
From Gas Phase Observations to Solid State Reality: The Identification and Isolation of Trinuclear Salicylaldoximato Copper Complexes

 , , , ,
, , , ,
Abstract
: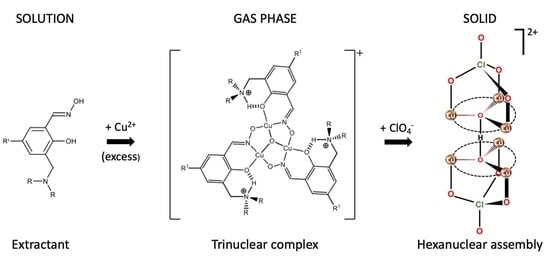
1. Introduction
2. Results and Discussion
2.1. ESI:MS Studies
2.2. UV/Vis Spectra
2.3. Solid State Samples and X-ray Structures of the Trinuclear Complexes

3. Materials and Methods
3.1. Materials and General Procedures
3.2. Synthesis of Proligands and Complexes
3.2.1. 5-t-Octyl-2-Hydroxyphenylethanone Oxime (L4H2)
3.2.2. 2-Hydroxy-5-t-Octylbenzaldehyde Oxime (L5H2)
3.2.3. (5-t-Butyl-2-Hydroxyphenyl)Ethanone Oxime (L7H2)
3.3. Syntheses of Complexes
3.3.1. [Cu(L5H)2]
3.3.2. [Cu(L8H)2]
3.3.3. [Cu3OH(L9H)3(ClO4)2]1.5H2O
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mackey, P.J. The changing landscape in copper in 2007. CIM MAGAZINE 2007, 2, 35. [Google Scholar]
- Kordosky, G.A. Proceedings of the International Solvent Extraction Conference, Cape Town, South Africa, 17–21 March 2002; South African Chemical Institute: Johanesburg, South Africa, 2002; pp. 853–862. [Google Scholar]
- International Copper Study Group. The World Copper Factbook 2021; ICSG: Lisbon, Portugal, 2021; p. 11. [Google Scholar]
- Szymanowski, J. Hydroxyoximes and Copper Hydrometallurgy; CRC Press: Boca Raton, FL, USA, 1993. [Google Scholar]
- Wilson, A.M.; Bailey, P.J.; Tasker, P.A.; Turkington, J.R.; Grant, R.A.; Love, J.B. Solvent extraction: The coordination chemistry behind extractive metallurgy. Chem. Soc. Rev. 2014, 43, 123–134. [Google Scholar] [CrossRef]
- Tasker, P.A.; Doidge, E.D. The coordination chemistry of extractive metallurgy. In Coordination Chemistry III; Elsevier: Amsterdam, The Netherlands, 2021; pp. 494–557. [Google Scholar]
- Mason, K.; Chang, J.; Prescimone, A.; Garlatti, E.; Carretta, S.; Tasker, P.A.; Brechin, E.K. Linking [MIII3] triangles with “double-headed” phenolic oximes. Dalton Trans. 2012, 41, 8777–8785. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.I.; Cheng, K.H.; Hung, S.P.; Nakano, M.; Tsai, H.L. Crystal packing effects within [MnIII3O]7+ single-molecule magnets: Controlling intermolecular antiferromagnetic interactions. Polyhedron 2011, 30, 3272–3278. [Google Scholar] [CrossRef]
- Inglis, R.; Jones, L.F.; Mason, K.; Collins, A.; Moggach, S.A.; Parsons, S.; Perlepes, S.P.; Wernsdorfer, W.; Brechin, E.K. Ground Spin State Changes and 3D Networks of Exchange Coupled [MnIII3] Single-Molecule Magnets. Chem. Eur. J. 2008, 14, 9117–9121. [Google Scholar] [CrossRef]
- Inglis, R.; Jones, L.F.; Milios, C.J.; Datta, S.; Collins, A.; Parsons, S.; Wernsdorfer, W.; Hill, S.; Perlepes, S.P.; Piligkos, S. Attempting to understand (and control) the relationship between structure and magnetism in an extended family of Mn6 single-molecule magnets. Dalton Trans. 2009, 18, 3403–3412. [Google Scholar] [CrossRef]
- Jones, L.F.; Inglis, R.; Cochrane, M.E.; Mason, K.; Collins, A.; Parsons, S.; Perlepes, S.P.; Brechin, E.K. New structural types and different oxidation levels in the family of Mn6-oxime single-molecule magnets. Dalton Trans. 2008, 44, 6205–6210. [Google Scholar] [CrossRef] [PubMed]
