
Metabolic Profiling of Inga Species with Antitumor Activity

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
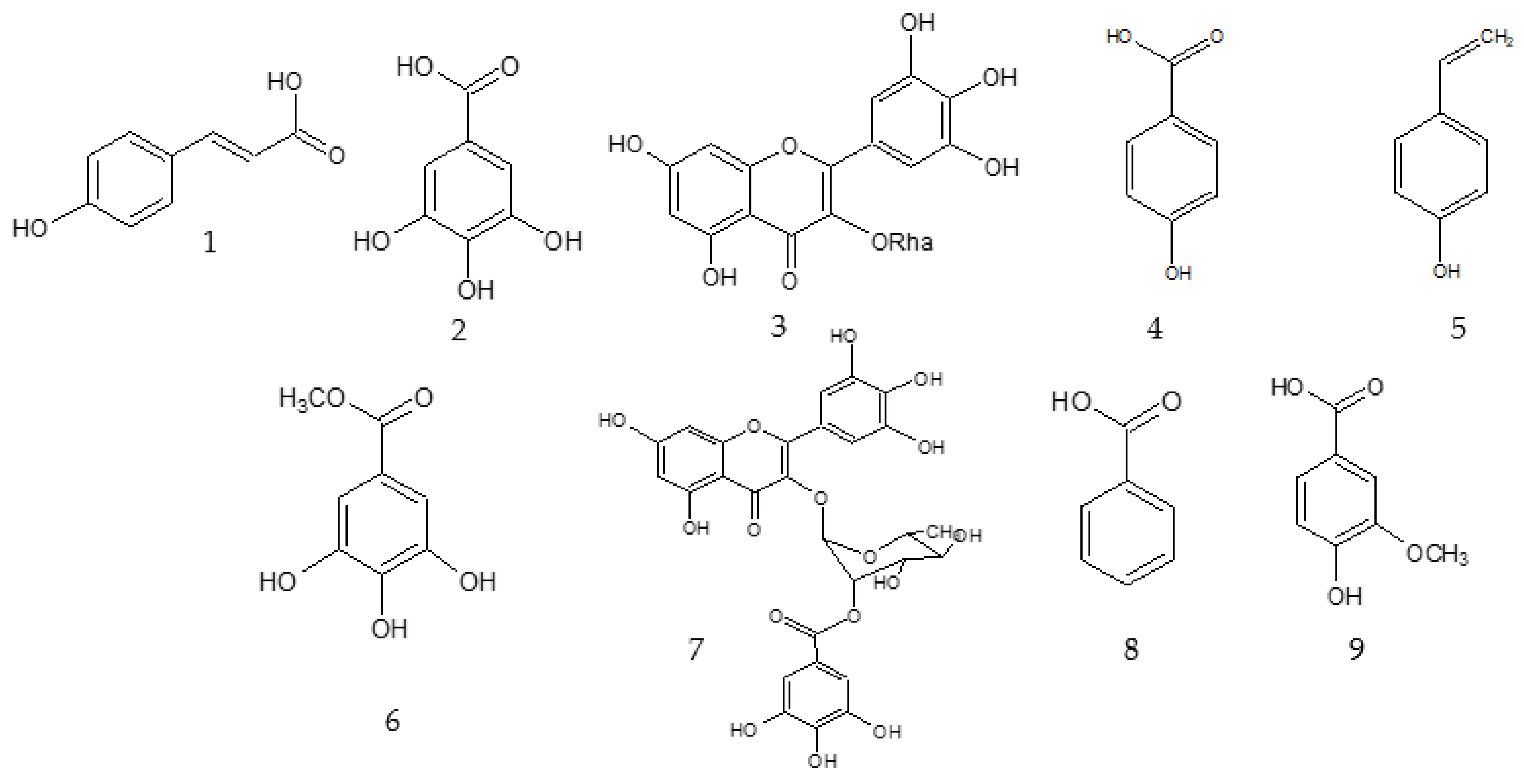
2.1. Bioassay Guided Fractionation
2.2. Quantification of Phenolic Compounds Isolated from Cytotoxic Extracts
2.3. Antigenotoxicity and Proteomic Approach from the Flavonoids
3. Materials and Methods
3.1. Plant Material
3.2. General Experimental Procedure
3.3. Extraction Procedures
3.4. HPLC-PDA Analysis of the Extracts and Fractions Bioactives
3.5. Purification of the Ethanolic Extracts of Inga Species
3.6. Description of the Isolated Compounds
3.7. Biological Assays
3.7.1. Cytotoxicity Assay
3.7.2. Antigenotoxicity Assessment
3.7.3. Proteomic Analysis
3.8. Quantification of Phenolic Compounds from Cytotoxic Extracts
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Agency for Research on Cancer. Latest Global Cancer Data: Cancer Burden Rises to 18.1 Million New Cases and 9.6 Million Cancer Deaths in 2018; World Health Organization: Geneva, Switzerland, 2018.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics. Ca-Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Amaral, R.G.; dos Santos, A.S.; Andrade, L.N.; Severino, P.; Carvalho, A.A. Natural products as treatment against cancer: A historical and current vision. Clin Oncol. 2019, 4, 1562. [Google Scholar]
- Lima, N.M.; Santos, V.N.C.; La Porta, F.A. Quimiodiversidade, Bioatividade e Quimiossistemática do Gênero Inga (FABACEAE): Uma Breve Revisão. Rev. Virtual Quim. 2018, 10, 459. [Google Scholar]
- Vivot, E.; Munoz, J.D.; Cruanes, M.D.C.; Cruanes, M.J.; Tapia, A.; Hirschmann, G.S.; Martinez, E.; Di Sapio, O.; Gattuso, M.; Zacchino, S. Inhibitory activity of xanthine-oxidase and superoxide scavenger properties of Inga verna subsp. affinis. Its morphological and micrographic characteristics. J. Ethnopharmacol. 2001, 76, 65. [Google Scholar]
- Silva, E.M.; Rogez, H.; Larondelle, Y. Optimization of extraction of phenolics from Inga edulis leaves using response surface methodology. Sep. Purif. Technol. 2007, 55, 381. [Google Scholar] [CrossRef]
- Lorenzi, H. Árvores Brasileiras: Manual de Identificação e cultivo de Plantas Arbóreas Nativas do Brasil, 2nd ed.; Instituto Plantarum: Nova Odessa, Brazil, 1998; p. 352. [Google Scholar]
- Edenharder, R.; Grünhage, D. Free radical scavenging abilities of flavonoids as mechanism of protection against mutagenicity induced by tert-butyl hydroperoxide or cumene hydroperoxide in Salmonella typhimurium TA102. Mutat. Res. 2003, 540, 1. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [Green Version]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2. [Google Scholar] [CrossRef] [Green Version]
