
Pro-Apoptotic Antitumoral Effect of Novel Acridine-Core Naphthoquinone Compounds against Oral Squamous Cell Carcinoma

 , , , ,
, , , ,  ,
, 
 and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Assays
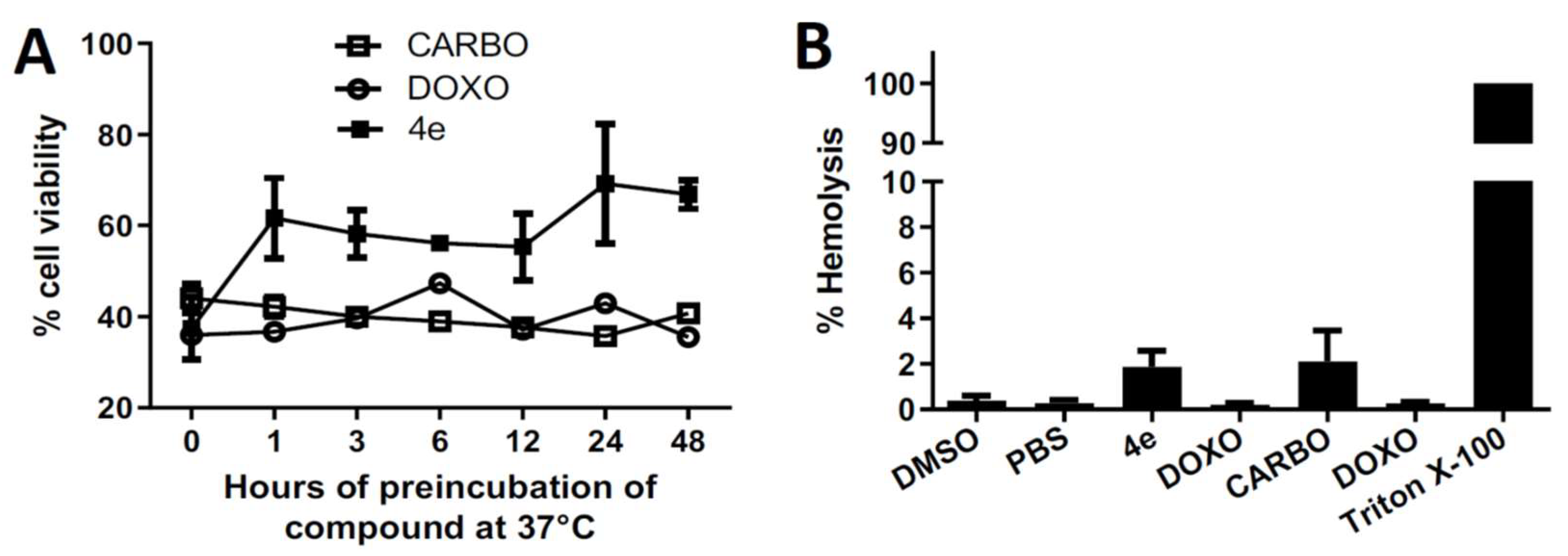
2.2.1. Cytotoxicity and Selectivity, Hemolytic Potential, and Biological Stability of the New Compounds
2.2.2. Acute Toxicity in Vivo
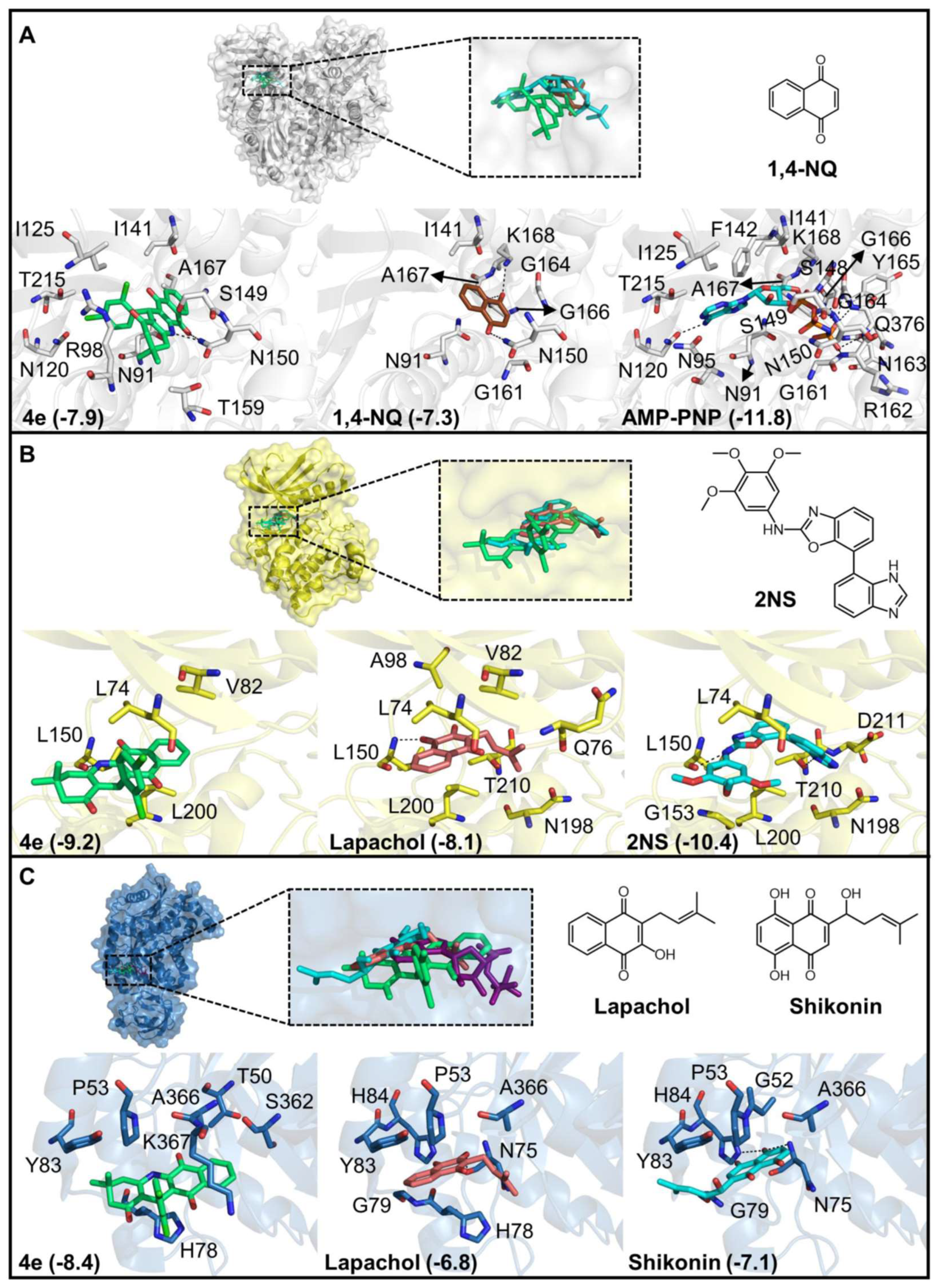
