

Analytical Approaches in Official Food Safety Control: An LC-Orbitrap-HRMS Screening Method for the Multiresidue Determination of Antibiotics in Cow, Sheep, and Goat Milk

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Method Validation
- Detection capability, ccβ
- Selectivity-Specificity
- Stability
- Robustness
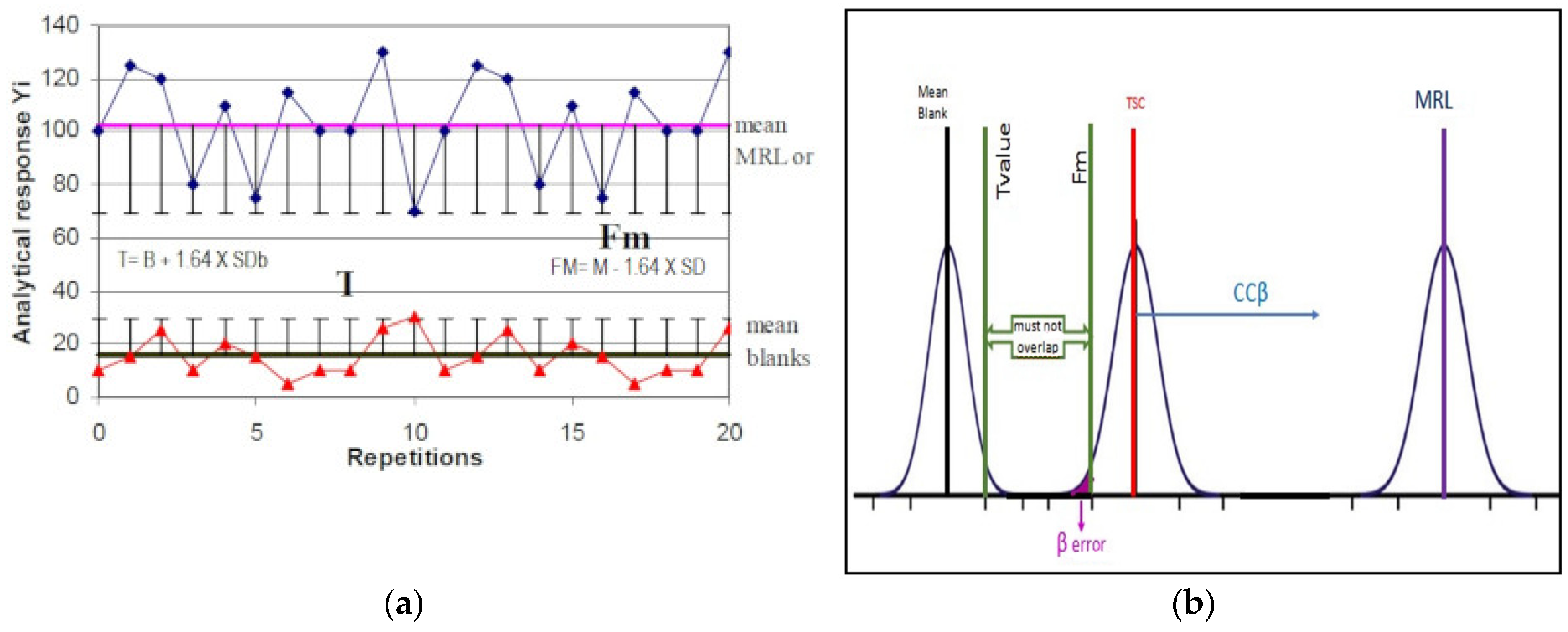
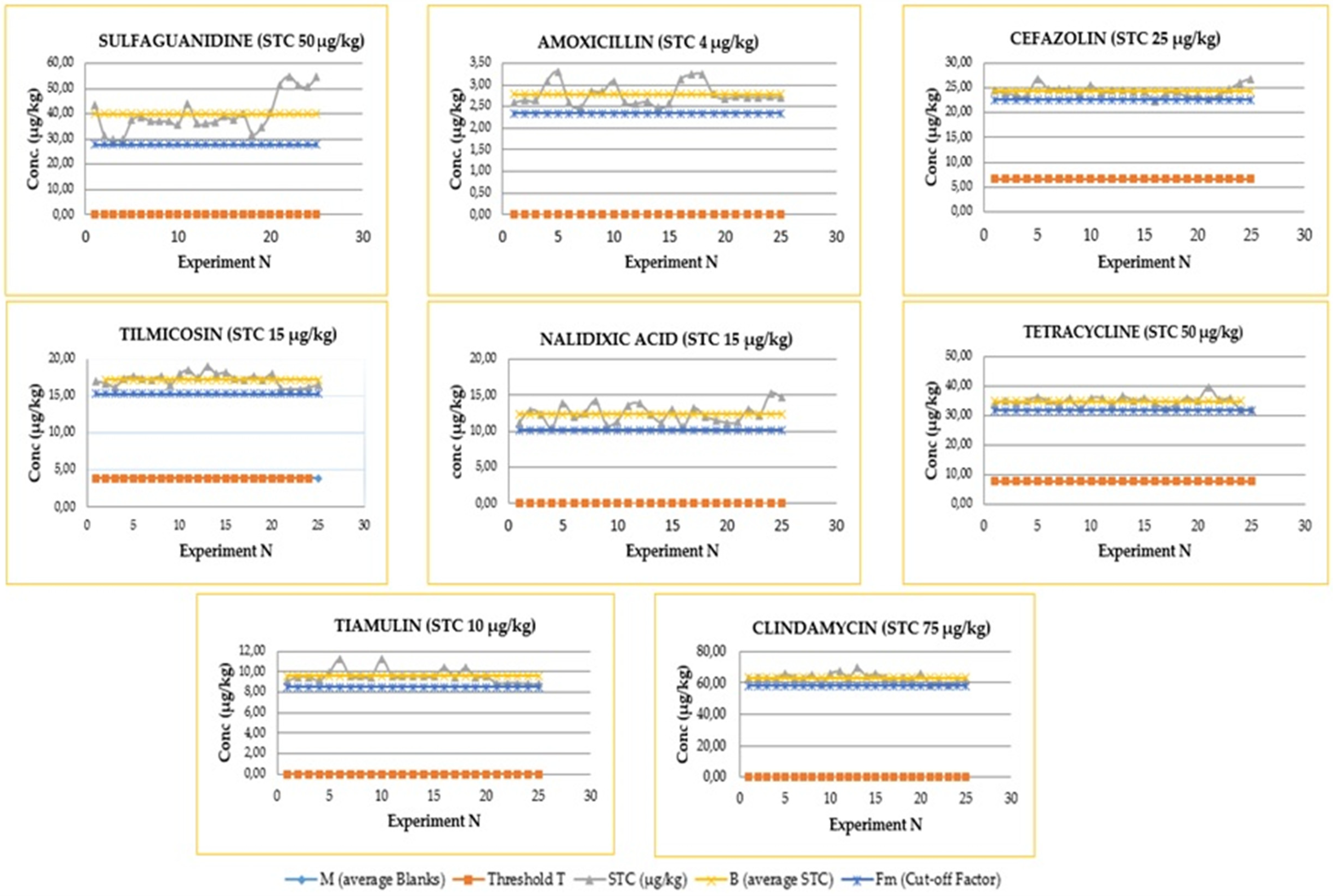
2.2. Detection Capability (ccβ)
- (a)
- the lowest concentration reasonably achievable for the detection of samples containing residues, for banned or unauthorized substances
- (b)
- below the permitted limit (MRL), for authorized substances
2.3. Specificity
- –
- exact mass accuracy ≤5 ppm
- –
- tolerance range of retention times associated to an exact mass ≤2.5%,
- Fm > T: is the optimal condition, corresponding to a percentage of false negatives less than 5%; therefore, the CCβ is less than the concentration of STC (less than or equal to MRL).
- Fm < T: the percentage of false negatives is greater than 5%, CCβ is greater than STC, and it is necessary to proceed with new experiments to determine the new ccβ.
2.4. Stability
2.5. Ruggedness
2.6. Quality Control
2.6.1. Internal Quality Control
- ensuring the quality of daily results,
- monitoring the efficiency of the extraction procedure
- checking for changes in retention times.
2.6.2. External Quality Control
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Work Solutions