- Saravanan, S.; Hairul Islam, V.I.; David, H.A.; Lakshmi Sundaram, R.; Chellappandian, M.; Balakrishna, K.; Ignacimuthu, S. Bioassay guided fractionation and identification of active anti-inflammatory constituent from Delonix elata flowers using RAW 264.7 cells. Pharm. Biol. 2015, 53, 174. [Google Scholar] [CrossRef]
- Sanzovo, T.O.R.; Lima, N.M.; Marqui, S.R.; Andrade, T.J.A.S.; Navegante, G.; Serafim, R.B.; Valente, V.; Silva, D.H.S.; Soares, C.P. Chemoprevention assessment, genotoxicity and cytotoxicity of flavonoids from Inga laurina leaves (FABACEAE). Nat. Prod. Res. 2021, 35, 3089. [Google Scholar] [CrossRef]
- Aourahoum, K.; Aissaoui, M.; Fazouane, F.; Larit, F.; Chalard, P.; Chalchat, J.C.; Figueredo, G.; Benayache, F.; Benayache, S. Essential oil of Cytisus triflorus L’Her. Pharm. Lett. 2013, 5, 276. [Google Scholar]
- Yang, D.P.; Ji, H.F.; Tang, G.Y.; Ren, W.; Zhang, H.Y. How many drugs are catecholics. Molecules 2007, 12, 878. [Google Scholar] [CrossRef]
- Matsubara, S.; Morosinotto, T.; Osmond, C.B.; Bassi, R. Short-and long-term operation of the lutein-epoxide cycle in light-harvesting antenna complexes. Plant Physiol. 2007, 144, 926–941. [Google Scholar] [CrossRef] [Green Version]
- Acton, Q.A. Patent issued for xanthophyll composition containing trans, meso-zeaxanthi, trans, R, R-zeaxanthin and trans, R, R-Lutein useful for nutrition and health care and a process for its preparation. In Carotenoids: Advances in Research and Application; ScholarlyEditions: Atlanta, GA, USA, 2013; Chapter 3; p. 79. [Google Scholar]
- Perveen, S.; Al-Taweel, A.M. Phenolic compounds from the natural sources and their cytotoxicity. In Phenolic Compounds Natural Sources, Importance and Applications; Soto-Hernandez, M., Palma Tenango, M., Del Rosario Garcia-Mateos, M., Eds.; Intech Open: London, UK, 2017; pp. 61–88. [Google Scholar]
- Csepregi, R.; Temesfői, V.; Das, S.; Alberti, Á.; Tóth, C.A.; Herczeg, R.; Papp, N.; Kőszegi, T. Cytotoxic, antimicrobial, antioxidant properties and effects on cell migration of phenolic compounds of selected transylvanian medicinal plants. Antioxidants 2020, 9, 166. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira Filho, J.W.G.; Dos Santos, T.D.J.A.; Tí, R.M.; Dos Reis, A.C.; Hameed, A.; de Oliveira Santos, J.V.; Cavalcante, A.A.D.C.M. Antitumor effects of citrinin in an animal model of Sarcoma 180 via cytogenetic mechanisms. Cell. Mol. Biol. 2020, 66, 120–126. [Google Scholar]
- Rezaei-Seresht, H.; Cheshomi, H.; Falanji, F.; Movahedi-Motlagh, F.; Hashemian, M.; Mireskandari, E. Cytotoxic activity of caffeic acid and gallic acid against MCF-7 human breast cancer cells: An in silico and in vitro study. Avicenna J. Phytomed. 2019, 9, 574. [Google Scholar]
