Chem. Rev. 2005, 74, 969–1013. [Google Scholar] [CrossRef]
- Jolly, J. Microwave assisted reactions in organic chemistry: A review of recent advances. Int. J. Chem. 2012, 4, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Dudley, G.B.; Stiegman, A.E. Changing perspectives on the strategic use of microwave heating in organic synthesis. Chem. Rec. 2018, 18, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Keglevich, G.; Mucsi, Z. Interpretation of the rate enhancing effect of microwaves. In Microwave Chemistry; Cravotto, G., Carnaroglio, D., Eds.; De Gruyter: Berlin, Germany, 2017; pp. 53–64. [Google Scholar] [CrossRef]
- Keglevich, G.; Novák, T.; Vida, L.; Greiner, I. Microwave irradiation as an alternative to phase transfer catalysis in the liquid-solid phase, solvent-free C-alkylation of active methylene containing substrates. Green Chem. 2006, 8, 1073–1075. [Google Scholar] [CrossRef]
- Keglevich, G.; Szekrényi, A. Eco-friendly accomplishment of the extended Kabachnik-Fields reaction; a solvent- and catalyst-free microwave-assisted synthesis of α-aminophosphonates and α-aminophosphine oxides. Lett. Org. Chem. 2008, 5, 616–622. [Google Scholar] [CrossRef]
- Kovács, T.; Urbanics, A.; Csatlós, F.; Keglevich, G. A study on the deoxygenation of trialkyl-, dialkyl-phenyl- and alkyl-diphenyl phosphine oxides by hydrosilanes. Heteroat. Chem. 2017, 28, e21376. [Google Scholar] [CrossRef]
- Keglevich, G.; Jablonkai, E.; Balázs, L.B. A “green” variation of the Hirao reaction: The P–C coupling of diethyl phosphite, alkyl phenyl-H-phosphinates and secondary phosphine oxides with bromoarenes using a P-ligand-free Pd(OAc)2 catalyst under microwave and solvent-free conditions. RSC Adv. 2014, 4, 22808–22816. [Google Scholar] [CrossRef] [Green Version]
- Keglevich, G.; Forintos, H.; Körtvélyesi, T. Synthesis and reactions of β-oxophosphoranes/ylides containing a cyclic or acyclic P-moiety. Curr. Org. Chem. 2004, 8, 1245–1261. [Google Scholar] [CrossRef]
- Quin, L.D. A Guide to Organophosphorus Chemistry; Wiley & Sons: New York, NY, USA, 2000; ISBN 978-0-471-31824-8. [Google Scholar]
- Barton, D.; Ollis, W.D. (Eds.) Comprehensive Organic Chemistry, Phosphorus Compounds; Pergamon: Oxford, UK, 1979; Volume 2. [Google Scholar]
- Kiss, N.Z.; Keglevich, G. Microwave-assisted direct esterification of cyclic phosphinic acids in the presence of ionic liquids. Tetrahedron Lett. 2016, 57, 971–974. [Google Scholar] [CrossRef]
- Harsági, N.; Henyecz, R.; Ábrányi-Balogh, P.; Drahos, L.; Keglevich, G. Microwave-assisted ionic liquid-catalyzed selective monoesterification of alkylphosphonic acids—An experimental and a theoretical study. Molecules 2021, 26, 5303. [Google Scholar] [CrossRef] [PubMed]
- Harsági, N.; Kiss, N.Z.; Keglevich, G. P-Chloride-free synthesis of phosphoric esters: Microwave-assisted esterification of alkyl- and dialkyl phosphoric ester-acids obtained from phosphorus pentoxide. Synthesis 2022, 54, 3047–3054. [Google Scholar] [CrossRef]
- Mucsi, Z.; Kiss, N.Z.; Keglevich, G. A quantum chemical study on the mechanism and energetics of the direct esterification, thioesterification and amidation of 1-hydroxy-3-methyl-3-phospholene 1-oxide. RSC Adv. 2014, 4, 11948–11954. [Google Scholar] [CrossRef] [Green Version]
- Zwierzak, A. Phase-transfer-catalysed phosphorylation of alcohols in a two-phase system. Synthesis 1976, 305–306. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}

