
Solvation of Large Polycyclic Aromatic Hydrocarbons in Helium: Cationic and Anionic Hexabenzocoronene

 , , , , , and
, , , , , and
Abstract
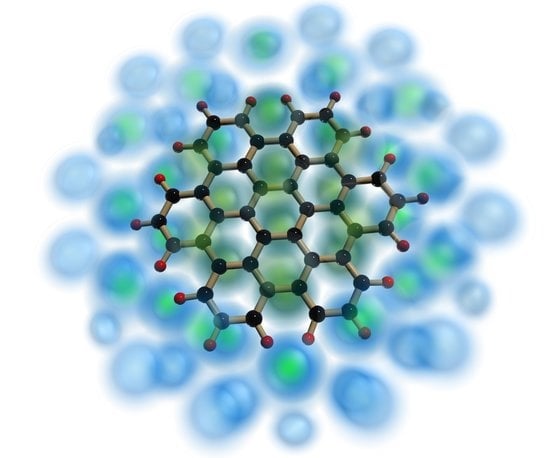
:
1. Introduction
2. Materials and Methods
2.1. Experimental Methods
2.2. Computational Methods
3. Results
3.1. Experimental Results
3.2. Theoretical Results
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Note
- Mauracher, A.; Echt, O.; Ellis, A.M.; Yang, S.; Bohme, D.K.; Postler, J.; Kaiser, A.; Denifl, S.; Scheier, P. Cold physics and chemistry: Collisions, ionization and reactions inside helium nanodroplets close to zero K. Phys. Rep. 2018, 751, 1–90. [Google Scholar] [CrossRef] [Green Version]
- Verma, D.; Tanyag, R.M.P.; O’Connell, S.M.O.; Vilesov, A.F. Infrared spectroscopy in superfluid helium droplets. Adv. Phys. X 2018, 4, 1553569. [Google Scholar] [CrossRef] [Green Version]
- Atkins, K.R. Ions in liquid helium. Phys. Rev. 1959, 116, 1339–1343. [Google Scholar] [CrossRef]
- González-Lezana, T.; Echt, O.; Gatchell, M.; Bartolomei, M.; Campos-Martínez, J.; Scheier, P. Solvation of ions in helium. Int. Rev. Phys. Chem. 2020, 39, 465–516. [Google Scholar] [CrossRef]
- Cole, M.W.; Bachman, R.A. Structure of positive impurity ions in liquid-helium. Phys. Rev. B 1977, 15, 1388–1394. [Google Scholar] [CrossRef]
- Mateo, D.; Eloranta, J. Solvation of intrinsic positive charge in superfluid helium. J. Phys. Chem. A 2014, 118, 6407–6415. [Google Scholar] [CrossRef] [Green Version]
- Bartl, P.; Leidlmair, C.; Denifl, S.; Scheier, P.; Echt, O. On the size and structure of helium snowballs formed around charged atoms and clusters of noble gases. J. Phys. Chem. A 2014, 118, 8050–8059. [Google Scholar] [CrossRef]
- Tramonto, F.; Salvestrini, P.; Nava, M.; Galli, D.E. Path integral Monte Carlo study confirms a highly ordered snowball in 4He nanodroplets doped with an Ar+ ion. J. Low Temp. Phys. 2015, 180, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Slavíček, P.; Lewerenz, M. Snowballs, quantum solvation and coordination: Lead ions inside small helium droplets. Phys. Chem. Chem. Phys. 2010, 12, 1152–1161. [Google Scholar] [CrossRef] [Green Version]
- Calvo, F. Size-induced melting and reentrant freezing in fullerene-doped helium clusters. Phys. Rev. B 2012, 85, 060502(R). [Google Scholar] [CrossRef]
- Harnisch, M.; Weinberger, N.; Denifl, S.; Scheier, P.; Echt, O. Adsorption of helium on isolated C60 and C70 anions. Mol. Phys. 2015, 113, 2191–2196. [Google Scholar] [CrossRef]
- Leidlmair, C.; Wang, Y.; Bartl, P.; Schöbel, H.; Denifl, S.; Probst, M.; Alcamí, M.; Martín, F.; Zettergren, H.; Hansen, K.; et al. Structures, energetics and dynamics of helium adsorbed on isolated fullerene ions. Phys. Rev. Lett. 2012, 108, 076101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.; Kwon, Y. Commensurate-incommensurate transition of 4He adsorbed on a single C60 molecule. J. Chem. Phys. 2012, 136, 064514. [Google Scholar] [CrossRef] [PubMed]