- Spilioti, E.; Jaakkola, M.; Tolonen, T.; Lipponen, M.; Virtanen, V.; Chinou, I.; Kassi, E.; Moutsatsou, P. Phenolic acid composition, antiatherogenic and anticancer potential of honeys derived from various regions in Greece. PLoS ONE 2014, 9, e94860. [Google Scholar] [CrossRef] [Green Version]
- Intisar, A.; Zhang, L.; Luo, H.; Kiazolu, J.B.; Zhang, R.; Zhang, W. Anticancer Constituents and Cytotoxic Activity of Methanol-Water Extract of Polygonum bistorta L. Afr. J. Tradit. Complement. Altern. Med. 2013, 10, 53. [Google Scholar]
- El Molla, S.G.; Motaal, A.A.; El Hefnawy, H.; El Fishawy, A. Cytotoxic activity of phenolic constituents from Echinochloa crus-galli against four human cancer cell lines. Rev. Bras. Farmacogn. 2016, 26, 62. [Google Scholar] [CrossRef] [Green Version]
- Han, D.S.; Park, Y.K.; Kang, J.I.; Baek, S.H. The cytotoxic effect of benzoic acid and related compounds against oral cancer cell lines. Yakhak Hoeji 2008, 52, 252. [Google Scholar]
- Lalthanpuii, P.B.; Lalchhandama, K. Analysis of Chemical Constituents and Antiparasitic Activities of the Extracts of Imperata cylindrica. Res. J. Pharm. Technol. 2020, 13, 653. [Google Scholar] [CrossRef]
- Jeong, J.B.; Jeong, H.J. 2-Methoxy-4-vinylphenol can induce cell cycle arrest by blocking the hyper-phosphorylation of retinoblastoma protein in benzo [a] pyrene-treated NIH3T3 cells. Biochem. Biophys. Res. Commun. 2010, 400, 752. [Google Scholar] [CrossRef] [PubMed]
- Krych, J.; Gebicka, L. Catalase is inhibited by flavonoids. Int. J. Biol. Macromol. 2013, 58, 148–153. [Google Scholar] [CrossRef]
- Huang, Z.; Fang, F.; Wang, J.; Wong, C.-W. Structural activity relationship of flavonoids with estrogen-related receptor gamma. FEBS Lett. 2010, 584, 22–26. [Google Scholar] [CrossRef] [Green Version]
- Murthy, K.C.; Kim, J.; Vikram, A.; Patil, B.S. Differential inhibition of human colon cancer cells by structurally similar flavonoids of citrus. Food Chem. 2012, 132, 27–34. [Google Scholar] [CrossRef]
- Kothandan, G.; Gadhe, C.G.; Madhavan, T.; Choi, C.H.; Cho, S.J. Docking and 3D-QSAR (quantitative structure activity relationship) studies of flavones, the potent inhibitors of p-glycoprotein targeting the nucleotide binding domain. Eur. J. Med. Chem. 2011, 46, 4078–4088. [Google Scholar] [CrossRef]
- Amrutha, K.; Nanjan, P.; Shaji, S.K.; Sunilkumar, D.; Subhalakshmi, K.; Rajakrishna, L.; Banerji, A. Discovery of lesser known flavones as inhibitors of NF-κB signaling in MDA-MB-231 breast cancer cells—A SAR study. Bioorg. Med. Chem. Lett. 2014, 24, 4735–4742. [Google Scholar] [CrossRef]
- Ribani, M.; Bottoli, C.B.G.; Collins, C.H.; Jardim, I.C.S.F.; Melo, L.F.C. Validação em métodos cromatográficos e eletroforéticos. Quim Nova 2004, 27, 771–780. [Google Scholar] [CrossRef]