- Stoumpos, C.C.; Inglis, R.; Karotsis, G.; Jones, L.F.; Collins, A.; Parsons, S.; Milios, C.J.; Papaefstathiou, G.S.; Brechin, E.K. Supramolecular entanglement from interlocked molecular nanomagnets. Cryst. Growth Des. 2009, 9, 24–27. [Google Scholar] [CrossRef]
- Milios, C.J.; Inglis, R.; Vinslava, A.; Bagai, R.; Wernsdorfer, W.; Parsons, S.; Perlepes, S.P.; Christou, G.; Brechin, E.K. Toward a magnetostructural correlation for a family of Mn6 SMMs. J. Am. Chem. Soc. 2007, 129, 12505–12511. [Google Scholar] [CrossRef]
- Verani, C.N.; Bothe, E.; Burdinski, D.; Weyhermüller, T.; Flörke, U.; Chaudhuri, P. Synthesis, structure, electrochemistry, and magnetism of [MnIIIMnIII], [MnIIIFeIII] and [FeIIIFeIII] cores: Generation of phenoxyl radical containing [FeIIIFeIII] species. Eur. J. Inorg. Chem. 2001, 2001, 2161–2169. [Google Scholar] [CrossRef]
- Frost, J.M.; Sanz, S.; Rajeshkumar, T.; Pitak, M.B.; Coles, S.J.; Rajaraman, G.; Wernsdorfer, W.; Schnack, J.; Lusby, P.J.; Brechin, E.K. A truncated [MnIII12] tetrahedron from oxime-based [MnIII3O] building blocks. Dalton Trans. 2014, 43, 10690–10694. [Google Scholar] [CrossRef] [Green Version]
- Mason, K.; Chang, J.; Garlatti, E.; Prescimone, A.; Yoshii, S.; Nojiri, H.; Schnack, J.; Tasker, P.A.; Carretta, S.; Brechin, E.K. Linking [FeIII3] triangles with “double-headed” phenolic oximes. Chem. Commun. 2011, 47, 6018–6020. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.; Prescimone, A.; Schau-Magnussen, M.; Piligkos, S.; Tasker, P.A.; Brechin, E.K. Salicylaldoxime-Supported Nona-and Tetrametallic FeIII Cages. Curr. Inorg. Chem. 2013, 3, 76–85. [Google Scholar] [CrossRef]
- Gass, I.A.; Milios, C.J.; Whittaker, A.G.; Fabiani, F.P.A.; Parsons, S.; Murrie, M.; Perlepes, S.P.; Brechin, E.K. A cube in a tetrahedron: Microwave-assisted synthesis of an octametallic FeIII cluster. Inorg. Chem. 2006, 45, 5281–5283. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.; Gass, I.A.; Parsons, S.; Collins, A.; White, F.J.; Slawin, A.M.Z.; Brechin, E.K.; Tasker, P.A. Building Fe(III) clusters with derivatised salicylaldoximes. Dalton Trans. 2010, 39, 2727–2734. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, J.M.; Beddoes, R.L.; Collison, D.; Garner, C.D.; Helliwell, M.; Holmes, J.M.; Tasker, P.A. Surface coordination chemistry: Corrosion inhibition by tetranuclear cluster formation of iron with salicylaldoxime. Angew. Chem. Int. Ed. 1999, 38, 1119–1121. [Google Scholar] [CrossRef]
- Wenzel, M.; Forgan, R.S.; Faure, A.; Mason, K.; Tasker, P.A.; Piligkos, S.; Brechin, E.K.; Plieger, P.G. A new polynuclear coordination type for (salicylaldoxime) copper(II) complexes: Structure and magnetic properties of an (oxime) Cu6 cluster. Eur. J. Inorg. Chem. 2009, 31, 4613–4617. [Google Scholar] [CrossRef]
- Curtis, N.F.; Morgan, K.R. A tri-nuclear oxygen centred (salicylaldoximato) copper(II) compound: The preparation and structure of [{{Cu(saloxH)}3O}3H1. 5]·Cl4·5:12DMSO·1.83H2O. J. Mol. Struct. 2011, 1006, 375–378. [Google Scholar] [CrossRef]
- Iasco, O.; Novitchi, G.; Jeanneau, E.; Luneau, D. Lanthanide Triangles Sandwiched by Tetranuclear Copper Complexes Afford a Family of Hendecanuclear Heterometallic Complexes [LnIII3CuII8] (Ln = La–Lu): Synthesis and Magnetostructural Studies. Inorg. Chem. 2013, 52, 8723–8731. [Google Scholar] [CrossRef] [PubMed]
