
Targeting Replication Stress Response Pathways to Enhance Genotoxic Chemo- and Radiotherapy

Abstract
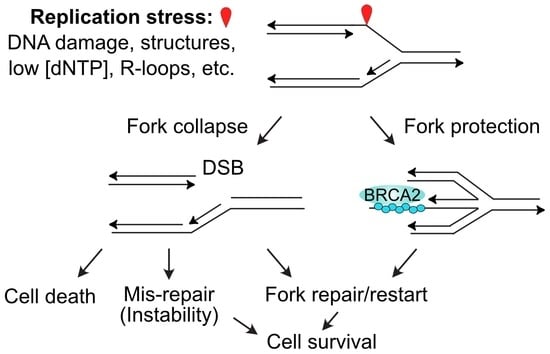
:
1. Introduction
“…regardless of the mechanism of administered therapies (either targeted agents, chemotherapy, or immunotherapy), resistance is a near-universal occurrence.”[5]
2. Key Features of the Cellular DNA Damage Response
3. Sources of DNA Replication Stress
4. Repair and Restart of Stressed Replication Forks
5. Targeting Replication Stress Signaling and Fork Repair/Restart Pathways in Cancer Therapy
5.1. Targeting Downstream DNA Damage Checkpoint Factors (Chk1, Chk2, Wee1)
5.2. Targeting the Upstream DNA Damage Checkpoint Kinase ATR and Its Activation Partner TopBP1
5.3. Targeting Replication Stress Nucleases (CtIP, MUS81, EEPD1, Metnase)
5.4. Targeting DSB Repair Proteins (RAD51, PARP1)
5.5. Exploiting Synthetic Lethality of TKIs Targeting Activated Oncogenes and ATM Inhibitors
6. Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Fattore, L.; Mancini, R.; Ciliberto, G. Cancer stem cells and the slow cycling phenotype: How to cut the gordian knot driving resistance to therapy in melanoma. Cancers 2020, 12, 3368. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Dong, Y.; Kumar, R.; Jeter, C.; Tang, D.G. Slow-cycling (dormant) cancer cells in therapy resistance, cancer relapse and metastasis. Semin. Cancer Biol. 2021, 78, 90–103. [Google Scholar] [CrossRef]
- Tang, H.; Shrager, J.B. CRISPR /Cas-mediated genome editing to treat EGFR -mutant lung cancer: A personalized molecular surgical therapy. EMBO Mol. Med. 2016, 8, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Loeb, L.A. Human cancers express a mutator phenotype: Hypothesis, origin, and consequences. Cancer Res. 2016, 76, 2057–2059. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, M.W.; Loeb, L.A.; Salk, J.J. The influence of subclonal resistance mutations on targeted cancer therapy. Nat. Rev. Clin. Oncol. 2016, 13, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020, 135, 41–55. [Google Scholar] [CrossRef]
- Gerlinger, M. Targeted drugs ramp up cancer mutability. Science 2019, 366, 1452–1453. [Google Scholar] [CrossRef]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magrì, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Fitzgerald, D.M.; Hastings, P.; Rosenberg, S.M. Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance. Annu. Rev. Cancer Biol. 2017, 1, 119–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daguenet, E.; Louati, S.; Wozny, A.-S.; Vial, N.; Gras, M.; Guy, J.-B.; Vallard, A.; Rodriguez-Lafrasse, C.; Magné, N. Radiation-induced bystander and abscopal effects: Important lessons from preclinical models. Br. J. Cancer 2020, 123, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Reynders, K.; Illidge, T.; Siva, S.; Chang, J.Y.; De Ruysscher, D. The abscopal effect of local radiotherapy: Using immunotherapy to make a rare event clinically relevant. Cancer Treat. Rev. 2015, 41, 503–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Dong, Y.; Kong, L.; Shi, F.; Zhu, H.; Yu, J. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Willers, H.; Azzoli, C.G.; Santivasi, W.; Xia, F. Basic mechanisms of therapeutic resistance to radiation and chemotherapy in lung cancer. Cancer J. 2013, 19, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Nickoloff, J.A.; Sharma, N.; Allen, C.P.; Taylor, L.; Allen, S.J.; Jaiswal, A.S.; Hromas, R. Roles of homologous recombination in response to ionizing radiation-induced DNA damage. Int. J. Radiat. Biol. 2021, 1–12. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Sharma, N.; Taylor, L. Clustered dna double-strand breaks: Biological effects and relevance to cancer radiotherapy. Genes 2020, 11, 99. [Google Scholar] [CrossRef] [Green Version]
- Trenner, A.; Sartori, A.A. Harnessing DNA double-strand break repair for cancer treatment. Front. Oncol. 2019, 9, 1388. [Google Scholar] [CrossRef]
- Hsieh, H.-J.; Peng, G. Cellular responses to replication stress: Implications in cancer biology and therapy. DNA Repair 2017, 49, 9–20. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Sharma, N.; Taylor, L.; Allen, S.J.; Hromas, R. The safe path at the fork: Ensuring replication-associated DNA double-strand breaks are repaired by homologous recombination. Front. Genet. 2021, 12, 748033. [Google Scholar] [CrossRef] [PubMed]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [Green Version]
- Nickoloff, J.A. Recombination: Mechanisms and roles in tumorigenesis. In Encyclopedia of Cancer, 2nd ed.; Bertino, J.R., Ed.; Elsevier Science (USA): San Diego, CA, USA, 2002; Volume 4, pp. 49–59. [Google Scholar]
- Tutt, A.; Bertwistle, D.; Valentine, J.; Gabriel, A.; Swift, S.; Ross, G.; Griffin, C.; Thacker, J.; Ashworth, A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001, 20, 4704–4716. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Hills, S.A.; Diffley, J.F. DNA Replication and Oncogene-Induced Replicative Stress. Curr. Biol. 2014, 24, R435–R444. [Google Scholar] [CrossRef] [Green Version]
- Primo, L.M.F.; Teixeira, L.K. DNA replication stress: Oncogenes in the spotlight. Genet. Mol. Biol. 2020, 43, e20190138. [Google Scholar] [CrossRef]
- Li, L.-Y.; Guan, Y.-D.; Chen, X.-S.; Yang, J.-M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11, 629266. [Google Scholar] [CrossRef] [PubMed]