2.2.3. Prediction of Anticancer Targets of 4e by Reverse Docking
2.2.4. Predicted Toxicity and Pharmacokinetic Properties of 4e
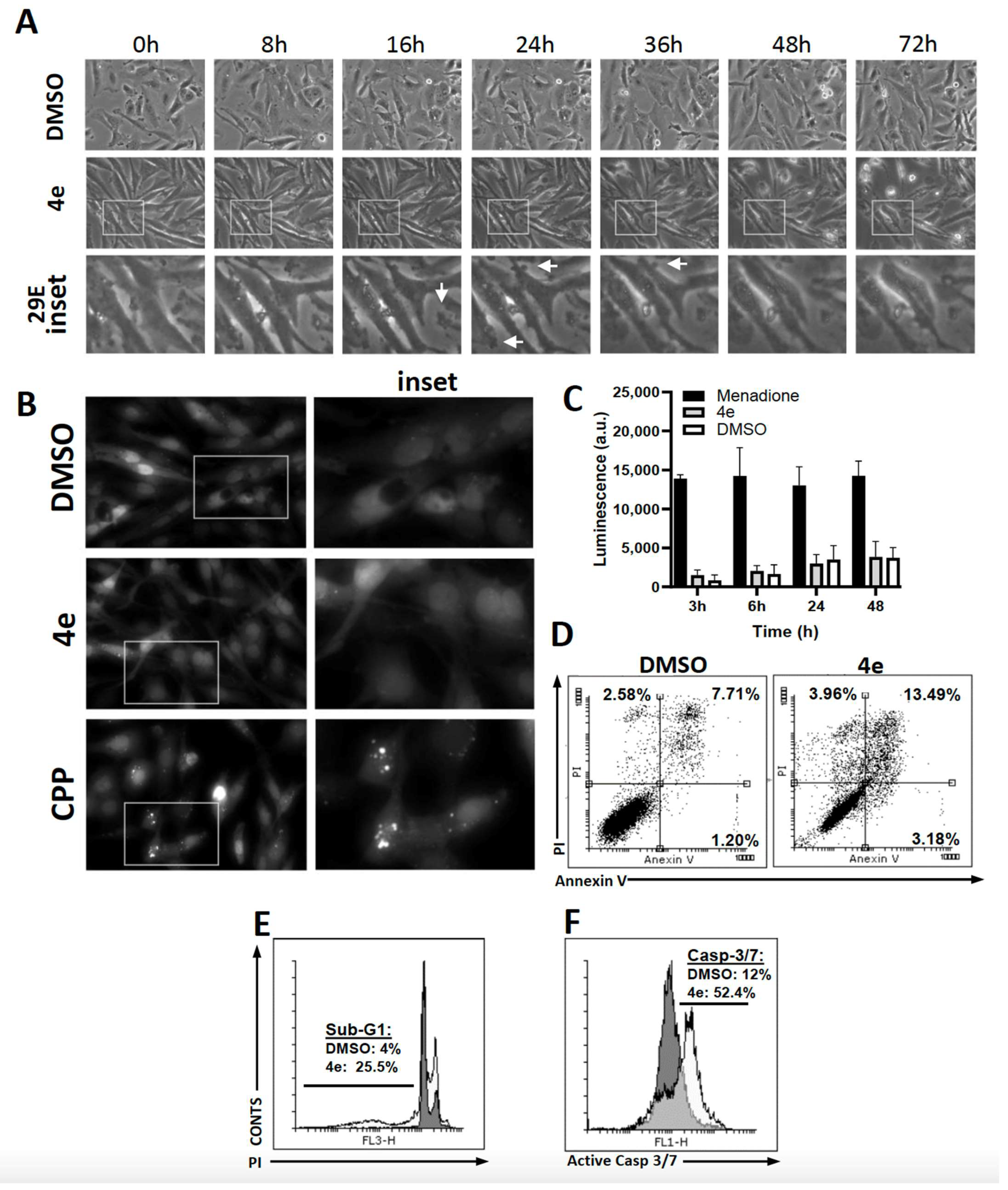
2.2.5. Cell Death Investigation
3. Materials and Methods
3.1. Chemistry
3.2. Biological Assays
3.3. In Silico Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Chai, A.W.Y.; Lim, K.P.; Cheong, S.C. Translational genomics and recent advances in oral squamous cell carcinoma. Semin. Cancer Biol. 2020, 61, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Li, C.C.; Shen, Z.; Bavarian, R.; Yang, F.; Bhattacharya, A. Oral Cancer: Genetics and the Role of Precision Medicine. Surg. Oncol. Clin. N. Am. 2020, 29, 127–144. [Google Scholar] [CrossRef]
- American Cancer Society. 2022. Treating Oral Cavity and Oropharyngeal Cancer. Available online: https://www.cancer.org/cancer/oral-cavity-and-oropharyngeal-cancer/treating.html (accessed on 15 June 2022).