- Zhekova, H.R.; Seth, M.; Ziegler, T. First Principle Simulation of the Temperature Dependent Magnetic Circular Dichroism of a Trinuclear Copper Complex in the Presence of Zero Field Splitting. J. Phys. Chem. A 2011, 115, 10323–10334. [Google Scholar] [CrossRef] [PubMed]
- Mariam, Y.H.; Musin, R.N. A B3LYP study of intramolecular hydrogen bonding and proton transfer in naphthazarin: A model system for daunomycin/adriamycin. J. Mol. Struct. THEOCHEM 2001, 549, 123–136. [Google Scholar] [CrossRef]
- Green, R.W.; Svasti, M.C.K. Copper(II) Complexes of 2-Pyridinaldoxime. Aust. J. Chem. 1963, 16, 356–359. [Google Scholar] [CrossRef]
- Speed, S.; Font-Bardía, M.; El Fallah, M.S.; Vicente, R. Four new trinuclear {Cu3(μ3-OH)(oximate)3}2+ clusters: Crystal structure and magnetic behaviour. Dalton Trans. 2014, 43, 16919–16927. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.K.; Duan, J.G.; Chai, L.Q.; Liu, G.L.; Wu, H.L. Synthesis and structural characterization of new trinuclear cobalt(II) and nickel(II) complexes possessing five-and six-coordinated geometry. J. Coord. Chem. 2008, 61, 1306–1315. [Google Scholar] [CrossRef]
- Dong, W.K.; Duan, J.G.; Guan, Y.H.; Shi, J.Y.; Zhao, C.Y. Synthesis, crystal structure and spectroscopic behaviors of Co(II) and Cu(II) complexes with Salen-type bisoxime ligands. Inorg. Chim. Acta 2009, 362, 1129–1134. [Google Scholar] [CrossRef]
- Martínez, L.; Bazzicalupi, C.; Bianchi, A.; Lloret, F.; González, R.; Kremer, C.; Chiozzone, R. Structural and magnetic properties of polynuclear oximate copper complexes with different topologies. Polyhedron 2017, 138, 125–132. [Google Scholar] [CrossRef]
- Chakraborty, A.; Escuer, A.; Ribas, J.; Maji, T.K. A discrete CuII6 cluster and a 3D MnII–CuII framework based on assembly of Mn2 Cu4 clusters: Synthesis, structure and magnetic properties. Dalton Trans. 2016, 45, 15523–15531. [Google Scholar] [CrossRef] [PubMed]
- Tarushi, A.; Raptopoulou, C.P.; Psycharis, V.; Kontos, C.K.; Kessissoglou, D.P.; Scorilas, A.; Tangoulis, V.; Psomas, G. Copper(II) Inverse-[9-Metallacrown-3] compounds accommodating nitrato or diclofenac ligands: Structure, magnetism, and biological activity. Eur. J. Inorg. Chem. 2016, 2016, 219–231. [Google Scholar] [CrossRef]
- Argyle, V.J.; Roxburgh, M.; Hanton, L.R. Structural control in Cu(II) coordination polymers through the conformational flexibility of a 2,3-dipyridyl ketone oxime ligand. Supramol. Chem. 2015, 27, 820–828. [Google Scholar] [CrossRef]
- Beckett, R.; Hoskins, B.F. Crystal and molecular structure of a trinuclear copper(II) complex: µ3-hydroxo-tri-µ-(pyridine-2-carbaldehyde oximato)-µ3-sulphato-tricopper(II)–16:3water. J. Chem. Soc. Dalton Trans. 1972, 3, 291–295. [Google Scholar] [CrossRef]
- Datta, D.; Chakravorty, A. Electron transfer in authentic triangular copper(II) trimers with Cu3X (X = oxygen or hydroxy) core. The CuII2CuIII-CuII3 and CuII3-CuCuICuII2 couples. Inorg. Chem. 1982, 21, 363–368. [Google Scholar] [CrossRef]