- Vesela, E.; Chroma, K.; Turi, Z.; Mistrik, M. Common Chemical Inductors of Replication Stress: Focus on Cell-Based Studies. Biomolecules 2017, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Ward, J. Complexity of Damage Produced by Ionizing Radiation. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Groth, P.; Orta, M.L.; Elvers, I.; Majumder, M.M.; Lagerqvist, A.; Helleday, T. Homologous recombination repairs secondary replication induced DNA double-strand breaks after ionizing radiation. Nucleic Acids Res. 2012, 40, 6585–6594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V.G. DNA Damage Response and Autophagy: A Meaningful Partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-H.; Tseng, W.H.-S.; Chi, J.-T. The Intersection of DNA Damage Response and Ferroptosis—A Rationale for Combination Therapeutics. Biology 2020, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Jette, N.; Lees-Miller, S.P. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 2014, 117, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.P.C.; Uematsu, N.; Kobayashi, J.; Lerenthal, Y.; Krempler, A.; Yajima, H.; Löbrich, M.; Shiloh, Y.; Chen, D.J. Ataxia Telangiectasia Mutated (ATM) Is Essential for DNA-PKcs Phosphorylations at the Thr-2609 Cluster upon DNA Double Strand Break. J. Biol. Chem. 2007, 282, 6582–6587. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Crowe, J.L.; Liu, X.; Nakajima, S.; Wang, Y.; Li, C.; Lee, B.J.; Dubois, R.L.; Liu, C.; Yu, X.; et al. Differential Phosphorylation of DNA-PKcs Regulates the Interplay between End-Processing and End-Ligation during Nonhomologous End-Joining. Mol. Cell 2015, 58, 172–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yajima, H.; Lee, K.-J.; Chen, B.P.C. ATR-dependent DNA-PKcs phosphorylation in response to UV-induced replication stress. Mol. Cell. Biol. 2006, 26, 7520–7528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickoloff, J.A.; Sharma, N.; Taylor, L.; Allen, S.J.; Hromas, R. Nucleases and Co-Factors in DNA Replication Stress Responses. DNA 2022, 2, 68–85. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2014, 25, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Rickman, K.; Smogorzewska, A. Advances in understanding DNA processing and protection at stalled replication forks. J. Cell Biol. 2019, 218, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickman, K.A.; Noonan, R.J.; Lach, F.; Sridhar, S.; Wang, A.; Abhyankar, A.; Huang, A.; Kelly, M.; Auerbach, A.D.; Smogorzewska, A. Distinct roles of BRCA2 in replication fork protection in response to hydroxyurea and DNA interstrand cross-links. Genes Dev. 2020, 34, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Zou, L. ATR: A master conductor of cellular responses to DNA replication stress. Trends Biochem. Sci. 2011, 36, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.M.; Zhang, X. Roles of ATM and ATR in DNA double strand breaks and replication stress. Prog. Biophys. Mol. Biol. 2021, 163, 109–119. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Kozlov, S.V.; Graham, M.E.; Peng, C.; Chen, P.; Robinson, P.J.; Lavin, M.F. Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 2006, 25, 3504–3514. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.V.; Graham, M.; Jakob, B.; Tobias, F.; Kijas, A.W.; Tanuji, M.; Chen, P.; Robinson, P.J.; Taucher-Scholz, G.; Suzuki, K.; et al. Autophosphorylation and ATM Activation: Additional sites add to the complexity. J. Biol. Chem. 2011, 286, 9107–9119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupré, A.; Boyer-Chatenet, L.; Gautier, J. Two-step activation of ATM by DNA and the Mre11–Rad50–Nbs1 complex. Nat. Struct. Mol. Biol. 2006, 13, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Paull, T.T. ATM Activation by DNA Double-Strand Breaks Through the Mre11-Rad50-Nbs1 Complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.A.; Pellegrini, M.; Lee, J.-H.; Paull, T.T.; Feigenbaum, L.; Nussenzweig, A. Multiple autophosphorylation sites are dispensable for murine ATM activation in vivo. J. Cell Biol. 2008, 183, 777–783. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, M.; Celeste, A.; Difilippantonio, S.; Guo, R.; Wang, W.; Feigenbaum, L.; Nussenzweig, A. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature 2006, 443, 222–225. [Google Scholar] [CrossRef]
- Chen, B.P.C.; Chan, D.W.; Kobayashi, J.; Burma, S.; Asaithamby, A.; Morotomi-Yano, K.; Botvinick, E.; Qin, J.; Chen, D.J. Cell Cycle Dependence of DNA-dependent Protein Kinase Phosphorylation in Response to DNA Double Strand Breaks. J. Biol. Chem. 2005, 280, 14709–14715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.W.; Chen, B.P.-C.; Prithivirajsingh, S.; Kurimasa, A.; Story, M.D.; Qin, J.; Chen, D.J. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002, 16, 2333–2338. [Google Scholar] [CrossRef] [Green Version]
- Haahr, P.; Hoffmann, S.; Tollenaere, M.A.X.; Ho, T.; Toledo, L.I.; Mann, M.; Bekker-Jensen, S.; Räschle, M.; Mailand, N. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 2016, 18, 1196–1207. [Google Scholar] [CrossRef] [Green Version]
- Nam, E.A.; Cortez, D. ATR signalling: More than meeting at the fork. Biochem. J. 2011, 436, 527–536. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldivar, J.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thada, V.; Cortez, D. ATR activation is regulated by dimerization of ATR activating proteins. J. Biol. Chem. 2021, 296, 100455. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G 2 checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bass, T.E.; Cortez, D. Quantitative phosphoproteomics reveals mitotic function of the ATR activator ETAA1. J. Cell Biol. 2019, 218, 1235–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, X.; Chastain, M.; Chai, W. Genome-wide mapping and profiling of γH2AX binding hotspots in response to different replication stress inducers. BMC Genom. 2019, 20, 579. [Google Scholar] [CrossRef] [Green Version]