4.3. Method Description
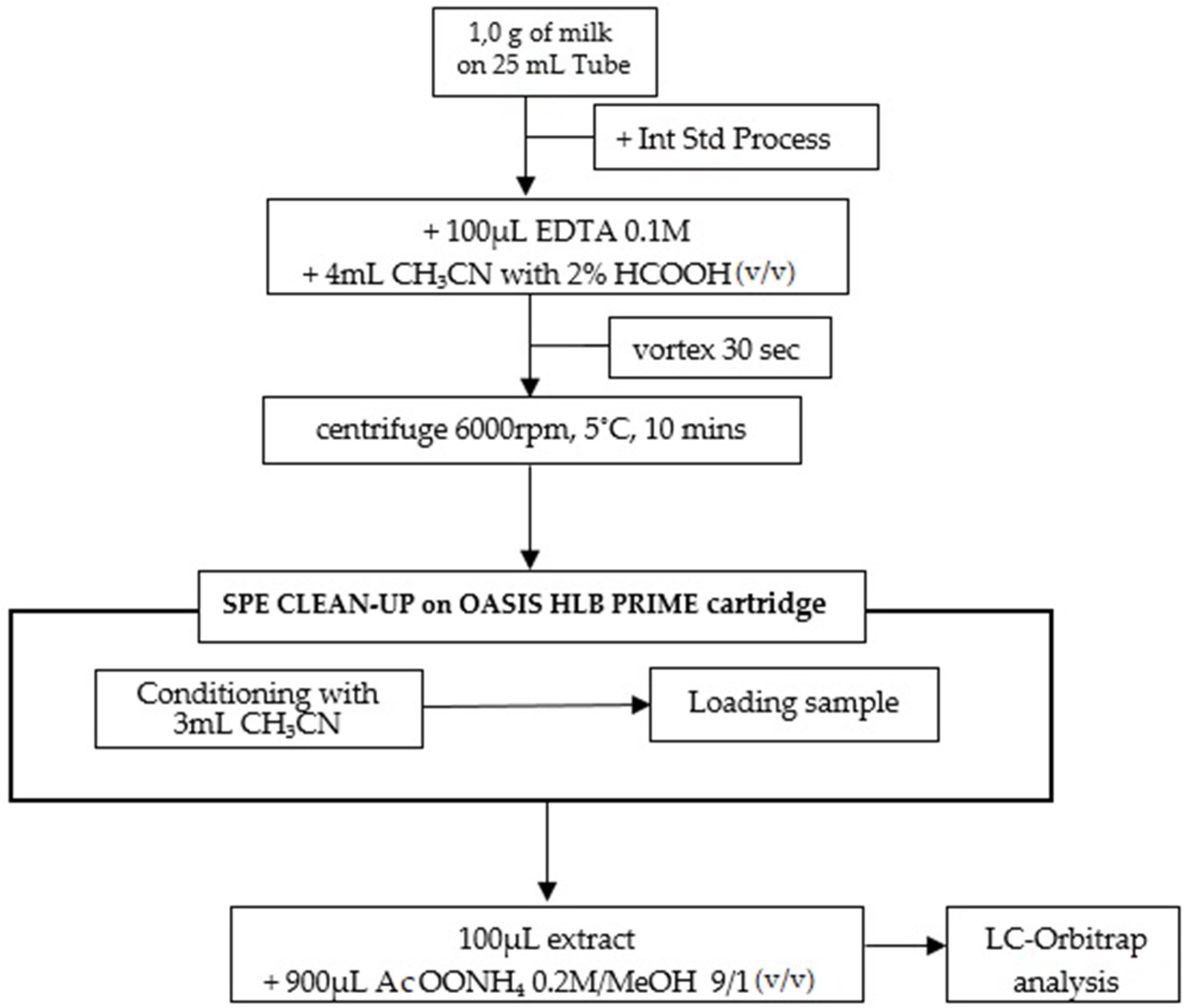
4.3.1. Sample Preparation
4.3.2. Instrumental Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Commission. Regulation 1831/2003/EC on additives for use in animal nutrition, replacing Directive 70/524/EEC on additives in feeding-stuffs. Off. J. Eur. Union 2003, 40, 238–252. [Google Scholar]
- Oliveira, N.A.; Gonçalves, B.L.; Lee, S.H.; Oliveira, C.A.F.; Corassin, C.H. Use of Antibiotics in Animal Production and Its Impact on Human Health. J. Food Chem. Nanotechnol. 2020, 6, 40–47. [Google Scholar] [CrossRef]
- Chen, J.; Ying, G.G.; Deng, W.J. Antibiotic Residues in Food: Extraction, Analysis, and Human Health Concerns. J. Agric. Food Chem. 2019, 67, 7569–7586. [Google Scholar] [CrossRef] [PubMed]
- Beyene, T. Veterinary Drug Residues in Food-animal Products: Its Risk Factors and Potential Effects on Public Health. J. Vet. Sci. Technol. 2016, 7, 285. [Google Scholar] [CrossRef]
- Schwarz, S.; Chaslus-Dancla, E. Use of antimicrobials in veterinary medicine and mechanisms of resistance. Vet. Res. 2001, 32, 201–225. [Google Scholar] [CrossRef]
- Lekshmi, M.; Ammini, P.; Kumar, S.; Varela, M.F. The Food Production Environment and the Development of Antimicrobial Resistance in Human Pathogens of Animal Origin. Microorganisms 2017, 5, 11. [Google Scholar] [CrossRef]