- Butcher, R.J.; O’Connor, C.J.; Sinn, E. Synthesis and relation between magnetism and structure of a trinuclear copper(II) hydroxo complex [Cu3OHL3(ClO4)]ClO4 and a hexanuclear copper(II) oxo complex [Cu3OL′3(ClO4)]2 (LH = 3-(phenylimino) butanone 2-oxime, L’H = 1, 2-diphenyl-2-(methylimino)ethanone-1-oxime). Inorg. Chem. 1981, 20, 537–545. [Google Scholar]
- Agnus, Y.; Louis, R.; Metz, B.; Boudon, C.; Gisselbrecht, J.P.; Gross, M. Trinuclear copper(II) hydroxo and hexanuclear copper(II) oxo complexes with the ligand 3-(benzylimino)butanone-2-oxime. Syntheses and spectral, structural and redox characteristics. Inorg. Chem. 1991, 30, 3155–3161. [Google Scholar] [CrossRef]
- Komori, H.; Sugiyama, R.; Kataoka, K.; Miyazaki, K.; Higuchi, Y.; Sakurai, T. New insights into the catalytic active-site structure of multicopper oxidases. Acta Crystallogr. Sect. D 2014, 70, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Vancoillie, S.; Chalupský, J.; Ryde, U.; Solomon, E.I.; Pierloot, K.; Neese, F.; Rulíšek, L. Multireference Ab Initio Calculations of g tensors for Trinuclear Copper Clusters in Multicopper Oxidases. J. Phys. Chem. B 2010, 114, 7692–7702. [Google Scholar] [CrossRef] [PubMed]
- Hannigan, S.F.; Arnoff, A.I.; Neville, S.E.; Lum, J.S.; Golen, J.A.; Rheingold, A.L.; Orth, N.; Ivanović-Burmazović, I.; Liebhäuser, P.; Rösener, T.; et al. On the Way to a Trisanionic {Cu3O2} Core for Oxidase Catalysis: Evidence of an Asymmetric Trinuclear Precursor Stabilized by Perfluoropinacolate Ligands. Chem. Eur. J. 2017, 23, 8212–8224. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.B.; Liang, L.; Zhang, J.; Sun, H.L.; Zhang, J.L. μ-Hydroxyl trinuclear copper(II) clusters: Reactivity and unusual formation in the three-component synthesis of 1,2,3-triazoles in aqueous media. Dalton Trans. 2013, 42, 5390–5400. [Google Scholar] [CrossRef]
- Chan, S.I.; Wang, V.C.C.; Lai, J.C.H.; Yu, S.S.F.; Chen, P.P.Y.; Chen, K.H.C.; Chen, C.-L.; Chan, M.K. Redox Potentiometry Studies of Particulate Methane Monooxygenase: Support for a Trinuclear Copper Cluster Active Site. Angew. Chem. Int. Ed. 2007, 46, 1992–1994. [Google Scholar] [CrossRef]
- Yoon, J.; Solomon, E.I. Ground-State Electronic and Magnetic Properties of a μ3-Oxo-Bridged Trinuclear Cu(II) Complex: Correlation to the Native Intermediate of the Multicopper Oxidases. Inorg. Chem. 2005, 44, 8076–8086. [Google Scholar] [CrossRef] [PubMed]
- Roach, B.D.; Forgan, R.S.; Tasker, P.A.; Swart, R.M.; Campbell, J.; McAllister, F.E.; Stopford, A.P.; Duncombe, B.J. Collision induced dissociation (CID) to probe the outer sphere coordination chemistry of bis-salicylaldoximate complexes. Dalton Trans. 2010, 39, 5614–5616. [Google Scholar] [CrossRef]
- Forgan, R.S.; Roach, B.D.; Wood, P.A.; White, F.J.; Campbell, J.; Henderson, D.K.; Kamenetzky, E.; McAllister, F.E.; Parsons, S.; Pidcock, E. Using the outer coordination sphere to tune the strength of metal extractants. Inorg. Chem. 2011, 50, 4515–4522. [Google Scholar] [CrossRef]