- Mazouzi, A.; Stukalov, A.; Müller, A.C.; Chen, D.; Wiedner, M.; Prochazkova, J.; Chiang, S.-C.; Schuster, M.; Breitwieser, F.P.; Pichlmair, A.; et al. A Comprehensive Analysis of the Dynamic Response to Aphidicolin-Mediated Replication Stress Uncovers Targets for ATM and ATMIN. Cell Rep. 2016, 15, 893–908. [Google Scholar] [CrossRef] [Green Version]
- Sarmiento-Salinas, F.L.; Perez-Gonzalez, A.; Acosta-Casique, A.; Ix-Ballote, A.; Diaz, A.; Treviño, S.; Rosas-Murrieta, N.H.; Millán-Perez-Peña, L.; Maycotte, P. Reactive oxygen species: Role in carcinogenesis, cancer cell signaling and tumor progression. Life Sci. 2021, 284, 119942. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, D.E.; Lindahl, T. Repair and Genetic Consequences of Endogenous DNA Base Damage in Mammalian Cells. Annu. Rev. Genet. 2004, 38, 445–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, A.R.; Yousefzadeh, M.J.; Rozgaja, T.A.; Wang, J.; Li, X.; Tilstra, J.S.; Feldman, C.H.; Gregg, S.Q.; Johnson, C.H.; Skoda, E.M.; et al. Spontaneous DNA damage to the nuclear genome promotes senescence, redox imbalance and aging. Redox Biol. 2018, 17, 259–273. [Google Scholar] [CrossRef]
- Billard, P.; Poncet, D.A. Replication Stress at Telomeric and Mitochondrial DNA: Common Origins and Consequences on Ageing. Int. J. Mol. Sci. 2019, 20, 4959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef]
- Gómez-González, B.; Aguilera, A. Transcription-mediated replication hindrance: A major driver of genome instability. Genes Dev. 2019, 33, 1008–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamperl, S.; Cimprich, K.A. Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 2016, 167, 1455–1467. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Muse, T.; Aguilera, A. Transcription–replication conflicts: How they occur and how they are resolved. Nat. Rev. Mol. Cell Biol. 2016, 17, 553–563. [Google Scholar] [CrossRef]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017, 170, 774–786.e19. [Google Scholar] [CrossRef] [Green Version]
- Freudenreich, C.H. R-loops: Targets for nuclease cleavage and repeat instability. Curr. Genet. 2018, 64, 789–794. [Google Scholar] [CrossRef]
- Poggi, L.; Richard, G.-F. Alternative DNA Structures In Vivo: Molecular Evidence and Remaining Questions. Microbiol. Mol. Biol. Rev. 2021, 85, e00110-20. [Google Scholar] [CrossRef]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, S.; Freudenreich, C.H. The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosom. Cancer 2018, 58, 270–283. [Google Scholar] [CrossRef] [Green Version]
- Gadaleta, M.C.; Noguchi, E. Regulation of DNA Replication through Natural Impediments in the Eukaryotic Genome. Genes 2017, 8, 98. [Google Scholar] [CrossRef] [Green Version]
- Berti, M.; Chaudhuri, A.R.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Emanuelli, M.; Sartini, D.; Molinelli, E.; Campagna, R.; Pozzi, V.; Salvolini, E.; Simonetti, O.; Campanati, A.; Offidani, A. The Double-Edged Sword of Oxidative Stress in Skin Damage and Melanoma: From Physiopathology to Therapeutical Approaches. Antioxidants 2022, 11, 612. [Google Scholar] [CrossRef]
- Yang, W. An Overview of Y-Family DNA Polymerases and a Case Study of Human DNA Polymerase η. Biochemistry 2014, 53, 2793–2803. [Google Scholar] [CrossRef]
- Ler, A.A.L.; Carty, M.P. DNA Damage Tolerance Pathways in Human Cells: A Potential Therapeutic Target. Front. Oncol. 2021, 11, 822500. [Google Scholar] [CrossRef]
- Goodman, M.F.; Woodgate, R. Translesion DNA Polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a010363. [Google Scholar] [CrossRef]
- Ma, X.; Tang, T.; Guo, C. Regulation of translesion DNA synthesis in mammalian cells. Environ. Mol. Mutagen. 2020, 61, 680–692. [Google Scholar] [CrossRef]
- Sale, J.E. Translesion DNA Synthesis and Mutagenesis in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: What makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef] [Green Version]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base excision repair and cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donigan, K.A.; Sun, K.-W.; Nemec, A.A.; Murphy, D.L.; Cong, X.; Northrup, V.; Zelterman, D.; Sweasy, J.B. Human POLB Gene Is Mutated in High Percentage of Colorectal Tumors. J. Biol. Chem. 2012, 287, 23830–23839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makridakis, N.M.; Reichardt, J.K.V. Translesion DNA Polymerases and Cancer. Front. Genet. 2012, 3, 174. [Google Scholar] [CrossRef] [Green Version]
- Quinet, A.; Tirman, S.; Cybulla, E.; Meroni, A.; Vindigni, A. To skip or not to skip: Choosing repriming to tolerate DNA damage. Mol. Cell 2021, 81, 649–658. [Google Scholar] [CrossRef]
- Conti, B.A.; Smogorzewska, A. Mechanisms of direct replication restart at stressed replisomes. DNA Repair 2020, 95, 102947. [Google Scholar] [CrossRef]
- Liu, W.; Krishnamoorthy, A.; Zhao, R.; Cortez, D. Two replication fork remodeling pathways generate nuclease substrates for distinct fork protection factors. Sci. Adv. 2020, 6, eabc3598. [Google Scholar] [CrossRef]