- WHO. Critically Important Antimicrobials for Human Medicine, 3rd ed.; World Health Organization: Geneva, Switzerland, 2011; Available online: http://apps.who.int/iris/bitstream/handle/10665/77376/9789241504485_eng.pdf (accessed on 2 March 2022).
- European Commission. Regulation (EC) No 178/2002 of the European Parliament and of the Council of 28 January 2002 laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety. Off. J. Eur. Union 2002, 6, 463–486. [Google Scholar]
- European Commission. Commission regulation (EU) No. 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin. Off. J. Eur. Union 2010, 32, 1–72. [Google Scholar]
- European Commission. Commission Decision 2002/657/EC implementing Council Directive 96/23/EC concerning the performances of analytical methods and the interpretation of results. N° L 221, 17/08/2002, Decision of 12 August 2002, European Commission, Directorate General for Public Health and Consumers Protection. Off. J. Eur. Comm. 2002, 36, 8–36. [Google Scholar]
- European Commission. Commission Implementing Regulation (EU) 2021/808 of 22 March 2021 on the Performance of Analytical Methods for Residues of Pharmacologically Active Substances Used in Food-Producing Animals and on the Interpretation of Results as Well as on the Methods to be Used for Sampling and Repealing Decisions 2002/657/EC and 98/179/EC. Available online: http://data.europa.eu/eli/reg_impl/2021/808/oj (accessed on 2 March 2022).