- Sharifi-Rad, J.; Ozleyen, A.; Boyunegmez Tumer, T.; Oluwaseun Adetunji, C.; El Omari, N.; Balahbib, A.; Taheri, Y.; Bouyahya, A.; Martorell, M.; Martins, N.; et al. Natural Products and Synthetic Analogs as a Source of Antitumor Drugs. Biomolecules 2019, 9, 679. [Google Scholar] [CrossRef]
- Aminin, D.; Polonik, S. 1,4-Naphthoquinones: Some biological properties and application. Chem. Pharm. Bull. 2020, 68, 46–57. [Google Scholar] [CrossRef]
- Pereyra, C.E.; Dantas, R.F.; Ferreira, S.B.; Gomes, L.P.; Silva, F.P. The diverse mechanisms and anticancer potential of naphthoquinones. Cancer Cell Int. 2019, 19, 207. [Google Scholar] [CrossRef]
- Zhang, B.; Li, X.; Li, B.; Gao, C.; Jiang, Y. Acridine and its derivatives: A patent review (2009–2013). Expert Opin. Ther. Pat. 2014, 24, 647–664. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, X. Current Scenario of Acridine Hybrids with Anticancer Potential. Curr. Top. Med. Chem. 2021, 21, 1773–1786. [Google Scholar] [CrossRef]
- Kozurkova, M.; Sabolova, D.; Kristian, P. A new look at 9-substituted acridines with various biological activities. J. Appl. Toxicol. 2021, 41, 175–189. [Google Scholar] [CrossRef]
- Fieser, L.F.; Hartwell, J.L. The Reaction of Hydrazoic Acid with the Naphthoquinones. J. Am. Chem. Soc. 1935, 57, 1482–1484. [Google Scholar] [CrossRef]
- Ho, G.Y.; Woodward, N.; Coward, J.I.G. Cisplatin versus carboplatin: Comparative review of therapeutic management in solid malignancies. Crit. Rev. Oncol. Hematol. 2016, 102, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Khasraw, M.; Bell, R.; Dang, C. Epirubicin: Is it like doxorubicin in breast cancer? A clinical review. Breast 2012, 21, 142–149. [Google Scholar] [CrossRef]
- Shafei, A.; El-Bakly, W.; Sobhy, A.; Wagdy, O.; Reda, A.; Aboelenin, O.; Marzouk, A.; el Habak, K.; Mostafa, R.; Ali, M.A.; et al. A review on the efficacy and toxicity of different doxorubicin nanoparticles for targeted therapy in metastatic breast cancer. Biomed. Pharmacother. 2017, 95, 1209–1218. [Google Scholar] [CrossRef]
- Cos.; Genovese, S.; Fiorito, S.; Mathieu, V.; Kiss, R. Lapachol and its congeners as anticancer agents: A review. Phytochem. Rev. 2014, 13, 37–49. [Google Scholar] [CrossRef]
- Boulos, J.C.; Rahama, M.; Hegazy, M.E.F.; Efferth, T. Shikonin derivatives for cancer prevention and therapy. Cancer Lett. 2019, 459, 248–267. [Google Scholar] [CrossRef] [PubMed]
- Basri, D.F.; Alamin, Z.A.Z.; Chan, K.M. Assessment of cytotoxicity and genotoxicity of stem bark extracts from Canarium odontophyllum Miq. (dabai) against HCT 116 human colorectal cancer cell line. BMC Complement. Altern. Med. 2016, 16, 36. [Google Scholar] [CrossRef] [PubMed]
- Chipoline, I.C.; da Fonseca, A.C.C.; da Costa, G.R.M.; de Souza, M.P.; Rabelo, V.W.-H.; de Queiroz, L.N.; de Souza, T.L.F.; de Almeida, E.C.P.; Abreu, P.A.; Pontes, B.; et al. Molecular mechanism of action of new 1,4-naphthoquinones tethered to 1,2,3-1H-triazoles with cytotoxic and selective effect against oral squamous cell carcinoma. Bioorg. Chem. 2020, 101, 103984. [Google Scholar] [CrossRef]