- Mukherjee, C.; Tripathi, V.; Manolika, E.M.; Heijink, A.M.; Ricci, G.; Merzouk, S.; De Boer, H.R.; Demmers, J.; Van Vugt, M.A.T.M.; Chaudhuri, A.R. RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat. Commun. 2019, 10, 3287. [Google Scholar] [CrossRef]
- Bhat, K.P.; Krishnamoorthy, A.; Dungrawala, H.; Garcin, E.B.; Modesti, M.; Cortez, D. RADX Modulates RAD51 Activity to Control Replication Fork Protection. Cell Rep. 2018, 24, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Pepe, A.; West, S.C. MUS81-EME2 Promotes Replication Fork Restart. Cell Rep. 2014, 7, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Pepe, A.; West, S.C. Substrate specificity of the MUS81-EME2 structure selective endonuclease. Nucleic Acids Res. 2013, 42, 3833–3845. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-S.; Nickoloff, J.A.; Wu, Y.; Williamson, E.A.; Sidhu, G.S.; Reinert, B.L.; Jaiswal, A.S.; Srinivasan, G.; Patel, B.; Kong, K.; et al. Endonuclease EEPD1 Is a Gatekeeper for Repair of Stressed Replication Forks. J. Biol. Chem. 2017, 292, 2795–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Speed, M.C.; Allen, C.P.; Maranon, D.G.; Williamson, E.; Singh, S.; Hromas, R.; Nickoloff, J.A. Distinct roles of structure-specific endonucleases EEPD1 and Metnase in replication stress responses. NAR Cancer 2020, 2, zcaa008. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lee, S.-H.; Williamson, E.A.; Reinert, B.L.; Cho, J.H.; Xia, F.; Jaiswal, A.S.; Srinivasan, G.; Patel, B.; Brantley, A.; et al. EEPD1 Rescues Stressed Replication Forks and Maintains Genome Stability by Promoting End Resection and Homologous Recombination Repair. PLoS Genet. 2015, 11, e1005675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Interthal, H.; Heyer, W.-D. MUS81 encodes a novel Helix-hairpin-Helix protein involved in the response to UV- and methylation-induced DNA damage in Saccharomyces cerevisiae. Mol. Gen. Genet. 2000, 263, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Sweasy, J.B. DNA polymerase κ: Friend or foe? Sci. Signal. 2020, 13, eabb2934. [Google Scholar] [CrossRef]
- Temprine, K.; Campbell, N.R.; Huang, R.; Langdon, E.M.; Simon-Vermot, T.; Mehta, K.; Clapp, A.; Chipman, M.; White, R.M. Regulation of the error-prone DNA polymerase Polκ by oncogenic signaling and its contribution to drug resistance. Sci. Signal. 2020, 13, eaau1453. [Google Scholar] [CrossRef]
- Nayak, S.; Calvo, J.A.; Cong, K.; Peng, M.; Berthiaume, E.; Jackson, J.; Zaino, A.M.; Vindigni, A.; Hadden, M.K.; Cantor, S.B. Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci. Adv. 2020, 6, eaaz7808. [Google Scholar] [CrossRef]
- Nayak, S.; Calvo, J.A.; Cantor, S.B. Targeting translesion synthesis (TLS) to expose replication gaps, a unique cancer vulnerability. Expert Opin. Ther. Targets 2021, 25, 27–36. [Google Scholar] [CrossRef]
- Tonzi, P.; Huang, T.T. Role of Y-family translesion DNA polymerases in replication stress: Implications for new cancer therapeutic targets. DNA Repair 2019, 78, 20–26. [Google Scholar] [CrossRef]
- Saha, P.; Mandal, T.; Talukdar, A.D.; Kumar, D.; Kumar, S.; Tripathi, P.P.; Wang, Q.; Srivastava, A.K. DNA polymerase eta: A potential pharmacological target for cancer therapy. J. Cell. Physiol. 2020, 236, 4106–4120. [Google Scholar] [CrossRef]
- Wilson, D.M.; Duncton, M.A.J.; Chang, C.; Luo, C.L.; Georgiadis, T.M.; Pellicena, P.; Deacon, A.M.; Gao, Y.; Das, D. Early Drug Discovery and Development of Novel Cancer Therapeutics Targeting DNA Polymerase Eta (POLH). Front. Oncol. 2021, 11, 778925. [Google Scholar] [CrossRef]
- Schrempf, A.; Slyskova, J.; Loizou, J.I. Targeting the DNA Repair Enzyme Polymerase θ in Cancer Therapy. Trends Cancer 2020, 7, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszek, J.L.; Chatterjee, N.; Najeeb, J.; Ramos, A.; Lee, M.; Bian, K.; Xue, J.Y.; Fenton, B.A.; Park, H.; Li, D.; et al. A Small Molecule Targeting Mutagenic Translesion Synthesis Improves Chemotherapy. Cell 2019, 178, 152–159.e11. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Chatterjee, N.; Hemann, M.T.; Walker, G.C. Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy? PLoS Genet. 2017, 13, e1006842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuse, E.; Tanii, H.; Kurata, N.; Kobayashi, H.; Shimada, Y.; Tamura, T.; Sasaki, Y.; Tanigawara, Y.; Lush, R.D.; Headlee, D.; et al. Unpredicted clinical pharmacology of UCN-01 caused by specific binding to human alpha1-acid glycoprotein. Cancer Res. 1998, 58, 3248–3253. [Google Scholar]
- Thompson, R.; Eastman, A. The cancer therapeutic potential of Chk1 inhibitors: How mechanistic studies impact on clinical trial design. Br. J. Clin. Pharmacol. 2013, 76, 358–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italiano, A.; Infante, J.; Shapiro, G.; Moore, K.; LoRusso, P.; Hamilton, E.; Cousin, S.; Toulmonde, M.; Postel-Vinay, S.; Tolaney, S.; et al. Phase I study of the checkpoint kinase 1 inhibitor GDC-0575 in combination with gemcitabine in patients with refractory solid tumors. Ann. Oncol. 2018, 29, 1304–1311. [Google Scholar] [CrossRef]
- Webster, J.A.; Tibes, R.; Morris, L.; Blackford, A.L.; Litzow, M.; Patnaik, M.; Rosner, G.L.; Gojo, I.; Kinders, R.; Wang, L.; et al. Randomized phase II trial of cytosine arabinoside with and without the CHK1 inhibitor MK-8776 in relapsed and refractory acute myeloid leukemia. Leuk. Res. 2017, 61, 108–116. [Google Scholar] [CrossRef]