- Cavalheiro, A.J.; Siqueira Silva, D.H.; Da Silva Bolzani, V.; Castro-Gamboa, I.; De Araújo, A.J.; Pytel, R.F.; Gesztesi, J.-L.; Hurtado Medina, S.P.; Di Mambro, V.M.; Nakamura, M.S. Standardized Plant Extract, Method for Preparing an Extract from Plants of the Sclerolobium Genus, Cosmetic Composition, Pharmaceutical Composition and Use of Said Extract. Patent WO2011038472A8, 11 August 2011. [Google Scholar]
- Tavender, T.J.; Bulleid, N.J. Molecular mechanisms regulating oxidative activity of the Ero1 family in the endoplasmic reticulum. Antioxid. Redox. Signal. 2010, 13, 1177. [Google Scholar] [CrossRef] [Green Version]
- Bechtel, T.J.; Weerapana, E. From structure to redox: The diverse functional roles of disulfides and implications in disease. Proteomics 2017, 17, 1600391. [Google Scholar] [CrossRef] [Green Version]
- Pablo, Y.; Nilsson, M.; Pekna, M.; Pekny, M. Intermediate filaments are important for astrocyte response to oxidative stress induced by oxygen-glucose deprivation and reperfusion. Histochem. Cell Biol. 2013, 140, 81. [Google Scholar] [CrossRef]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005, 19, 316. [Google Scholar] [CrossRef] [Green Version]
- Liang, D.; Burkhart, S.L.; Singh, R.K.; Kabbaj, M.H.M.; Gunjan, A. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Nucleic Acids Res. 2012, 40, 9604. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.N.Y.; Oka, T.; Ito, H.; Tamura, T.; Tagawa, K.; Sasabe, T.; Katsuta, A.; Motoki, K.; Shiwaku, H.; Sone, M.; et al. A functional deficiency of TERA/VCP/p97 contributes to impaired DNA repair in multiple polyglutamine diseases. Nat. Commun. 2013, 4, 1816. [Google Scholar] [CrossRef] [Green Version]
- Sekhar, K.R.; Reddy, Y.T.; Reddy, P.N.; Crooks, P.A.; Venkateswaran, A.; McDonald, W.H.; Geng, L.; Sasi, S.; Waal, R.P.V.D.; Roti, J.L.R.; et al. The novel chemical entity YTR107 inhibits recruitment of nucleophosmin to sites of DNA damage, suppressing repair of DNA double-strand breaks and enhancing radiosensitization. Clin. Cancer Res. 2011, 17, 6490. [Google Scholar] [CrossRef] [Green Version]
- Moller, P. Genotoxicity of environmental agents assessed by the alkaline comet assay. Basic Clin. Pharm. Toxico.l 2005, 96, 1–42. [Google Scholar]
- Izumi, C.; Laure, H.J.; Barbosa, N.G.; Hassibe Thomé, C.; Aguiar Ferreira, G.; Paulo Barreto Sousa, J.; Lopes, N.P.; César Rosa, J. Sequesterpene lactones isolated from a Brazilian cerrado plant (eremanthus spp.) as anti-proliferative compounds, characterized by functional and proteomic analysis, are candidates for new therapeutics in glioblastoma. Int. J. Mol. Sci. 2020, 21, 4713. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Extracts | I. laurina | I. edulis | I. marginata |
|---|---|---|---|
| Branches | >20 | >20 | 13.8 |