- Zorzanelli, B.C.; de Queiroz, L.N.; Santos, R.M.A.; Menezes, L.M.; Gomes, F.C.; Ferreira, V.F.; da Silva, F.C.; Robbs, B.K. Potential cytotoxic and selective effect of new benzo[b]xanthenes against oral squamous cell carcinoma. Fut. Med. Chem. 2018, 10, 1141–1157. [Google Scholar] [CrossRef]
- Pereira, V.S.S.; de Oliveira, C.B.S.; Fumagalli, F.; Emery, F.S.; da Silva, N.B.; de Andrade-Neto, V.F. Cytotoxicity, hemolysis and in vivo acute toxicity of 2-hydroxy-3-anilino-1,4-naphthoquinone derivatives. Toxicol. Rep. 2016, 3, 756–762. [Google Scholar] [CrossRef]
- Parasuraman, S. Toxicological screening. J. Pharmacol. Pharmacother. 2011, 2, 74–79. [Google Scholar] [PubMed]
- Lassota, P.; Singh, G.; Kramer, R. Mechanism of Topoisomerase II Inhibition by Staurosporine and Other Protein Kinase Inhibitors. J. Biol. Chem. 1996, 271, 26418–26423. [Google Scholar] [CrossRef] [PubMed]
- Gurbani, D.; Kukshal, V.; Laubenthal, J.; Kumar, A.; Pandey, A.; Tripathi, S.; Arora, A.; Jain, S.K.; Ramachandran, R.; Anderson, D.; et al. Mechanism of inhibition of the ATpase domain of human topoisomerase IIα by 1,4-benzoquinone, 1,2-naphthoquinone, 1,4-naphthoquinone, and 9,10-phenanthroquinone. Toxicol. Sci. 2012, 126, 372–390. [Google Scholar] [CrossRef]
- Zu, X.; Xie, X.; Zhang, Y.; Liu, K.; Bode, A.M.; Dong, Z.; Kim, D.J. Lapachol is a novel ribosomal protein S6 kinase 2 inhibitor that suppresses growth and induces intrinsic apoptosis in esophageal squamous cell carcinoma cells. Phytother. Res. 2019, 33, 2337–2346. [Google Scholar] [CrossRef]
- Costales, A.; Mathur, M.; Ramurthy, S.; Lan, J.; Subramanian, S.; Jain, R.; Atallah, G.; Setti, L.; Lindvall, M.; Appleton, B.A. 2-Amino-7-substituted benzoxazole analogs as potent RSK2 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 1592–1596. [Google Scholar] [CrossRef] [PubMed]
- Aronchik, I.; Appleton, B.A.; Basham, S.E.; Crawford, K.; del Rosario, M.; Doyle, L.V.; Estacio, W.F.; Lan, J.; Lindvall, M.K.; Luu, C.A.; et al. Novel potent and selective inhibitors of p90 ribosomal S6 kinase reveal the heterogeneity of RSK function in MAPK-driven cancers. Mol. Cancer Res. 2014, 12, 803–812. [Google Scholar] [CrossRef]
- Jain, R.; Mathur, M.; Lan, J.; Costales, A.; Atallah, G.; Ramurthy, S.; Subramanian, S.; Setti, L.; Feucht, P.; Warne, B.; et al. Discovery of Potent and Selective RSK Inhibitors as Biological Probes. J. Med. Chem. 2015, 58, 6766–6783. [Google Scholar] [CrossRef]
- Chen, J.; Xie, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar] [CrossRef]
- Babu, M.S.; Mahanta, S.; Lakhter, A.J.; Hato, T.; Paul, S.; Naidu, S.R. Lapachol inhibits glycolysis in cancer cells by targeting pyruvate kinase M2. PLoS ONE 2018, 13, e0191419. [Google Scholar]