- Byers, L.A.; Navarro, A.; Schaefer, E.; Johnson, M.; Özgüroğlu, M.; Han, J.-Y.; Bondarenko, I.; Cicin, I.; Dragnev, K.H.; Abel, A.; et al. A Phase II Trial of Prexasertib (LY2606368) in Patients with Extensive-Stage Small-Cell Lung Cancer. Clin. Lung Cancer 2021, 22, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Cash, T.; Fox, E.; Liu, X.; Minard, C.G.; Reid, J.M.; Scheck, A.C.; Weigel, B.J.; Wetmore, C. A phase 1 study of prexasertib (LY2606368), a CHK1/2 inhibitor, in pediatric patients with recurrent or refractory solid tumors, including CNS tumors: A report from the Children’s Oncology Group Pediatric Early Phase Clinical Trials Network (ADVL1515). Pediatr. Blood Cancer 2021, 68, e29065. [Google Scholar] [CrossRef] [PubMed]
- Gatti-Mays, M.E.; Karzai, F.H.; Soltani, S.N.; Zimmer, A.; Green, J.E.; Lee, M.-J.; Trepel, J.B.; Yuno, A.; Lipkowitz, S.; Nair, J.; et al. A Phase II Single Arm Pilot Study of the CHK1 Inhibitor Prexasertib (LY2606368) in BRCA Wild-Type, Advanced Triple-Negative Breast Cancer. Oncologist 2020, 25, 1013–e1824. [Google Scholar] [CrossRef]
- Hong, D.; Infante, J.; Janku, F.; Jones, S.; Nguyen, L.M.; Burris, H.A.; Naing, A.; Bauer, T.M.; Piha-Paul, S.; Johnson, F.M.; et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients with Advanced Cancer. J. Clin. Oncol. 2016, 34, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Estevez-Diz, M.D.P.; Grischke, E.-M.; Hall, M.; Marmé, F.; Provencher, D.M.; Uyar, D.S.; Weberpals, J.I.; Wenham, R.M.; Laing, N.; et al. A Biomarker-enriched, Randomized Phase II Trial of Adavosertib (AZD1775) Plus Paclitaxel and Carboplatin for Women with Platinum-sensitive TP53-mutant Ovarian Cancer. Clin. Cancer Res. 2020, 26, 4767–4776. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Tan, D.S.P.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, G.B.C.V.; Haris, N.R.M.; Bashir, S.; Drew, Y.; et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Krebs, M.G.; Postel-Vinay, S.; El-Khouiery, A.; Soria, J.-C.; Lopez, J.; Berges, A.; Cheung, S.Y.A.; Irurzun-Arana, I.; Goldwin, A.; et al. Ceralasertib (AZD6738), an Oral ATR Kinase Inhibitor, in Combination with Carboplatin in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2021, 27, 5213–5224. [Google Scholar] [CrossRef]
- Chowdhury, P.; Lin, G.E.; Liu, K.; Song, Y.; Lin, F.-T.; Lin, W.-C. Targeting TopBP1 at a convergent point of multiple oncogenic pathways for cancer therapy. Nat. Commun. 2014, 5, 5476. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Liu, R.; Xie, S.; Zheng, X.; Mao, J.; Cai, Y.; Chen, W. Calcein-acetoxymethy ester enhances the antitumor effects of doxorubicin in nonsmall cell lung cancer by regulating the TopBP1/p53RR pathway. Anti-Cancer Drugs 2017, 28, 861–868. [Google Scholar] [CrossRef]
- Kuster, A.; Mozaffari, N.L.; Wilkinson, O.J.; Wojtaszek, J.L.; Zurfluh, C.; Przetocka, S.; Zyla, D.; von Aesch, C.; Dillingham, M.S.; Williams, R.S.; et al. A stapled peptide mimetic of the CtIP tetramerization motif interferes with double-strand break repair and replication fork protection. Sci. Adv. 2021, 7, eabc6381. [Google Scholar] [CrossRef]
- Zhang, C.; Zhou, B.; Gu, F.; Liu, H.; Wu, H.; Yao, F.; Zheng, H.; Fu, H.; Chong, W.; Cai, S.; et al. Micropeptide PACMP inhibition elicits synthetic lethal effects by decreasing CtIP and poly(ADP-ribosyl)ation. Mol. Cell 2022, 82, 1297–1312.e8. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, P.; Li, C.; Liu, W.; Shen, Q.; Yang, L.; Xie, G.; Bai, J.; Li, R.; Tao, K.; et al. MUS81 Inhibition Enhances the Anticancer Efficacy of Talazoparib by Impairing ATR/CHK1 Signaling Pathway in Gastric Cancer. Front. Oncol. 2022, 12, 844135. [Google Scholar] [CrossRef]
- Williamson, E.A.; Damiani, L.; Leitao, A.; Hu, C.; Hathaway, H.; Oprea, T.; Sklar, L.; Shaheen, M.; Bauman, J.; Wang, W.; et al. Targeting the Transposase Domain of the DNA Repair Component Metnase to Enhance Chemotherapy. Cancer Res. 2012, 72, 6200–6208. [Google Scholar] [CrossRef] [Green Version]
- Balbous, A.; Cortes, U.; Guilloteau, K.; Rivet, P.; Pinel, B.; Duchesne, M.; Godet, J.; Boissonnade, O.; Wager, M.; Bensadoun, R.J.; et al. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem-like cells. BMC Cancer 2016, 16, 604. [Google Scholar] [CrossRef] [Green Version]
- King, H.O.; Brend, T.; Payne, H.L.; Wright, A.; Ward, T.A.; Patel, K.; Egnuni, T.; Stead, L.F.; Patel, A.; Wurdak, H.; et al. RAD51 Is a Selective DNA Repair Target to Radiosensitize Glioma Stem Cells. Stem Cell Rep. 2017, 8, 125–139. [Google Scholar] [CrossRef] [Green Version]
- Berte, N.; Piée-Staffa, A.; Piecha, N.; Wang, M.; Borgmann, K.; Kaina, B.; Nikolova, T. Targeting Homologous Recombination by Pharmacological Inhibitors Enhances the Killing Response of Glioblastoma Cells Treated with Alkylating Drugs. Mol. Cancer Ther. 2016, 15, 2665–2678. [Google Scholar] [CrossRef] [Green Version]
- Alagpulinsa, D.A.; Ayyadevara, S.; Reis, R.J.S. A Small-Molecule Inhibitor of RAD51 Reduces Homologous Recombination and Sensitizes Multiple Myeloma Cells to Doxorubicin. Front. Oncol. 2014, 4, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghafoor, A.; Mian, I.; Wagner, C.; Mallory, Y.; Agra, M.G.; Morrow, B.; Wei, J.S.; Khan, J.; Thomas, A.; Sengupta, M.; et al. Phase 2 Study of Olaparib in Malignant Mesothelioma and Correlation of Efficacy with Germline or Somatic Mutations in BAP1 Gene. JTO Clin. Res. Rep. 2021, 2, 100231. [Google Scholar] [CrossRef] [PubMed]