- Blasco, C.; Picó, Y.; Torres, C.M. Progress in analysis of residual antibacterials in food. TrAC Trends Anal. Chem. 2007, 26, 895–913. [Google Scholar] [CrossRef]
- Bohm, D.A.; Stachel, C.S.; Gowik, P. Multi-method for the determination of antibiotics of different substance groups in milk and validation in accordance with Commission Decision 2002/657/EC. J. Chromatogr. A 2009, 1216, 8217–8223. [Google Scholar] [CrossRef]
- Junza, A.; Amatya, R.; Barron, D.; Barbosa, J. Comparative study of the LC-MS/MS and UPLC-MS/MS for the multi-residue analysis of quinolones, penicillins and cephalosporins in cow milk, and validation according to the regulation 2002/657/EC. J. Chromatogr. B 2011, 879, 2601–2610. [Google Scholar] [CrossRef]
- Schwaiger, B.; König, J.; Lesueur, C. Development and validation of amulti-class UHPLC-MS/MS method for determination of antibiotic residues in dairy products. Food Anal. Methods 2018, 11, 1417–1434. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Liu, X.; Zhang, J.; Cao, Y.; Shi, Z.; Sun, H. Multi-class, multi-residue analysis of trace veterinary drugs in milk by rapid screening and quantification using ultra-performance liquid chromatography-quadrupole time-of-flight mass spectrometry. J. Dairy Sci. 2015, 98, 8433–8444. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Xua, J.J.; Cong, J.M.; Cai, Z.X.; Zhang, J.S.; Wang, J.L.; Ren, Y.P. Optimization for quick, easy, cheap, effective, rugged and safe extraction of mycotoxins and veterinary drugs by response surface methodology for application to egg and milk. J. Chromatogr. A 2018, 1532, 20–29. [Google Scholar] [CrossRef]
- Young, M.S.; Shia, J.C. Oasis PRiME HLB Cartridge for Rapid and Effective Cleanup of Avocado, A High Fat Matrix, Prior to APGC-MS/MS Analysis, Waters Application Note 720005816EN; Waters Corporation: Milford, MA, USA, 2017; Volume 1, pp. 38–42. [Google Scholar]
- Huang, D.; van Tran, K.; Young, M.S. A simple cleanup protocol using a novel SPE device for UPLC-MS/MS analysis of multi-residue veterinary drugs in milk. In Waters Application Note; Waters Corporation: Milford, MA, USA, 2017; Available online: http://www.waters.com/webassets/cms/library/docs/720005414en.pdf (accessed on 2 March 2022).
- Chen, Z.Y.; Ying, S.; Liu, J.H.; Zhan, P.P.; Zhao, Y.G. PRiME pass-through cleanup for the fast determination of aflatoxins in human serum by using LC-MS/MS. Anal. Methods 2016, 8, 1457–1462. [Google Scholar] [CrossRef]
- Jadhav, M.R.; Pudale, A.; Raut, P.; Utture, S.; Ahammed Shabeer, T.P.; Banerjee, K. A unified approach for high-throughput quantitative analysis of the residues of multi-class veterinary drugs and pesticides in bovine milk using LC-MS/MS and GC–MS/MS. Food Chem. 2019, 272, 292–305. [Google Scholar] [CrossRef]
- Holstege, D.M.; Puschner, B.; Whitehead, G.; Galey, F.D. Screening and Mass Spectral Confirmation of β-Lactam Antibiotic Residues in Milk Using LC-MS/MS. J. Agric. Food Chem. 2002, 50, 406–441. [Google Scholar] [CrossRef]
- Alija, G.; Hajrulai-Musliu, Z.; Uzunov, R. Development and validation of confirmatory LC–MS/MS method for multi-residue analysis of antibiotic drugs in bovine milk. SN Appl. Sci. 2020, 2, 1563. [Google Scholar] [CrossRef]
- Moretti, S.; Cruciani, G.; Romanelli, S.; Rossi, R.; Saluti, G.; Galarini, R. Multiclass method for the determination of 62 antibiotics in milk. J. Mass Spectrom. 2016, 51, 792–804. [Google Scholar] [CrossRef]
- Gaugain-Juhel, M.; Delépine, B.; Gautier, S.; Fourmond, M.P.; Gaudin, V.; Hurtaud-Pessel, D.; Verdon, E.; Sanders, P. Validation of a liquid chromatography-tandem mass spectrometry screening method to monitor 58 antibiotics in milk: A qualitative approach. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2009, 26, 1459. [Google Scholar] [CrossRef] [PubMed]
- Community Reference Laboratory (CRL). Guideline for the Validation of Screening Methods for Residues of Veterinary Medicines (Initial Validation and Transfer). 2010. Available online: http://ec.europa.eu/food/food/chemicalsafety/residues/lab_analysis_en.htm/ (accessed on 2 March 2022).