- Martini, P.; Albertini, S.; Laimer, F.; Meyer, M.; Gatchell, M.; Echt, O.; Zappa, F.; Scheier, P. Splashing of large helium nanodroplets upon surface collisions. Phys. Rev. Lett. 2021, 127, 263401. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, A.; Postler, J.; Ončák, M.; Kuhn, M.; Renzler, M.; Spieler, S.; Simpson, M.; Gatchell, M.; Beyer, M.K.; Wester, R.; et al. Isomeric broadening of C60+ electronic excitation in helium droplets: Experiments meet theory. J. Phys. Chem. Lett. 2018, 9, 1237–1242. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M.; Renzler, M.; Postler, J.; Ralser, S.; Spieler, S.; Simpson, M.; Linnartz, H.; Tielens, A.G.G.M.; Cami, J.; Mauracher, A.; et al. Atomically resolved phase transition of fullerene cations solvated in helium droplets. Nat. Commun. 2016, 7, 13550. [Google Scholar] [CrossRef] [Green Version]
- Heidenreich, A.; Even, U.; Jortner, J. Nonrigidity, delocalization, spatial confinement and electronic-vibrational spectroscopy of anthracene-helium clusters. J. Chem. Phys. 2001, 115, 10175–10185. [Google Scholar] [CrossRef]
- Xu, M.; Bacic, Z. Wave function delocalization and large-amplitude vibrations of helium on corrugated aromatic microsurfaces: Tetracene·He and pentacene·He van der Waals complexes. J. Phys. Chem. A 2007, 111, 7653–7663. [Google Scholar] [CrossRef]
- Whitley, H.D.; DuBois, J.L.; Whaley, K.B. Theoretical analysis of the anomalous spectral splitting of tetracene in 4He droplets. J. Phys. Chem. A 2011, 115, 7220–7233. [Google Scholar] [CrossRef]
- Kurzthaler, T.; Rasul, B.; Kuhn, M.; Lindinger, A.; Scheier, P.; Ellis, A.M. The adsorption of helium atoms on coronene cations. J. Chem. Phys. 2016, 145, 064305. [Google Scholar] [CrossRef]
- Rodriguez-Cantano, R.; de Tudela, R.P.; Bartolomei, M.; Hernandez, M.I.; Campos-Martinez, J.; Gonzalez-Lezana, T.; Villarreal, P.; Hernandez-Rojas, J.; Breton, J. Coronene molecules in helium clusters: Quantum and classical studies of energies and configurations. J. Chem. Phys. 2015, 143, 224306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, F. Shell completion of helium atoms around the coronene cation. Comput. Theor. Chem. 2017, 1107, 2–6. [Google Scholar] [CrossRef]
- Bergmeister, S.; Kollotzek, S.; Calvo, F.; Gruber, E.; Zappa, F.; Scheier, P.; Echt, O. Adsorption of helium and hydrogen on triphenylene and 1,3,5-triphenylbenzene. Molecules 2022, 27, 4937. [Google Scholar] [CrossRef] [PubMed]
- Kollotzek, S.; Calvo, F.; Krasnokutski, S.; Zappa, F.; Scheier, P.; Echt, O. Adsorption of helium on a charged propeller molecule: Hexaphenylbenzene. Eur. Phys. J. D 2021, 75, 299. [Google Scholar] [CrossRef]
- The strain in the adsorbate layer can be eased by moving the outer carbon rings further away from the central ring, as in triphenylbenzene (C24H18) and hexaphenylbenzene (C42H30), but in these propeller-shaped molecules, the orientation of the highly fluxional phenyl rings confounds the usefulness of simple geometric models.