- Poveda, A.M.; Davidson, R.; Blakeley, C.; Milner, A. Olaparib maintenance monotherapy in platinum-sensitive, relapsed ovarian cancer without germline BRCA mutations: OPINION Phase IIIb study design. Future Oncol. 2019, 15, 3651–3663. [Google Scholar] [CrossRef]
- Gelmon, K.A.; Fasching, P.A.; Couch, F.J.; Balmaña, J.; Delaloge, S.; Labidi-Galy, I.; Bennett, J.; McCutcheon, S.; Walker, G.; O’Shaughnessy, J.; et al. Clinical effectiveness of olaparib monotherapy in germline BRCA-mutated, HER2-negative metastatic breast cancer in a real-world setting: Phase IIIb LUCY interim analysis. Eur. J. Cancer 2021, 152, 68–77. [Google Scholar] [CrossRef]
- Cadoo, K.; Simpkins, F.; Mathews, C.; Liu, Y.L.; Provencher, D.; McCormick, C.; ElNaggar, A.C.; Altman, A.D.; Gilbert, L.; Black, D.; et al. Olaparib treatment for platinum-sensitive relapsed ovarian cancer by BRCA mutation and homologous recombination deficiency status: Phase II LIGHT study primary analysis. Gynecol. Oncol. 2022. [Google Scholar] [CrossRef]
- Fasching, P.; Link, T.; Hauke, J.; Seither, F.; Jackisch, C.; Klare, P.; Schmatloch, S.; Hanusch, C.; Huober, J.; Stefek, A.; et al. Neoadjuvant paclitaxel/olaparib in comparison to paclitaxel/carboplatinum in patients with HER2-negative breast cancer and homologous recombination deficiency (GeparOLA study). Ann. Oncol. 2020, 32, 49–57. [Google Scholar] [CrossRef]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.-A.; Park, Y.H.; Delord, J.-P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Arrieta, O.; Giménez-Capitán, A.; Aldeguer, E.; Drozdowskyj, A.; Chaib, I.; Reguart, N.; Garcia-Campelo, R.; Chen, J.-H.; Molina-Vila, M.A.; et al. BRCA1 Expression and Outcome in Patients With EGFR-Mutant NSCLC Treated with Gefitinib Alone or in Combination with Olaparib. JTO Clin. Res. Rep. 2021, 2, 100113. [Google Scholar] [CrossRef] [PubMed]
- Penson, R.T.; Valencia, R.V.; Cibula, D.; Colombo, N.; Leath, C.A., 3rd; Bidziński, M.; Kim, J.-W.; Nam, J.H.; Madry, R.; Hernández, C.; et al. Olaparib Versus Nonplatinum Chemotherapy in Patients with Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J. Clin. Oncol. 2020, 38, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men with Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients with Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Kristeleit, R.; Shapiro, G.I.; Burris, H.A.; Oza, A.M.; LoRusso, P.; Patel, M.R.; Domchek, S.M.; Balmaña, J.; Drew, Y.; Chen, L.-M.; et al. A Phase I–II Study of the Oral PARP Inhibitor Rucaparib in Patients with Germline BRCA1/2-Mutated Ovarian Carcinoma or Other Solid Tumors. Clin. Cancer Res. 2017, 23, 4095–4106. [Google Scholar] [CrossRef] [Green Version]
- Molin, G.Z.D.; Omatsu, K.; Sood, A.K.; Coleman, R.L. Rucaparib in ovarian cancer: An update on safety, efficacy and place in therapy. Ther. Adv. Med. Oncol. 2018, 10, 1758835918778483. [Google Scholar] [CrossRef]
- Okamoto, A.; Kondo, E.; Nakamura, T.; Yanagida, S.; Hamanishi, J.; Harano, K.; Hasegawa, K.; Hirasawa, T.; Hori, K.; Komiyama, S.; et al. Phase 2 single-arm study on the efficacy and safety of niraparib in Japanese patients with heavily pretreated, homologous recombination-deficient ovarian cancer. J. Gynecol. Oncol. 2021, 32, e16. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Thara, E.; Awad, M.M.; Dowlati, A.; Haque, B.; Stinchcombe, T.E.; Dy, G.K.; Spigel, D.R.; Lu, S.; Ms, N.I.S.; et al. JASPER: Phase 2 trial of first-line niraparib plus pembrolizumab in patients with advanced non–small cell lung cancer. Cancer 2021, 128, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined with Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.R.; Lundqvist, E.; Birrer, M.J.; Christensen, R.D.; Nyvang, G.-B.; Malander, S.; Anttila, M.; Werner, T.L.; Lund, B.; Lindahl, G.; et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): A randomised, phase 2, superiority trial. Lancet Oncol. 2019, 20, 1409–1419. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Ali, M.; Lu, M.; Ang, H.X.; Soderquist, R.S.; Eyler, C.E.; Hutchinson, H.M.; Glass, C.; Bassil, C.F.; Lopez, O.M.; Kerr, D.L.; et al. Small-molecule targeted therapies induce dependence on DNA double-strand break repair in residual tumor cells. Sci. Transl. Med. 2022, 14, eabc7480. [Google Scholar] [CrossRef]
- Luo, L.; Keyomarsi, K. PARP inhibitors as single agents and in combination therapy: The most promising treatment strategies in clinical trials for BRCA-mutant ovarian and triple-negative breast cancers. Expert Opin. Investig. Drugs 2022, 31, 607–631. [Google Scholar] [CrossRef]
- Barayan, R.; Ran, X.; Lok, B.H. PARP inhibitors for small cell lung cancer and their potential for integration into current treatment approaches. J. Thorac. Dis. 2020, 12, 6240–6252. [Google Scholar] [CrossRef]
- Rao, A.; Antonarakis, E.S. The growing role of rucaparib in contemporary treatment of metastatic prostate cancer: A review of efficacy and guidance for side effect management. Expert Rev. Anticancer Ther. 2022, 22, 671–679. [Google Scholar] [CrossRef]
- Xia, M.; Guo, Z.; Hu, Z. The Role of PARP Inhibitors in the Treatment of Prostate Cancer: Recent Advances in Clinical Trials. Biomolecules 2021, 11, 722. [Google Scholar] [CrossRef]
- Kamel, D.; Gray, C.; Walia, J.S.; Kumar, V. PARP Inhibitor Drugs in the Treatment of Breast, Ovarian, Prostate and Pancreatic Cancers: An Update of Clinical Trials. Curr. Drug Targets 2018, 19, 21–37. [Google Scholar] [CrossRef]