- Larsen, T.M.; Benning, M.M.; Rayment, I.; Reed, G.H. Structure of the Bis(Mg2+)-ATP-oxalate complex of the rabbit muscle pyruvate kinase at 2.1 Å resolution: ATP binding over a barrel. Biochemistry 1988, 37, 6247–6255. [Google Scholar] [CrossRef]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Ruthenburg, A.J.; Bechis, S.K.; Verdine, G.L. Nucleotide-dependent Domain Movement in the ATPase Domain of a Human Type IIA DNA Topoisomerase. J. Biol. Chem. 2005, 280, 37041–37047. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-Y. Molecular Targeting of ERKs/RSK2 Signaling in Cancers. Curr. Pharm. Des. 2017, 23, 4247–4258. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Hodos, R.A.; Kidd, B.A.; Shameer, K.; Readhead, B.P.; Dudley, J.T. In silico methods for drug repurposing and pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 186–210. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Palm, K.; Stenberg, P.; Luthman, K.; Artursson, P. Polar Molecular Surface Properties Predict the Intestinal Absorption of Drugs in Humans. Pharm. Res. 1997, 14, 568–571. [Google Scholar] [CrossRef]
- Wu, F.; Zhou, Y.; Li, L.; Shen, X.; Chen, G.; Wang, X.; Liang, X.; Tan, M.; Huang, Z. Computational Approaches in Preclinical Studies on Drug Discovery and Development. Front. Chem. 2020, 8, 726. [Google Scholar] [CrossRef]
- Alrushaid, S.; Sayre, C.L.; Yáñez, J.A.; Forrest, M.L.; Senadheera, S.N.; Burczynski, F.J.; Löbenberg, R.; Davies, N.M. Pharmacokinetic and Toxicodynamic Characterization of a Novel Doxorubicin Derivative. Pharmaceutics 2017, 9, 35. [Google Scholar] [CrossRef]
- Oguri, S.; Sakakibara, T.; Mase, H.; Shimizu, T.; Ishikawa, K.; Kimura, K.; Smyth, R.D. Clinical Pharmacokinetics of Carboplatin. J. Clin. Pharmacol. 1988, 28, 208–215. [Google Scholar] [CrossRef]
- Hamaguchi, K.; Godwin, A.K.; Yakushiji, M.; O’Dwyer, P.J.; Ozols, R.F.; Hamilton, T.C. Cross-resistance to diverse drugs is associated with primary cisplatin resistance in ovarian cancer cell lines. Cancer Res. 1993, 53, 5225–5232. [Google Scholar] [PubMed]
- Mansilla, S.; Llovera, L.; Portugal, J. Chemotherapeutic Targeting of Cell Death Pathways. Anti-Cancer Agents Med. Chem. 2012, 12, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lai, Y.; Hua, Z.C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 39, BSR20180992. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, M.; Chen, D.; Gao, W.; Guan, J.L.; Komatsu, M.; Yin, X.M. Autophagy Induced by Calcium Phosphate Precipitates Involves Endoplasmic Reticulum Membranes in Autophagosome Biogenesis. PLoS ONE 2012, 7, e52347. [Google Scholar] [CrossRef] [PubMed]
- Jamier, V.; Ba, L.A.; Jacob, C. Selenium- and tellurium-containing multifunctional redox agents as biochemical redox modulators with selective cytotoxicity. Chem. Eur. J. 2010, 16, 10920–10928. [Google Scholar] [CrossRef]
- Li, K.; Wang, B.; Zheng, L.; Yang, K.; Li, Y.; Hu, M.; He, D. Target ROS to induce apoptosis and cell cycle arrest by 5,7-dimethoxy-1,4-naphthoquinone derivative. Bioorg. Med. Chem. Lett. 2018, 28, 273–277. [Google Scholar] [CrossRef]
- Machado, T.Q.; Felisberto, J.R.S.; Guimarães, E.F.; de Queiroz, G.A.; da Fonseca, A.C.C.; Ramos, Y.J.; Marques, A.M.; Moreira, D.L.; Robbs, B.K. Apoptotic effect of β-pinene on oral squamous cell carcinoma as one of the major compounds from essential oil of medicinal plant Piper rivinoides Kunth. Nat. Prod. Res. 2022, 36, 1636–1640. [Google Scholar] [CrossRef]