- Berendsen, B.J.A.; Elbers, I.J.W.; Stolker, A.A.M. Determination of the stability of antibiotics in matrix and reference solutions using a straightforward procedure applying mass spectrometric detection. Food Addit. Contam. Part A 2011, 28, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shushan, B. A review of clinical diagnostic applications of liquid chromatography-tandem mass spectrometry. Mass Spectrom. Rev. 2010, 29, 930. [Google Scholar] [CrossRef] [PubMed]
- Maurer, H.H. Current role of liquid chromatography-mass spectrometry in clinical and forensic toxicology. Anal. Bioanal. Chem. 2007, 388, 1315. [Google Scholar] [CrossRef]
- Jiwan, J.L.; Wallemacq, P.; Hérent, M.F. HPLC-high resolution mass spectrometry in clinical laboratory. Clin. Biochem. 2011, 44, 136. [Google Scholar] [CrossRef]
- Moulard, Y.; Bailly-Chouriberry, L.; Boyer, S.; Garcia, P.; Popot, M.A.; Bonnaire, Y. Use of benchtop exactive high resolution and high mass accuracy orbitrap mass spectro-meter for screening in horse doping control. Anal. Chim. Acta 2011, 700, 126. [Google Scholar] [CrossRef]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Quantitative and confirmative performance of liquid chromatography coupled to high-resolution mass spectrometry compared to tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 979. [Google Scholar] [CrossRef]
- Zhang, N.R.; Yu, S.; Tiller, P.; Yeh, S.; Mahan, E.; Emary, W.B. Quantitation of small molecules using high-resolution accurate mass spectrometers - a different approach for analysis of biological samples. Rapid Commun. Mass Spectrom. 2009, 23, 1085. [Google Scholar] [CrossRef]
- Bateman, K.P.; Kellmann, M.; Muenster, H.; Papp, R.; Taylor, L. Quantitative-qualitative data acquisition using a benchtop Orbitrap mass spectrometer. J. Am. Soc. Mass Spectrom. 2009, 81, 1881. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Sample Matrix | Validation Samples | Samples N | Indicator | Experiments N |
|---|---|---|---|---|---|
| Detection Capability ccβ | milk | 10 sheep, 10 cow, 5 goat | 25 spiked milk | Cutt-off factor (Fm) | 25 |
| Specificity | milk | 10 sheep, 10 cow, 5 goat | 25 blank milk | Threshold value Tv | 25 |
| Analyte’s Stability | Standard solution of calibration curve | 5 | y/x Ratio | 10 | |
| Ruggedness | milk | Variation on 2 levels for 4 factors (*) | 10 blank + 10 STC spiked | Ccβ | 20 |
| Analyte | MRL (µg/kg) | STC (µg/kg) | Analyte | MRL (µg/kg) | STC (µg/kg) |
|---|---|---|---|---|---|
| Amoxicillin | 4 | 4 (a) | Lincomycin | - | 75 (b) |
| Ampicillin | 4 | 4 (a) | Oxolinic Acid | - | 15 (b) |
| Cloxacillin | 30 | 15 (b) | Ciprofloxacin | 100 | 15 (b) |
| Dicloxacillin | 30 | 15 (b) | Danofloxacin | 30 | 15 (b) |
| Nafcillin | 30 | 15 (b) | Difloxacin | - | 15 (b) |
| Oxacillin | 30 | 15 (b) | Enrofloxacin | 100 | 15 (b) |
| Penicillin G | 4 | 4 (a) | Flumequin | 50 | 15 (b) |
| Penicillin V | 4 | 4 (a) | Marbofloxacin | 75 | 15 (b) |
| Cefalexin | 100 | 25 (b) | Norfloxacin | - | 15 (b) |
| Cefazolin | 50 | 25 (b) | Ofloxacin | - | 15 (b) |
| Cefapirin | 60 | 25 (b) | Sarafloxacin | - | 15 (b) |
| Cefquinome | 20 | 10 (b) | Sulfaquinoxaline | 100 | 50 (b) |
| Cefoperazone | 50 | 25 (b) | Sulfachloropyridazine | 100 | 50 (b) |
| Ceftiofur | 100 | 50 (b) | Sulfadiazine | 100 | 50 (b) |
| Chlortetracycline | 100 | 50 (b) | Sulfadimethoxin | 100 | 50 (b) |