| Entry | R | T (°C) | t (h) | Product Composition (%) * | Yield of 2 (%) | |
|---|---|---|---|---|---|---|
| 2 | 3 | |||||
| 1 | Bu (a) | 200 | 2 | 96 | 4 | 83 [18] |
| 2 | Pent (b) | 200 | 2 | 95 | 5 | 84 |
| 3 | Pr (c) | 200 | 2.5 | 96 | 4 | 87 |
| 4 | Et (d) | 175 | 4.5 | 95 | 5 | 83 |
| R | 1/2 | 4 | TS1 | 5 | TS2 | 6 | 2/3 |
|---|---|---|---|---|---|---|---|
| Et (mono → di) | 0.0 | −151.6 | −56.9 | −60.9 | 7.2 | −150.4 | −10.4 |
| Bu (mono → di) | 0.0 | −160.9 | −67.9 | −71.9 | −6.6 | −165.3 | −16.3 |
| Et (di → tri) | 0.0 | −143.3 | −45.0 | −70.9 | 30.5 | −121.0 | −7.6 |
| Bu (di → tri) | 0.0 | −144.1 | −36.0 | −52.7 | 24.6 | −107.8 | −13.7 |

| Entry | R | X | t (h) | Yield of 3 (%) |
|---|---|---|---|---|
| 1 | Bu (a) | Br | 2.5 | 85 [18] |
| 2 | Pent (b) | Br | 2.5 | 84 |
| 3 | Pr (c) | Br | 3.5 | 86 |
| 4 | Et (d) | I | 5 | 84 |

| Entry | RX | T (°C) | t (h) | Conversion (%) | Conversion to 7 (%) | Yield of 7 (%) |
|---|---|---|---|---|---|---|
| 1 | EtI | 135 | 3 | 92 | 71 | 65 (7a) |
| 2 | PrBr | 135 | 3 | 94 | 71 | 63 (7b) |
| 3 | iPrBr | 150 | 3 | 100 | 67 | 58 (7c) |
| 4 | PentBr | 135 | 2 | 98 | 98 | 89 (7d) |

| Entry | RX | T (°C) | t (h) | Conversion (%) | Conversion to 8 (%) | Yield of 8 (%) |
|---|---|---|---|---|---|---|
| 1 | EtBr | 135 | 4.5 | 83 | 67 | 59 (8a) |
| 2 | PrBr | 150 | 3 | 89 | 51 | 48 (8b) |
| 3 | iPrBr | 150 | 3 | 45 | 27 | 19 (8c) |
| 4 | BuBr | 150 | 3 | 91 | 49 | 44 (8d) |
| Compound | δP (CDCl3) | δP(lit) (CDCl3) [18] | HRMS [M+Na]+ | |
|---|---|---|---|---|
| Found | Calculated | |||
| 2a | 0.032 | 0.029 | 233.0912 | 233.0919 |
| 2b | 0.15 | 0.10 | 239.1412 * | 239.1413 * |
| 2c | 1.94 | 2.0 | 205.0606 | 205.0601 |
| 2d | 0.52 | 0.55 | 177.0293 | 177.0290 |
| Compound | δP (CDCl3) | δP(lit) (CDCl3) [18] | HRMS [M+Na]+ | |
|---|---|---|---|---|
| Found | Calculated | |||
| 3a | −0.92 | −0.89 | 267.1725 * | 267.1717 * |
| 3b | −1.02 | −0.99 | 331.2014 | 331.2013 |
| 3c | −0.89 | −0.88 | 247.1075 | 247.1074 |
| 3d | −1.05 | −1.0 | 205.0606 | 205.0602 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ábrányi-Balogh, P.; Harsági, N.; Drahos, L.; Keglevich, G. A Study on the Direct Esterification of Monoalkylphosphates and Dialkylphosphates; The Conversion of the Latter Species to Trialkylphosphates by Alkylating Esterification. Molecules 2022, 27, 4674. https://doi.org/10.3390/molecules27154674
Ábrányi-Balogh P, Harsági N, Drahos L, Keglevich G. A Study on the Direct Esterification of Monoalkylphosphates and Dialkylphosphates; The Conversion of the Latter Species to Trialkylphosphates by Alkylating Esterification. Molecules. 2022; 27(15):4674. https://doi.org/10.3390/molecules27154674
Chicago/Turabian StyleÁbrányi-Balogh, Péter, Nikoletta Harsági, László Drahos, and György Keglevich. 2022. "A Study on the Direct Esterification of Monoalkylphosphates and Dialkylphosphates; The Conversion of the Latter Species to Trialkylphosphates by Alkylating Esterification" Molecules 27, no. 15: 4674. https://doi.org/10.3390/molecules27154674
APA StyleÁbrányi-Balogh, P., Harsági, N., Drahos, L., & Keglevich, G. (2022). A Study on the Direct Esterification of Monoalkylphosphates and Dialkylphosphates; The Conversion of the Latter Species to Trialkylphosphates by Alkylating Esterification. Molecules, 27(15), 4674. https://doi.org/10.3390/molecules27154674