- Clar, E. The Aromatic Sextet; Wiley Interscience: London, UK, 1972. [Google Scholar]
- Sola, M. Forty years of Clar’s aromatic π-sextet rule. Front. Chem. 2013, 1, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karunathilake, A.A.K.; Chang, J.; Thompson, C.M.; Nguyen, C.U.; Nguyen, D.Q.; Rajan, A.; Sridharan, A.; Vyakaranam, M.; Adegboyega, N.; Kim, S.J.; et al. Hexaphenylbenzene and hexabenzocoronene-based porous polymers for the adsorption of volatile organic compounds. RSC Adv. 2016, 6, 65763–65769. [Google Scholar] [CrossRef]
- Kumar, S.; Tao, Y.-T. Coronenes, benzocoronenes and beyond: Modern aspects of their syntheses, properties, and applications. Chem. Asian J. 2021, 16, 621–647. [Google Scholar] [CrossRef]
- Wong, W.W.H.; Subbiah, J.; Puniredd, S.R.; Purushothaman, B.; Pisula, W.; Kirby, N.; Müllen, K.; Jones, D.J.; Holmes, A.B. Liquid crystalline hexa-peri-hexabenzocoronene-diketopyrrolopyrrole organic dyes for photovoltaic applications. J. Mater. Chem. 2012, 22, 21131–21137. [Google Scholar] [CrossRef]
- Le Page, V.; Snow, T.P.; Bierbaum, V.M. Hydrogenation and charge states of polycyclic aromatic hydrocarbons in diffuse clouds. II. Results. Astrophys. J. 2003, 584, 316–330. [Google Scholar] [CrossRef]
- Montillaud, J.; Joblin, C.; Toublanc, D. Evolution of polycyclic aromatic hydrocarbons in photodissociation regions. Hydrogenation and charge states. Astron. Astrophys. 2013, 552, A15. [Google Scholar] [CrossRef]
- Wenzel, G.; Joblin, C.; Giuliani, A.; Castillo, S.R.; Mulas, G.; Ji, M.; Sabbah, H.; Quiroga, S.; Pena, D.; Nahon, L. Astrochemical relevance of VUV ionization of large PAH cations. Astron. Astrophys. 2020, 641, A98. [Google Scholar] [CrossRef]
- Calvo, F. Coating polycyclic aromatic hydrocarbon cations with helium clusters: Snowballs and slush. J. Phys. Chem. A 2015, 119, 5959–5970. [Google Scholar] [CrossRef]
- Schiller, A.; Meyer, M.; Martini, P.; Zappa, F.; Krasnokutskiy, S.; Calvo, F.; Scheier, P. Adsorption of helium on small cationic PAHs: Influence of hydrocarbon structure on the microsolvation pattern. J. Phys. Chem. A 2021, 125, 7813–7824. [Google Scholar] [CrossRef]
- Rempala, P.; Kroulik, J.; King, B.T. Investigation of the mechanism of the intramolecular Scholl reaction of contiguous phenylbenzenes. J. Org. Chem. 2006, 71, 5067–5081. [Google Scholar] [CrossRef]
- Meyer, M.; Martini, P.; Schiller, A.; Zappa, F.; Krasnokutski, S.A.; Scheier, P. Electronic spectroscopy of anthracene cations and protonated anthracene in the search for carriers of diffuse interstellar bands. Astrophys. J. 2021, 913, 136. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Rouille, G.; Steglich, M.; Huisken, F.; Henning, T.; Müllen, K. UV/visible spectroscopy of matrix-isolated hexa-peri-hexabenzocoronene: Interacting electronic states and astrophysical context. J. Chem. Phys. 2009, 131, 204311. [Google Scholar] [CrossRef]