| Doxycycline | 100 | 50 (b) | Sulfaguanidine | 100 | 50 (b) |
| Oxytetracycline | 100 | 50 (b) | Sulfamerazine | 100 | 50 (b) |
| Tetracycline | 100 | 50 (b) | Sulfametazine | 100 | 50 (b) |
| Epi- Chlortetracycline | 100 | 50 (b) | Sulfamethizole | 100 | 50 (b) |
| Epi- Doxycycline | 100 | 50 (b) | Sulfamethoxazole | 100 | 50 (b) |
| Epi- Oxytetracycline | 100 | 50 (b) | Sulfamethoxipyridazine | 100 | 50 (b) |
| Epi-Tetracycline | 100 | 50 (b) | Sulfamonomethoxin | 100 | 50 (b) |
| Tiamulin | - | 10 (c) | Sulfanilamide | 100 | 50 (b) |
| Valnemulin | - | 10 (c) | Sulfapyridin | 100 | 50 (b) |
| Tilmicosin | - | 20 (b) | Sulfathiazole | 100 | 50 (b) |
| Tylosin | 50 | 20 (b) | Trimethoprim | 50 | 50 (b) |
| Azithromycin | 50 | 20 (b) | |||
| Erythromycin | 40 | 20 (b) | |||
| Spiramycin | 200 | 20 (b) | |||
| Clindamycin | 150 | 75 (b) | |||
| Nalidixic Acid | - | 15 (b) | |||
| Analyte | STC (µg/kg) | CVR (%) | Analyte | STC (µg/kg) | CVR (%) | Analyte | STC (µg/kg) | CVR (%) |
|---|---|---|---|---|---|---|---|---|
| Amoxicillin | 4 | 9.7 | Epi- Oxytetracycline | 50 | 5.6 | Ofloxacin | 15 | 5.9 |
| Ampicillin | 4 | 7.1 | Epi-Tetracycline | 50 | 3.7 | Sarafloxacin | 15 | 3.4 |
| Cloxacillin | 15 | 3.6 | Tiamulin | 10 | 6.7 | Sulfaquinoxaline | 50 | 6.2 |
| Dicloxacillin | 15 | 6.6 | Valnemulin | 10 | 11.5 | Sulfachloropyridazine | 50 | 4.9 |
| Nafcillin | 15 | 7.0 | Tilmicosin | 20 | 7.0 | Sulfadiazine | 50 | 6.4 |
| Oxacillin | 15 | 4.9 | Tylosin | 20 | 8.6 | Sulfadimethoxin | 50 | 7.7 |
| Penicillin G | 4 | 14.2 | Azithromycin | 20 | 2.9 | Sulfaguanidine | 50 | 18.5 |
| Penicillin V | 4 | 16.2 | Erythromycin | 20 | 4.4 | Sulfamerazine | 50 | 6.4 |
| Cefalexin | 25 | 7.1 | Spiramycin | 20 | 4.3 | Sulfametazine | 50 | 6.0 |
| Cefazolin | 25 | 4.5 | Clindamycin | 75 | 4.8 | Sulfamethizole | 50 | 5.7 |
| Cefapirin | 25 | 6.3 | Lincomycin | 75 | 4.2 | Sulfamethoxazole | 50 | 5.0 |
| Cefquinome | 10 | 11.2 | Nalidixic Acid | 15 | 10.9 | Sulfamethoxipyridazine | 50 | 3.2 |
| Cefoperazone | 25 | 4.3 | Oxolinic Acid | 15 | 5.1 | Sulfamonomethoxin | 50 | 3.5 |
| Ceftiofur | 50 | 5.1 | Ciprofloxacin | 15 | 4.7 | Sulfanilamide | 50 | 6.2 |
| Chlortetracycline | 50 | 4.9 | Danofloxacin | 15 | 4.1 | Sulfapyridin | 50 | 3.5 |
| Doxycycline | 50 | 8.3 | Difloxacin | 15 | 6.0 | Sulfathiazole | 50 | 3.9 |
| Oxytetracycline | 50 | 7.8 | Enrofloxacin | 15 | 4.5 | Trimethoprim | 50 | 7.1 |
| Tetracycline | 50 | 5.3 | Flumequin | 15 | 6.4 | |||
| Epi- Chlortetracycline | 50 | 3.3 | Marbofloxacin | 15 | 5.1 | |||
| Epi- Doxycycline | 50 | 2.7 | Norfloxacin | 15 | 6.5 |
| Time (min) | A% Formic Acid 0.1% | B% Methanol | Flow mL/min |
|---|---|---|---|
| 0 | 95 | 5 | 0.250 |
| 1.00 | 95 | 5 | 0.250 |
| 20.00 | 5 | 95 | 0.250 |
| 25.00 | 5 | 95 | 0.250 |
| 26.00 | 95 | 5 | 0.250 |
| 30.00 | 95 | 5 | 0.250 |
| Calibration Point | Concentration Level (µg/L) | Volume of Stock Standard Solution at 10 µg/mL | Final Volume AcOONH4 0.2 M:MeOH 9:1 |
|---|---|---|---|
| 1 | 0.1 | 10 µL | 10 mL |
| 2 | 0.5 | 50 µL | |
| 3 | 1.0 | 100 µL | |
| 4 | 5.0 | 500 µL | |
| 5 | 10.0 | 1000 µL |
| Full MS | dd-MS2 | |
|---|---|---|
| Resolution: 70,000 | Scan Range: 150–1000 m/z | Resolution: 35,000 |
| AGC Target: 3e6 | Auxiliary Gas: 15 | AGC target: 1e6 |
| Maximum IT: 100 ms | Polarity: ES+ | Maximum IT: 100 ms |