| Flowers | >20 | - | - |
| Fruit peel | >20 | - | |
| Fruit pulp | >20 | - | - |
| Seeds | 15.2 | - | - |
| Doxorubicin | 1.25 | ||
| Extracts | I. laurina | I. edulis | I. marginata |
|---|---|---|---|
| Branches | >20 | >20 | >20 |
| Flowers | >20 | >20 | - |
| Fruit pulp | >20 | >20 | - |
| Fruit peel | >20 | 18.6 | - |
| Seeds | 14.4 | >20 | - |
| Doxorubicin | 2.10 | ||
| Compound | Line Equation y = ax + b | R2 | LOD | LOQ | DP |
|---|---|---|---|---|---|
| 2 | y = 963,313.28x + 14,384.16 | 0.992 | 3.27 × 10−3 | 1.09 × 10−2 | 4500.44 |
| 6 | y = 999,970.55x + 20,108.94 | 0.995 | 0.045 | 0.15 | 14,533.01 |
| 3 | y = 523,4447.0x + 18,893.8 | 0.994 | 0.01 | 0.04 | 19,471.80 |
| 7 | y = 3,410,900.0x + 10,0491.6 | 0.991 | 0.09 | 0.30 | 10,1824.1 |
| Myricetin-3-Rhamnoside | |||
|---|---|---|---|
| Prot Acc | Protein | Score | 116/114 |
| K6PP_HUMAN | 6-phosphofructokinase type C OS = Homo sapiens GN = PFKP PE = 1 SV = 2 | 208 | 0.00 |
| CH60_HUMAN | 60 kDa heat shock protein, mitochondrial OS = Homo sapiens GN = HSPD1 PE = 1 SV = 2 | 141 | 0.00 |
| G6PI_HUMAN | Glucose-6-phosphate isomerase OS = Homo sapiens GN = GPI PE = 1 SV = 4 | 82 | 0.00 |
| CYB5B_HUMAN | Cytochrome b5 type B OS = Homo sapiens GN = CYB5B PE = 1 SV = 2 | 44 | 0.00 |
| QCR6_HUMAN | Cytochrome b-c1 complex subunit 6, mitochondrial OS = Homo sapiens GN = UQCRH PE = 1 SV = 2 | 38 | 0.00 |
| ST1A3_HUMAN | Sulfotransferase 1A3/1A4 OS = Homo sapiens GN = SULT1A3 PE = 1 SV = 1 | 37 | 0.00 |
| NQO1_HUMAN | NAD(P)H dehydrogenase [quinone] 1 OS = Homo sapiens GN = NQO1 PE = 1 SV = 1 | 45 | 0.20 |
| ALDR_HUMAN | Aldose reductase OS = Homo sapiens GN = AKR 1B1 PE = 1 SV = 3 | 65 | 0.22 |
| ERO1A_HUMAN | ERO1-like protein alpha OS = Homo sapiens GN = ERO1L PE = 1 SV = 2 | 36 | 0.27 |
| CHSP1_HUMAN | Calcium-regulated heat stable protein 1 OS = Homo sapiens GN = CARHSP1 PE = 1 SV = 2 | 60 | 0.28 |
| ANXA2_HUMAN | Annexin A2 OS = Homo sapiens GN = ANXA2 PE = 1 SV = 2 | 185 | 0.45 |
| TERA_HUMAN | Transitational endoplasmic reticulum ATPase OS = Homo sapiens GN = VCP PE = 1 SV = 4 | 86 | 0.48 |
| FLNA_HUMAN | Filamin-A OS = Homo sapiens GN = FLNA PE = 1 SV = 1 | 48 | 0.49 |
| PROF1_HUMAN | Profilin-1 OS = Homo sapiens GN = PFN1 PE = 1 SV = 2 | 48 | 0.49 |
| PSA7_HUMAN | Proteasome subunit alpha type-7 OS = Homo sapiens GN = PSMA7 PE = 1 SV = 1 | 54 | 1.66 |
| VIME_HUMAN | Vimentin OS = Homo sapiens GN = VIM PE = 1 SV = 4 | 278 | 1.68 |
| GFAP_HUMAN | Glial fibrillary acidic protein OS = Homo sapiens GN = GFAP PE = 1 SV = 1 | 46 | 1.71 |
| PDIA1_HUMAN | Protein disulfide-isomerase OS = Homo sapiens GN = P4HB PE = 1 SV = 3 | 42 | 1.79 |
| myricetin-3-O-(2″-O-galloyl)-α rhamnopyranoside | |||
| Prot Acc | Protein | Score | 117/114 |