- Lucena, P.I.; Faget, D.V.; Pachulec, E.; Robaina, M.C.; Klumb, C.E.; Robbs, B.K.; Viola, J.P.B. NFAT2 Isoforms Differentially Regulate Gene Expression, Cell Death, and Transformation through Alternative N-Terminal Domains. Mol. Cell. Biol. 2016, 36, 119–131. [Google Scholar] [CrossRef]
- Faget, D.V.; Lucena, P.I.; Robbs, B.K.; Viola, J.P.B. NFAT1 C-Terminal Domains Are Necessary but Not Sufficient for Inducing Cell Death. PLoS ONE 2012, 7, e47868. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Costa, D.C.S.; de Almeida, G.S.; Rabelo, V.W.-H.; Cabral, L.M.; Sathler, P.C.; Abreu, P.A.; Ferreira, V.F.; da Silva, L.C.R.P.; da Silva, F.C. Synthesis and evaluation of the cytotoxic activity of Furanaphthoquinones tethered to 1H-1,2,3-triazoles in Caco-2, Calu-3, MDA-MB231 cells. Eur. J. Med. Chem. 2018, 156, 524–533. [Google Scholar] [CrossRef]
- Lee, A.; Kim, D. CRDS: Consensus Reverse Docking System for target fishing. Bioinformatics 2020, 36, 959–960. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Lee, K.; Kim, D. Using reverse docking for target identification and its applications for drug discovery. Exp. Opin. Drug Discov. 2016, 11, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Lüpertz, R.; Wätjen, W.; Kahl, R.; Chovolou, Y. Dose- and time-dependent effects of doxorubicin on cytotoxicity, cell cycle and apoptotic cell death in human colon cancer cells. Toxicology 2010, 271, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yin, X.; Liu, H.; Chen, Q.; Feng, Y.; Ma, X.; Liu, W. Antiproliferative activity of plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) in human gastric arcinoma cells is facilitated via activation of autophagic pathway, mitochondrialmediated programmed cell death and inhibition of cell migration and invasion. J. BUON 2019, 24, 2000–2005. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | SCC9—Oral Cancer | |
|---|---|---|
| IC50 (µM) | S.D. | |
| 4c | 14.69 | 0.11 |
| 4d | 51.57 | 0.18 |
| 4e | 14.71 | 0.12 |
| 4h | 5.86 | 0.15 |
| 4j | 4.10 | 0.07 |
| 4k | N.D. | N.D. |
| 4l | 4.46 | 0.07 |
| 4m | 2.06 | 0.07 |
| 5c | 8.93 | 0.09 |
| 5d | 2.21 | 0.12 |
| 5e | 4.75 | 0.06 |
| 5h | 7.63 | 0.09 |
| 5j | 6.84 | 0.14 |
| 5k | 7.20 | 0.08 |
| 5l | 5.90 | 0.14 |
| 5m | 3.32 | 0.13 |
| Doxorubicin | 7.8 | 0.10 |
| Carboplatine | 250.0 | 0.14 |
| Lapachol | 58.32 | 0.06 |
| Shikonin | 1.22 | 0.02 |
| COMPOUND | Oral Tumor Cells | Primary Gingival Fibroblast | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SCC9 | SCC25 | SCC4 | |||||||||
| IC50 | S.D. | S.I. | IC50 | S.D. | S.I. | IC50 | S.D. | S.I. | IC50 | S.D. | |
| 4e | 14.71 | 0.12 | 6.0 | 21.52 | 0.07 | 4.1 | 53.75 | 0.09 | 1.6 | 87.99 | 0.07 |
| 4j | 4.10 | 0.07 | 5.7 | 11.50 | 0.05 | 2.0 | 18.86 | 0.09 | 1.2 | 23.56 | 0.08 |
| 4m | 2.06 | 0.07 | 3.8 | 7.41 | 0.06 | 1.1 | 10.99 | 0.65 | 0.7 | 7.86 | 0.04 |
| Doxorubicin | 7.8 | 0.10 | 1.6 | 1.27 | 0.06 | 9.7 | 4.56 | 0.03 | 2.7 | 12.30 | 0.20 |
| Carboplatin | 250.0 | 0.14 | 2.2 | 186.3 | 0.06 | 3.0 | 180.4 | 0.03 | 3.1 | 559.6 | 0.06 |