- Smith, A.G.; Tasker, P.A.; White, D.J. The structures of phenolic oximes and their complexes. Coord. Chem. Rev. 2003, 241, 61–85. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Strassner, T.; Taige, M.A. Evaluation of Functionals O3LYP, KMLYP, and MPW1K in Comparison to B3LYP for Selected Transition-Metal Compounds. J. Chem. Theory Comput. 2005, 1, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Yu, L.; Dai, Y. Density functional theory and topological analysis on the hydrogen bonds in cysteine—Propanoic acid complexes. Struct. Chem. 2010, 21, 855–862. [Google Scholar] [CrossRef]
- Ruangpornvisuti, V. A DFT investigation of dissociation of nitrous acid, transformation of its monomeric isomers, their dimeric isomers, and dimerization. Int. J. Quantum Chem. 2010, 110, 1019–1029. [Google Scholar] [CrossRef]
- Himmel, H.-J. Calculated Enthalpies for Dimerisation of Binary, Unsaturated, Main-Group Element Hydrides as a Means to Analyse Their Potential for Multiple Bonding. Eur. J. Inorg. Chem. 2003, 2003, 2153–2163. [Google Scholar] [CrossRef]
- Aloisio, S.; Francisco, J.S. Existence of a Hydroperoxy and Water (HO2·H2O) Radical Complex. J. Phys. Chem. A 1998, 102, 1899–1902. [Google Scholar] [CrossRef]
- Troitiño, D.; Bailey, L.; Peral, F. Formation of hydrogen-bonded dimers of uracil and amino acids: Cysteine, lysine and phenylalanine. A density functional theory study. J. Mol. Struct. THEOCHEM 2006, 767, 131–138. [Google Scholar] [CrossRef]
- Borisenko, V.E.; Krekov, S.A.; Fomenko, M.Y.; Koll, A.; Lipkovski, P. Influence of methoxy- and nitro-substitutions in the aromatic ring on proton donation ability in hydrogen bond and on the amino group parameters of free and H-bonded molecules of 2-aminopyrimidine. J. Mol. Struct. 2008, 882, 9–23. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Forgan, R.S.; Davidson, J.E.; Fabbiani, F.P.A.; Galbraith, S.G.; Henderson, D.K.; Moggach, S.A.; Parsons, S.; Tasker, P.A.; White, F.J. Cation and anion selectivity of zwitterionic salicylaldoxime metal salt extractants. Dalton Trans. 2010, 39, 1763–1770. [Google Scholar] [CrossRef]
- Forgan, R.S.; Davidson, J.E.; Galbraith, S.G.; Henderson, D.K.; Parsons, S.; Tasker, P.A.; White, F.J. Transport of metal salts by zwitterionic ligands; simple but highly efficient salicylaldoxime extractants. Chem. Commun. 2008, 34, 4049–4051. [Google Scholar] [CrossRef]
- van der Sluis, P.; Spek, A.L. BYPASS: An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Crystallogr. Sect. A 1990, 46, 194–201. [Google Scholar] [CrossRef]
- Roach, B.D.; White, F.J.; Tasker, P.A. CSD Communication 2015; Cambridge Crystallographic Data Centre: Cambridge, UK, 2015. [Google Scholar] [CrossRef]
- Whyte, A.M.; Roach, B.D.; Henderson, D.K.; Tasker, P.A.; Matsushita, M.M.; Awaga, K.; White, F.J.; Richardson, P.; Robertson, N. Structural, Magnetic, and Electronic Properties of Phenolic Oxime Complexes of Cu and Ni. Inorg. Chem. 2011, 50, 12867–12876. [Google Scholar] [CrossRef] [PubMed] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LnH2 | LnH2:Cu2+ | Monoisotopic m/z for [Cu3(L)3O]− | Calculated Monoisotopic m/z for [Cu3(L)3O]− | Observed Intensity (%) |
|---|---|---|---|---|
| L4H2 | 1:3 | 990.01 | 990.30 | 100 |
| L5H2 | 1:2 | 948.49 | 948.25 | 100 |
| L6H2 | 1:2 | 779.93 | 780.07 | 100 |
| L7H2 | 1:2 | 822.02 | 822.11 | 100 |
| L8H2 | 1:2 | 1015.47 | 1015.80 | 100 |