- Buege, M.; Mahajan, P. Clinical Trials of Poly(ADP-Ribose) Polymerase Inhibitors for Cancer Therapy: A Review. Rev. Recent Clin. Trials 2015, 10, 326–339. [Google Scholar] [CrossRef] [PubMed]
- McCann, K.E.; Hurvitz, S.A. Advances in the use of PARP inhibitor therapy for breast cancer. Drugs Context 2018, 7, 212540. [Google Scholar] [CrossRef] [PubMed]
- King, C.; Diaz, H.B.; McNeely, S.; Barnard, D.; Dempsey, J.; Blosser, W.; Beckmann, R.; Barda, D.; Marshall, M.S. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer Ther. 2015, 14, 2004–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, S.-B. The expanding role of WEE1. Cell. Signal. 2022, 94, 110310. [Google Scholar] [CrossRef]
- Day, M.; Oliver, A.W.; Pearl, L.H. Phosphorylation-dependent assembly of DNA damage response systems and the central roles of TOPBP1. DNA Repair 2021, 108, 103232. [Google Scholar] [CrossRef] [PubMed]
- Moudry, P.; Watanabe, K.; Wolanin, K.M.; Bartkova, J.; Wassing, I.E.; Watanabe, S.; Strauss, R.; Pedersen, R.T.; Oestergaard, V.; Lisby, M.; et al. TOPBP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. J. Cell Biol. 2016, 212, 281–288. [Google Scholar] [CrossRef]
- Wang, J.; Gong, Z.; Chen, J. MDC1 collaborates with TopBP1 in DNA replication checkpoint control. J. Cell Biol. 2011, 193, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.-P.; Yan, X.-B.; Liu, L.-G.; Tian, C.; Han, K.; Zhang, H.; Min, D.-L. TopBP1 promotes malignant progression and correlates with poor prognosis in osteosarcoma. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4022–4031. [Google Scholar]
- Liu, K.; Graves, J.D.; Lin, F.-T.; Lin, W.-C. Overexpression of TopBP1, a canonical ATR/Chk1 activator, paradoxically hinders ATR/Chk1 activation in cancer. J. Biol. Chem. 2021, 296, 100382. [Google Scholar] [CrossRef]
- Cruz-García, A.; López-Saavedra, A.; Huertas, P. BRCA1 Accelerates CtIP-Mediated DNA-End Resection. Cell Rep. 2014, 9, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Makharashvili, N.; Paull, T.T. CtIP: A DNA damage response protein at the intersection of DNA metabolism. DNA Repair 2015, 32, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Przetocka, S.; Porro, A.; Bolck, H.; Walker, C.; Lezaja, A.; Trenner, A.; von Aesch, C.; Himmels, S.-F.; D’Andrea, A.D.; Ceccaldi, R.; et al. CtIP-Mediated Fork Protection Synergizes with BRCA1 to Suppress Genomic Instability upon DNA Replication Stress. Mol. Cell 2018, 72, 568–582.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Geng, X.; Syeda, M.Z.; Huang, Z.; Zhang, C.; Ying, S. Human MUS81: A Fence-Sitter in Cancer. Front. Cell Dev. Biol. 2021, 9, 657305. [Google Scholar] [CrossRef]
- Ward, A.; Khanna, K.K.; Wiegmans, A.P. Targeting homologous recombination, new pre-clinical and clinical therapeutic combinations inhibiting RAD51. Cancer Treat. Rev. 2015, 41, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Budke, B.; Logan, H.L.; Kalin, J.; Zelivianskaia, A.S.; McGuire, W.C.; Miller, L.L.; Stark, J.M.; Kozikowski, A.P.; Bishop, D.K.; Connell, P.P. RI-1: A chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res. 2012, 40, 7347–7357. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Mazina, O.M.; Zentner, I.J.; Cocklin, S.; Mazin, A.V. Inhibition of Homologous Recombination in Human Cells by Targeting RAD51 Recombinase. J. Med. Chem. 2012, 55, 3011–3020. [Google Scholar] [CrossRef]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target. Oncol. 2021, 16, 255–282. [Google Scholar] [CrossRef]
- del Rivero, J.; Kohn, E.C. PARP inhibitors: The cornerstone of DNA repair-targeted therapies. Oncology 2017, 31, 265–273. [Google Scholar]
- Shkundina, I.; Gall, A.; Dick, A.; Cocklin, S.; Mazin, A. New RAD51 Inhibitors to Target Homologous Recombination in Human Cells. Genes 2021, 12, 920. [Google Scholar] [CrossRef]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Shen, Y.; Aoyagi-Scharber, M.; Wang, B. Trapping Poly(ADP-Ribose) Polymerase. J. Pharmacol. Exp. Ther. 2015, 353, 446–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-H.; Oshige, M.; Durant, S.T.; Rasila, K.K.; Williamson, E.A.; Ramsey, H.; Kwan, L.; Nickoloff, J.A.; Hromas, R. The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc. Natl. Acad. Sci. USA 2005, 102, 18075–18080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Shi, Y.; Mulligan, P.; Gay, F.; Landry, J.; Liu, H.; Lu, J.; Qi, H.; Wang, W.; Nickoloff, J.A.; et al. A YY1–INO80 complex regulates genomic stability through homologous recombination–based repair. Nat. Struct. Mol. Biol. 2007, 14, 1165–1172. [Google Scholar] [CrossRef] [Green Version]
- Chambers, A.L.; Downs, J.A. The RSC and INO80 Chromatin-Remodeling Complexes in DNA Double-Strand Break Repair. Prog. Mol. Biol. Transl. Sci. 2012, 110, 229–261. [Google Scholar] [CrossRef]
- Bao, Y.; Shen, X. Chromatin remodeling in DNA double-strand break repair. Curr. Opin. Genet. Dev. 2007, 17, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 2018, 72, 250–262.e6. [Google Scholar] [CrossRef] [Green Version]
- Price, B.D.; D’Andrea, A.D. Chromatin Remodeling at DNA Double-Strand Breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Morrison, A.J. Genome maintenance functions of the INO80 chromatin remodeller. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160289. [Google Scholar] [CrossRef] [Green Version]