| Lapachol | 58.32 | 0.06 | 1.8 | 93.81 | 0.03 | 1.1 | 76.93 | 0.08 | 1.4 | 105.6 | 0.15 |
| Shikonin | 1.22 | 0.02 | 2.0 | 2.47 | 0.03 | 1.0 | 3.19 | 0.03 | 0.8 | 2.48 | 0.03 |
| Treatment | Dose mg/kg a | Change in Body Weight | Change in Food Consumption | Morbidity b | Mortality | Gross Necropsy c | Histology d |
|---|---|---|---|---|---|---|---|
| Control | 0 N = 3 | Absent | Absent | Absent | Absent | No alteration | Normal |
| 4e | 100 N = 3 | Absent | Absent | Absent | Absent | No alteration | No significant alteration in comparison to control |
| 200 N = 4 | Absent | Absent | Absent | Absent | No alteration | Presence of mild Perivascular and periportal lymphocyte focus and Bronchus-Associated Lymphoid Tissue (BALT) | |
| 400 N = 4 | Absent | Absent | Absent | Absent | No alteration | Presence of mild Perivascular and periportal lymphocyte focus and Bronchus-Associated Lymphoid Tissue (BALT) |
| Compounds | cLogP | nON | nOH/NH | MW | Lipinski’s Violations a | TPSA (Å2) |
|---|---|---|---|---|---|---|
| 4e | 3.37 | 3 | 1 | 452.3 | 0 | 63.2 |
| Doxorubicin | −2.10 | 12 | 6 | 543.3 | 3 | 206.1 |
| Carboplatin | −1.79 | 6 | 4 | 371.2 | 0 | 126.6 |
| ADMET | 4e | Carboplatin | Doxoribicin |
|---|---|---|---|
| Oral Bioavailability | +0.66 | −0.60 | −0.91 |
| P-glycoprotein inhibitor | −0.44 | −0.99 | −0.92 |
| P-glycoprotein substrate | −0.72 | −0.99 | +0.95 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zorzanelli, B.C.; Ouverney, G.; Pauli, F.P.; da Fonseca, A.C.C.; de Almeida, E.C.P.; de Carvalho, D.G.; Possik, P.A.; Rabelo, V.W.-H.; Abreu, P.A.; Pontes, B.; et al. Pro-Apoptotic Antitumoral Effect of Novel Acridine-Core Naphthoquinone Compounds against Oral Squamous Cell Carcinoma. Molecules 2022, 27, 5148. https://doi.org/10.3390/molecules27165148
Zorzanelli BC, Ouverney G, Pauli FP, da Fonseca ACC, de Almeida ECP, de Carvalho DG, Possik PA, Rabelo VW-H, Abreu PA, Pontes B, et al. Pro-Apoptotic Antitumoral Effect of Novel Acridine-Core Naphthoquinone Compounds against Oral Squamous Cell Carcinoma. Molecules. 2022; 27(16):5148. https://doi.org/10.3390/molecules27165148
Chicago/Turabian StyleZorzanelli, Bruna Costa, Gabriel Ouverney, Fernanda P. Pauli, Anna Carolina Carvalho da Fonseca, Elan Cardozo Paes de Almeida, Danielle Gonçalves de Carvalho, Patricia Abrão Possik, Vitor Won-Held Rabelo, Paula Alvarez Abreu, Bruno Pontes, and et al. 2022. "Pro-Apoptotic Antitumoral Effect of Novel Acridine-Core Naphthoquinone Compounds against Oral Squamous Cell Carcinoma" Molecules 27, no. 16: 5148. https://doi.org/10.3390/molecules27165148
APA StyleZorzanelli, B. C., Ouverney, G., Pauli, F. P., da Fonseca, A. C. C., de Almeida, E. C. P., de Carvalho, D. G., Possik, P. A., Rabelo, V. W. -H., Abreu, P. A., Pontes, B., Ferreira, V. F., Forezi, L. d. S. M., da Silva, F. d. C., & Robbs, B. K. (2022). Pro-Apoptotic Antitumoral Effect of Novel Acridine-Core Naphthoquinone Compounds against Oral Squamous Cell Carcinoma. Molecules, 27(16), 5148. https://doi.org/10.3390/molecules27165148