| Cu-Noximate | Cu-Ophenolate | Cu-Ooximate | Cu-Ohydroxide | |
| Bond lengths in A | 1.949(3)–1.970(3) | 1.872(3)–1.921(2) | 1.906(3)–1.963(2) | 1.932(3)–1.962(3) |
| and in B | 1.941(2)–1.956(2) | 1.882(2)–1.9207(19) | 1.915(2)–1.9443(19) | 1.950(2)–1.9791(19) |
| Noximate-Cu-Ophenolate | Ophenolate-Cu-Ooximate | Ooximate-Cu-Ohydroxide | Ohydroxide-Cu-Noximate | |
| Bond angles in A | 92.16(12)–93.94(11) | 85.07(11)–88.04(11) | 89.82(11)–91.38(10) | 89.72(11)–93.94(11) |
| and in B | 93.18(10)–94.95(9) | 84.72(8)–86.07(8) | 87.99(8)–91.57(8) | 88.75(9)–91.05(9) |
| Complex | (a) [Cu3(L2)3OH]− | (b) [Cu3(L2)3OH] | (c) [Cu3(L2)3O]2− | (d) [Cu3(L2)3O]− |
| Charge | −1 | 0 | −2 | −1 |
| Multiplicity | 4 | 3 | 4 | 3 |
| Enthalpy (Hartree) | −6421.9645 | −6421.8295 | −6421.3514 | −6421.3277 |
| Enthalpy (J mol−1) | −16,860,868 | −16,860,513 | −16,859,258 | −16,859,196 |
| Edeprotonation [Cu3(L2)3OH]− (a) → [Cu3(L2)3O]2− (c) | 1610 | |||
| Edeprotonation [Cu3(L2)3OH] (b) → [Cu3(L2)3O]− (d) | 1317 | |||
| Eionisation [Cu3(L2)3OH]− (a) → [Cu3(L2)3OH]2− (b) | 355 | |||
| Eionisation [Cu3(L2)3O]2− (c) → [Cu3(L2)3O]− (d) | 62 | |||
| Sample Flow Rate | 5 µL/min |
|---|---|
| Sheath gas flow rate | 46 (Arb. Units) |
| Aux. gas flow rate | 0 (Arb. Units) |
| Capillary temperature | 190 °C |
| RF amplitude | 560 V |
| Spray Voltage | 4.5 kV |
| Capillary Voltage | −26.5 V |
| Octapole 1 offset | 6.8 V |
| Lens Voltage | 13 V |
| Octapole 2 offset | 10.5 V |
| Tube lens offset | 0 V |
| Sample Injection Volume | 6 µL |
|---|---|
| Desolvation gas flow rate | 800 L/h |
| Aux. gas flow rate | 30 L/h |
| Source temperature | 90 °C |
| Desolvation Temperature | 350 °C |
| Spray Voltage | 2.2 kV |
| Extraction Cone | 4.0 |
| Sampling Cone | 80 |
| Ion Guide | 2.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roach, B.D.; Forgan, R.S.; Kamenetzky, E.; Parsons, S.; Plieger, P.G.; White, F.J.; Woodhouse, S.; Tasker, P.A. From Gas Phase Observations to Solid State Reality: The Identification and Isolation of Trinuclear Salicylaldoximato Copper Complexes. Molecules 2022, 27, 6421. https://doi.org/10.3390/molecules27196421
Roach BD, Forgan RS, Kamenetzky E, Parsons S, Plieger PG, White FJ, Woodhouse S, Tasker PA. From Gas Phase Observations to Solid State Reality: The Identification and Isolation of Trinuclear Salicylaldoximato Copper Complexes. Molecules. 2022; 27(19):6421. https://doi.org/10.3390/molecules27196421
Chicago/Turabian StyleRoach, Benjamin D., Ross S. Forgan, Eduardo Kamenetzky, Simon Parsons, Paul G. Plieger, Fraser J. White, Sidney Woodhouse, and Peter A. Tasker. 2022. "From Gas Phase Observations to Solid State Reality: The Identification and Isolation of Trinuclear Salicylaldoximato Copper Complexes" Molecules 27, no. 19: 6421. https://doi.org/10.3390/molecules27196421
APA StyleRoach, B. D., Forgan, R. S., Kamenetzky, E., Parsons, S., Plieger, P. G., White, F. J., Woodhouse, S., & Tasker, P. A. (2022). From Gas Phase Observations to Solid State Reality: The Identification and Isolation of Trinuclear Salicylaldoximato Copper Complexes. Molecules, 27(19), 6421. https://doi.org/10.3390/molecules27196421