| Capillary Temperature: 300 °C | Capillary (kV): 3.0 | Source temperature (°C): 320 |
| Sheath Gas: 35 |
| Technique | Identification Points |
|---|---|
| Separation (UPLC) | 1.0 |
| HR-MS Precursor Ion | 1.5 |
| Ion Product (HR-MSn) | 2.5 |
| Chemical Class | Analyte | Formula | Specie | RT (min) | Precursor (m/z) | Fragment1 | Fragment2 | N(CE) |
|---|---|---|---|---|---|---|---|---|
| Betalattamics Penicillins (8) | Amoxicillin | C16H19N3O5S | [M + H]+ | 7.22 | 366.1118 | 208.0 | 349.1 | 10 |
| Ampicillin | C16H19N3O4S | [M + H]+ | 11.02 | 350.1169 | 106.1 | 192.0 | 20 | |
| Cloxacillin | C19H18ClN3O5S | [M + H]+ | 17.24 | 436.0728 | 277.0 | 160.0 | 10 | |
| Dicloxacillin | C19H17Cl2N3O5S | [M + H]+ | 17.89 | 470.0339 | 160.0 | 311.0 | 15 | |
| Nafcillin | C21H22N2O5S | [M + H]+ | 17.95 | 415.1322 | 199.1 | 256.1 | 20 | |
| Oxacillin | C19H19N3O5S | [M + H]+ | 16.91 | 402.1118 | 160.0 | 243.1 | 15 | |
| Penicillin G | C16H18N2O4S | [M + Na]+ | 15.90 | 357.0882 | 160.0 | 176.1 | 10 | |
| Penicillin V | C16H18N2O5S | [M + Na]+ | 16.93 | 373.0829 | 160.0 | 192.1 | 15 | |
| Betalattamics Cephalosporins (6) | Cefalexin | C16H17N3O4S | [M + H]+ | 10.37 | 348.1013 | 158.0 | 174.1 | 40 |
| Cefazolin | C14H14N8O4S3 | [M + H]+ | 10.80 | 455.0373 | 156.0 | 153.0 | 15 | |
| Cefapirin | C17H17N3O6S2 | [M + H]+ | 8.08 | 424.0632 | 152.0 | 292.1 | 25 | |
| Cefquinome | C23H24N6O5S2 | [M + 2H]+ | 8.66 | 265.0695 | 134.1 | 324.1 | 16 | |
| Cefoperazone | C25H27N9O8S2 | [M + H]+ | 11.37 | 646.1497 | 143.1 | 290.1 | 16 | |
| Ceftiofur | C19H17N5O7S3 | [M + H]+ | 13.84 | 524.0363 | 241.0 | 210.0 | 25 | |
| Tetracyclines Epi-tetrcyclines (4+4) | Chlortetracycline | C22H23ClN2O8 | [M + H]+ | 12.55 | 479.1216 | 444.1 | 154.0 | 26 |
| Doxycycline | C22H24N2O8 | [M + H]+ | 14.21 | 445.1621 | 428.1 | 410.1 | 30 | |
| Oxytetracycline | C22H24N2O9 | [M + H]+ | 10.53 | 461.1555 | 426.1 | 337.1 | 30 | |
| Tetracycline | C22H24N2O8 | [M + H]+ | 10.27 | 445.1605 | 154.0 | 410.1 | 30 | |
| Epi- Chlortetracycline | C22H23ClN2O8 | [M + H]+ | 11.58 | 479.1216 | 444.1 | 154.0 | 26 | |
| Epi- Doxycycline | C22H24N2O8 | [M + H]+ | 13.57 | 445.1605 | 428.1 | 410.1 | 30 | |
| Epi- Oxytetracycline | C22H24N2O9 | [M + H]+ | 10.07 | 461.1555 | 426.1 | 201.1 | 30 | |
| Epi-Tetracycline | C22H24N2O8 | [M + H]+ | 9.38 | 445.1605 | 410.1 | 392.1 | 30 | |
| Pleuromutilins (2) | Tiamulin | C28H47NO4S | [M + H]+ | 15.70 | 494.3299 | 192.1 | 119.0 | 30 |
| Valnemulin | C31H52N2O5S | [M + H]+ | 17.62 | 565.3670 | 263.1 | 164.1 | 30 | |
| Macrolides (5) | Tilmicosin | C46H80N2O13 | [M + 2H]+ | 13.94 | 435.2903 | 174.1 | 696.5 | 32 |
| Tylosin | C46H77NO17 | [M + H]+ | 15.93 | 916.5264 | 174.1 | 101.1 | 25 | |
| Azithromycin | C38H72N2O12 | [M + H]+ | 13.05 | 749.5171 | 158.1 | 83.0 | 28 | |
| Erythromycin | C37H67NO13 | [M + H]+ | 16.14 | 734.4685 | 158.1 | 83.0 | 20 | |
| Spiramycin | C43H74N2O14 | [M + 2H]+ | 12.48 | 422.2643 | 540.3 | 699.4 | 30 | |
| Lincosamides (2) | Clindamycin | C18H33ClN2O5S | [M + H]+ | 14.46 | 425.1872 | 126.1 | 377.2 | 30 |
| Lincomycin | C18H34N2O6S | [M + H]+ | 9.17 | 407.2210 | 126.1 | 359.2 | 30 | |
| Quinolones (11) | nalidixic Acid | C12H12N2O3 | [M + H]+ | 15.51 | 233.0921 | 205.1 | 159.1 | 70 |
| oxolinic Acid | C13H11NO5 | [M + H]+ | 13.86 | 262.0710 | 160.0 | 234.0 | 80 | |
| Ciprofloxacin | C17H18FN3O3 | [M + H]+ | 10.60 | 332.1405 | 231.1 | 203.1 | 65 | |