- Mavrinskaya, N.; Räder, H.J.; Müllen, K. Nonlinear behavior during semi-quantitative analysis of thin organic layers by laser desorption mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 2196–2200. [Google Scholar] [CrossRef]
- Reed, R.I.; Tennent, A. Mass spectrum of 1-12, 2-3, 4-5, 6-7, 8-9, 10-11-hexabenzocoronene. Org. Mass Spectrom. 1971, 5, 619–621. [Google Scholar] [CrossRef]
- Ralser, S.; Postler, J.; Harnisch, M.; Ellis, A.M.; Scheier, P. Extracting cluster distributions from mass spectra: Isotopefit. Int. J. Mass Spectrom. 2015, 379, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Bjørnholm, S.; Borggreen, J.; Echt, O.; Hansen, K.; Pedersen, J.; Rasmussen, H.D. The influence of shells, electron thermodynamics, and evaporation on the abundance spectra of large sodium metal clusters. Z. Phys. D 1991, 19, 47–50. [Google Scholar] [CrossRef]
- Brack, M.; Genzken, O.; Hansen, K. Thermal electronic properties of alkali clusters. Z. Phys. D 1991, 19, 51–53. [Google Scholar] [CrossRef] [Green Version]
- Greywall, D.S. Heat capacity and the commensurate-incommensurate transition of 4He adsorbed on graphite. Phys. Rev. B 1993, 47, 309–318. [Google Scholar] [CrossRef]
- Gronowski, M.; Koza, A.M.; Tomza, M. Ab initio properties of the NaLi molecule in the a3Σ+ electronic state. Phys. Rev. A 2020, 102, 020801. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cations | Anions | |
|---|---|---|
| Experiment | 14, 26, 38, 68, 82 | 14, 26, 60, 62, 84 |
| Theory (PIMD) | 14, 24, 38, 68 | 14, 24, 38, 68 |
| Theory (classical) | 14, 24, 38, 86 | 14, 38, 68, 86 |
| Sphere packing | 14, 26, 38, 50, 62, 74 | 14, 26, 38, 50, 62, 74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kappe, M.; Calvo, F.; Schöntag, J.; Bettinger, H.F.; Krasnokutski, S.; Kuhn, M.; Gruber, E.; Zappa, F.; Scheier, P.; Echt, O. Solvation of Large Polycyclic Aromatic Hydrocarbons in Helium: Cationic and Anionic Hexabenzocoronene. Molecules 2022, 27, 6764. https://doi.org/10.3390/molecules27196764
Kappe M, Calvo F, Schöntag J, Bettinger HF, Krasnokutski S, Kuhn M, Gruber E, Zappa F, Scheier P, Echt O. Solvation of Large Polycyclic Aromatic Hydrocarbons in Helium: Cationic and Anionic Hexabenzocoronene. Molecules. 2022; 27(19):6764. https://doi.org/10.3390/molecules27196764
Chicago/Turabian StyleKappe, Miriam, Florent Calvo, Johannes Schöntag, Holger F. Bettinger, Serge Krasnokutski, Martin Kuhn, Elisabeth Gruber, Fabio Zappa, Paul Scheier, and Olof Echt. 2022. "Solvation of Large Polycyclic Aromatic Hydrocarbons in Helium: Cationic and Anionic Hexabenzocoronene" Molecules 27, no. 19: 6764. https://doi.org/10.3390/molecules27196764
APA StyleKappe, M., Calvo, F., Schöntag, J., Bettinger, H. F., Krasnokutski, S., Kuhn, M., Gruber, E., Zappa, F., Scheier, P., & Echt, O. (2022). Solvation of Large Polycyclic Aromatic Hydrocarbons in Helium: Cationic and Anionic Hexabenzocoronene. Molecules, 27(19), 6764. https://doi.org/10.3390/molecules27196764