| TPIS_HUMAN | Triosephosphate isomerase OS = Homo sapiens GN = TPI1 PE = 1 SV = 3 | 105 | 0.00 |
| G6PI_HUMAN | Glucose-6-phosphate isomerase OS = Homo sapiens GN = GPI PE = 1 SV = 4 | 82 | 0.00 |
| TYPH_HUMAN | Thymidine phos’phorylase OS = Homo sapiens GN = TYMP PE = 1 SV = 2 | 59 | 0.00 |
| PSA7_HUMAN | Proteasome subunit alpha type-7 OS = Homo sapiens GN = PSMA7 PE = 1 SV = 1 | 54 | 0.00 |
| H4_HUMAN | Histone H4 OS = Homo sapiens GN = HIST1H4A PE = 1 SV = 2 | 36 | 0.00 |
| FLNA_HUMAN | Filamin-A OS = Homo sapiens GN = FLNA PE = 1 SV = 4 | 48 | 0.09 |
| PROF1_HUMAN | Profilin-1 OS = Homo sapiens GN = PFN1 PE = 1 SV = 2 | 48 | 0.20 |
| MVP_HUMAN | Major vault protein OS = Homo sapiens GN = MVP PE = 1 SV = 4 | 80 | 0.36 |
| CHSP1_HUMAN | Calcium-regulated heat stable protein 1 OS = Homo sapiens GN = CARHSP1 PE = 1 SV = 2 | 60 | 0.39 |
| PYGB_HUMAN | Glycogen phosphorylase, brain form OS = Homo sapiens GN = PYGB PE = 1 SV = 5 | 39 | 0.43 |
| TBB3_HUMAN | Tubulin beta-3 chain OS = Homo sapiens GN = TUBB3 PE = 1 SV = 2 | 160 | 0.45 |
| BLVRB_HUMAN | Flavin reductase (NADPH) OS = Homo sapiens GN = BLVRB PE = 1 SV = 3 | 85 | 0.50 |
| CYB5B_HUMAN | Cytochrome b5 type B OS = Homo sapiens GN = CYB5B PE = 1 SV = 2 | 44 | 1.65 |
| ERO1A_HUMAN | ERO1-like protein alpha OS = Homo sapiens GN = ERO1L PE = 1 SV = 2 | 36 | 1.72 |
| MYOF_HUMAN | Myoferlin OS = Homo sapiens GN = MYOF PE = 1 SV = 1 | 61 | 1.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, N.M.; Preet, G.; Marqui, S.R.; Falcoski, T.d.O.R.S.; Navegante, G.; Soares, C.P.; Andrade, T.d.J.A.S.; La Porta, F.A.; Rakotondraie, H.L.R.; Jaspars, M.; et al. Metabolic Profiling of Inga Species with Antitumor Activity. Molecules 2022, 27, 4695. https://doi.org/10.3390/molecules27154695
Lima NM, Preet G, Marqui SR, Falcoski TdORS, Navegante G, Soares CP, Andrade TdJAS, La Porta FA, Rakotondraie HLR, Jaspars M, et al. Metabolic Profiling of Inga Species with Antitumor Activity. Molecules. 2022; 27(15):4695. https://doi.org/10.3390/molecules27154695
Chicago/Turabian StyleLima, Nerilson M., Gagan Preet, Sara R. Marqui, Thaís de O. R. S. Falcoski, Geovana Navegante, Christiane P. Soares, Teresinha de Jesus A. S. Andrade, Felipe A. La Porta, Harinantenaina Liva R. Rakotondraie, Marcel Jaspars, and et al. 2022. "Metabolic Profiling of Inga Species with Antitumor Activity" Molecules 27, no. 15: 4695. https://doi.org/10.3390/molecules27154695
APA StyleLima, N. M., Preet, G., Marqui, S. R., Falcoski, T. d. O. R. S., Navegante, G., Soares, C. P., Andrade, T. d. J. A. S., La Porta, F. A., Rakotondraie, H. L. R., Jaspars, M., & Silva, D. H. S. (2022). Metabolic Profiling of Inga Species with Antitumor Activity. Molecules, 27(15), 4695. https://doi.org/10.3390/molecules27154695