| Danofloxacin | C19H20FN3O3 | [M + H]+ | 10.75 | 358.1562 | 82.1 | 255.1 | 70 | |
| Difloxacin | C21H19F2N3O3 | [M + H]+ | 11.03 | 400.1467 | 299.1 | 58.1 | 65 | |
| Enrofloxacin | C19H22FN3O3 | [M + H]+ | 10.66 | 360.1718 | 203.1 | 245.1 | 60 | |
| Flumequin | C14H12FNO3 | [M + H]+ | 16.01 | 262.0874 | 238.1 | 220.0 | 80 | |
| Marbofloxacin | C17H19FN4O4 | [M + H]+ | 9.54 | 363.1463 | 72.1 | 320.1 | 25 | |
| Norfloxacin | C16H18FN3O3 | [M + H]+ | 10.40 | 320.1405 | 231.1 | 203.1 | 80 | |
| Ofloxacin | C18H20FN3O4 | [M + H]+ | 10.07 | 362.1511 | 261.1 | 221.1 | 50 | |
| Sarafloxacin | C20H17F2N3O3 | [M + H]+ | 11.34 | 386.1311 | 299.1 | 338.1 | 60 | |
| Sulfonamides (15) | Sulfaquinoxaline | C14H12N4O2S | [M + H]+ | 13.56 | 301.0754 | 156.0 | 108.0 | 38 |
| Sulfachloropyridazine | C10H9ClN4O2S | [M + H]+ | 10.75 | 285.0208 | 156.0 | 108.0 | 35 | |
| Sulfadiazine | C10H10N4O2S | [M + H]+ | 7.39 | 251.0597 | 156.0 | 108.0 | 35 | |
| Sulfadimethoxin | C12H14N4O4S | [M + H]+ | 13.16 | 311.0809 | 156.1 | 108.0 | 42 | |
| Sulfaguanidine | C7H10N4O2S | [M + H]+ | 3.21 | 215.0597 | 156.0 | 108.0 | 40 | |
| Sulfamerazine | C11H12N4O2S | [M + Na]+ | 8.69 | 287.0573 | 156.0 | 190.0 | 42 | |
| Sulfametazine | C12H14N4O2S | [M + H]+ | 9.80 | 279.0910 | 124.1 | 156.0 | 42 | |
| Sulfamethizole | C9H10N4O2S2 | [M + H]+ | 9.78 | 271.0318 | 156.0 | 108.0 | 40 | |
| Sulfamethoxazole | C10H11N3O3S | [M + H]+ | 10.91 | 254.0594 | 156.0 | 108.0 | 40 | |
| S.methoxipyridazine | C11H12N4O3S | [M + H]+ | 10.20 | 281.0703 | 126.1 | 108.0 | 50 | |
| Sulfamonomethoxin | C11H12N4O3S | [M + H]+ | 11.17 | 281.0710 | 156.0 | 108.0 | 41 | |
| Sulfanilamide | C6H8N2O2S | [M + H][NH3]+ | 3.65 | 156.0114 | 65.0 | 92.0 | 70 | |
| Sulfapyridin | C11H11N3O2S | [M + H]+ | 8.27 | 250.0645 | 156.0 | 184.1 | 43 | |
| Sulfathiazole | C9H9N3O2S2 | [M + H]+ | 7.97 | 256.0209 | 156.0 | 108.0 | 38 | |
| Trimethoprim | C14H18N4O3 | [M + H]+ | 9.37 | 291.1452 | 123.1 | 261.1 | 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salis, S.; Rubattu, N.; Rubattu, F.; Cossu, M.; Sanna, A.; Chessa, G. Analytical Approaches in Official Food Safety Control: An LC-Orbitrap-HRMS Screening Method for the Multiresidue Determination of Antibiotics in Cow, Sheep, and Goat Milk. Molecules 2022, 27, 6162. https://doi.org/10.3390/molecules27196162
Salis S, Rubattu N, Rubattu F, Cossu M, Sanna A, Chessa G. Analytical Approaches in Official Food Safety Control: An LC-Orbitrap-HRMS Screening Method for the Multiresidue Determination of Antibiotics in Cow, Sheep, and Goat Milk. Molecules. 2022; 27(19):6162. https://doi.org/10.3390/molecules27196162
Chicago/Turabian StyleSalis, Severyn, Nicola Rubattu, Federica Rubattu, Maurizio Cossu, Andrea Sanna, and Giannina Chessa. 2022. "Analytical Approaches in Official Food Safety Control: An LC-Orbitrap-HRMS Screening Method for the Multiresidue Determination of Antibiotics in Cow, Sheep, and Goat Milk" Molecules 27, no. 19: 6162. https://doi.org/10.3390/molecules27196162
APA StyleSalis, S., Rubattu, N., Rubattu, F., Cossu, M., Sanna, A., & Chessa, G. (2022). Analytical Approaches in Official Food Safety Control: An LC-Orbitrap-HRMS Screening Method for the Multiresidue Determination of Antibiotics in Cow, Sheep, and Goat Milk. Molecules, 27(19), 6162. https://doi.org/10.3390/molecules27196162