- Fournier, L.-A.; Kumar, A.; Stirling, P.C. Chromatin as a Platform for Modulating the Replication Stress Response. Genes 2018, 9, 622. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Oma, Y.; Schleker, T.; Kugou, K.; Ohta, K.; Harata, M.; Gasser, S.M. Ino80 Chromatin Remodeling Complex Promotes Recovery of Stalled Replication Forks. Curr. Biol. 2008, 18, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Mognato, M.; Burdak-Rothkamm, S.; Rothkamm, K. Interplay between DNA replication stress, chromatin dynamics and DNA-damage response for the maintenance of genome stability. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108346. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Grewal, K.; Grewal, K.; Tabbara, I.A. PARP Inhibitors in Prostate Cancer. Anticancer Res. 2021, 41, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Gonzalez-Martin, A.; Matulonis, U.A.; Korach, J.; Mirza, M.R.; Moore, K.N.; Wu, X.; York, W.; Gupta, D.; Lechpammer, S.; Monk, B.J. Niraparib treatment for patients with BRCA-mutated ovarian cancer: Review of clinical data and therapeutic context. Future Oncol. 2022, 18, 2505–2536. [Google Scholar] [CrossRef]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Aubry, A.; Galiacy, S.; Allouche, M. Targeting ALK in Cancer: Therapeutic Potential of Proapoptotic Peptides. Cancers 2019, 11, 275. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.-Y.; Bang, Y.-J. HER2-targeted therapies—a role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Mustachio, L.; Chelariu-Raicu, A.; Szekvolgyi, L.; Roszik, J. Targeting KRAS in Cancer: Promising Therapeutic Strategies. Cancers 2021, 13, 1204. [Google Scholar] [CrossRef] [PubMed]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, E.D.S.; Nogueira, K.A.B.; Fernandes, L.C.C.; Martins, J.R.P.; Reis, A.V.F.; Neto, J.D.B.V.; Júnior, I.J.D.S.; Pessoa, C.; Petrilli, R.; Eloy, J.O. EGFR targeting for cancer therapy: Pharmacology and immunoconjugates with drugs and nanoparticles. Int. J. Pharm. 2021, 592, 120082. [Google Scholar] [CrossRef]
- Woo, E.-J.; Kim, Y.-G.; Kim, M.-S.; Han, W.-D.; Shin, S.; Robinson, H.; Park, S.-Y.; Oh, B.-H. Structural Mechanism for Inactivation and Activation of CAD/DFF40 in the Apoptotic Pathway. Mol. Cell 2004, 14, 531–539. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Jones, D.; Lee, S.-H.; Williamson, E.A.; Hromas, R. Drugging the Cancers Addicted to DNA Repair. J. Natl. Cancer Inst. 2017, 109, djx059. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Target Protein | Inhibitor | Recent/Ongoing Clinical Trials * | Tumor/Cell Targets ** | References |
|---|---|---|---|---|
| Chk1/Chk2 | UCN-01 | Solid tumors, leukemia | [116] | |
| LY2603618 | NCT01341457 | Solid tumors | [117] | |
| GDC-0575 | NCT01564251 | Solid tumors, lymphoma | [118] | |
| MK-8776 | NCT01870596 | Acute myeloid leukemia | [119] | |
| NCT00779584 | Solid tumors, lymphoma | |||
| LY2606368 | NCT02735980 | Lung cancer | [120] | |
| NCT02778126 | Solid/CNS tumors | [121] | ||
| NCT02203513 | TNBC | [122] | ||
| NCT01115790 | Advanced solid tumors | [123] | ||
| Wee1 | MK1775 | NCT01357161 | Ovarian cancer | [124] |
| ATR | BAY1895344 | NCT03188965 | Advanced solid tumors | [125] |
| AZD6738 | NCT02264678 | Advanced solid tumors | [126] | |
| TopBP1 | Calcein AM | Breast, lung xenografts | [127,128] | |
| CtIP | SP(18–28) | Breast cancer cells | [129] | |
| PACMP | siRNA-lnc15.2 | Breast, ovarian, lung, OS, stomach cancer cells | [130] | |
| MUS81 | siRNA-MUS81 | Gastric cancer cells | [131] | |
| EEPD1 | siRNA-MUS81 | Lung, OS, fibrosarcoma, cervical cancer cells | [103,104] | |
| Metnase | Ciprofloxacin | Embryonic kidney cells | [132] | |
| RAD51 | RI-1 | Glioblastoma, glioma cells | [133,134,135] | |
| B02 | Glioblastoma, multiple myeloma cells | [135,136] | ||
| PARP1 | Olaparib | NCT03531840 | Mesothelioma | [137] |
| NCT03402841 | Ovarian cancer | [138] | ||
| NCT03286842 | Breast cancer | [139] | ||
| NCT02983799 | Ovarian cancer | [140] | ||
| NCT02789332 | Breast cancer | [141] | ||
| NCT02734004 | Breast cancer | [142] | ||
| NCT02477644 | Ovarian cancer | [143] | ||
| NCT01513174 | Lung cancer | [144] | ||
| NCT02282020 | Ovarian cancer | [145] | ||
| Rucaparib | NCT02952534 | Prostate cancer | [146] | |
| NCT02042378 | Pancreatic cancer | [147] | ||
| NCT01891344 | Ovarian cancer | [148] | ||
| NCT01074970 | Ovarian, solid tumors | [149] | ||
| NCT01891344 | Ovarian cancer | [150] | ||
| Niraparib | NCT03759600 | Ovarian cancer | [151] | |
| NCT04475939 | Lung cancer | [152] | ||
| NCT02657889 | TNBC | [153] | ||
| NCT02354131 | Ovarian cancer | [154] | ||
| NCT02354586 | Ovarian cancer | [155] | ||
| ATM | AZD0156 | NCT02588105 | Advanced solid tumors | [156] |
| ATM/ATR | AZD1390 | NCT03215381 | Healthy volunteers | [156] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nickoloff, J.A. Targeting Replication Stress Response Pathways to Enhance Genotoxic Chemo- and Radiotherapy. Molecules 2022, 27, 4736. https://doi.org/10.3390/molecules27154736
Nickoloff JA. Targeting Replication Stress Response Pathways to Enhance Genotoxic Chemo- and Radiotherapy. Molecules. 2022; 27(15):4736. https://doi.org/10.3390/molecules27154736
Chicago/Turabian StyleNickoloff, Jac A. 2022. "Targeting Replication Stress Response Pathways to Enhance Genotoxic Chemo- and Radiotherapy" Molecules 27, no. 15: 4736. https://doi.org/10.3390/molecules27154736
APA StyleNickoloff, J. A. (2022). Targeting Replication Stress Response Pathways to Enhance Genotoxic Chemo- and Radiotherapy. Molecules, 27(15), 4736. https://doi.org/10.3390/molecules27154736