
Syntheses of New Multisubstituted 1-Acyloxyindole Compounds

Abstract
:
1. Introduction
2. Results and Discussion
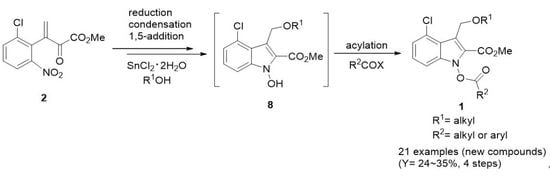
2.1. Synthesis of Conjugate Nitro Ketoester 2
2.2. Optimization for Formation of 1-Acyloxyindoles 1
2.3. Synthesis of New Derivatives of 1-Acyloxyindole 1
2.4. Mechanistic Investigations on Reaction Pathways
3. Experimental
3.1. General
3.2. Substrate Synthesis
- Methyl 3-(2′-Chloro-6′-Nitrophenyl)-2-Oxopropanoate (4) [32]
- 2-Chloro-6-nitrotoluene (3, 1.17 g, 6.8 mmol, 1.0 eq) and dimethyl oxalate (4.02 g, 34.0 mmol, 5.0 eq) were dissolved in anhydrous DMF (8.2 mL). To a stirred mixture of NaH (60% in mineral oil, 1.09 g, 27.2 mmol, 4.0 eq) in anhydrous DMF (4.1 mL) at 0 °C was added dropwise a solution of dimethyl oxalate and 2-chloro-6-nitrotoluene. The reaction mixture was stirred at 0 °C for 1 h and at room temperature for 4.5 h. The reaction mixture was quenched with saturated NH4Cl (15 mL) at 0 °C, extracted with methylene chloride (2 × 50 mL), and washed with H2O (2 × 50 mL). The organic layer was dried over MgSO4 and concentrated. The residue was purified by column chromatography (1:4 → 1:2 EtOAc/hexanes) to obtain compound 4 (1.68 g, 96%) as a pale-yellow solid. Spectral data are in accordance with literature information [32].
- Methyl 3-(2′-chloro-6′-nitrophenyl)-2-oxobut-3-enoate (2) [32]
- Ketoester (4, 1.53 g, 5.95 mmol, 1.0 eq) was dissolved in anhydrous THF (50 mL). To a stirred mixture of NaH (60% in mineral oil, 262 mg, 6.54 mmol, 1.1 eq) in anhydrous THF (100 mL) at 0 °C was added dropwise a solution of ketoester. After stirring for 1 h at 0 °C, N,N-dimethylmethyleneiminium chloride (1.85 g, 17.84 mmol, 3.0 eq) was added and the reaction mixture was stirred for 1 h at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred for additional 5 h. The reaction mixture was quenched with saturated NH4Cl (10 mL) at 0 °C, extracted with EtOAc (2 × 250 mL), and washed with H2O (2 × 250 mL). The organic layer was dried over MgSO4 and concentrated. The residue was purified by column chromatography (1:4 → 1:2 EtOAc/hexanes) to obtain compound 2 (1.36 g, 85%) as a pale-yellow solid. Spectral data are in accordance with literature information [32].
3.3. General Procedure for Synthesis of 1-Acyloxyindoles 1
- Methyl 4-chloro-1-acetoxy-3-[(methoxy)methyl]-1H-indole-2-carboxylate (1ax)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), methanol (9 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2.5 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and acetic anhydride (32 μL, 0.22 mmol, 2.0 eq) for 4 h in general procedure afforded the title compound 1ax (9.6 mg, 28%) as a yellow solid. Mp 104–106 °C; Rf 0.35 (1:2 EtOAc/hexanes); HPLC tR 12.1 min; UV–Vis (CH3CN-H2O) λmax 213, 232, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.30–7.14 (m, 3H, Ar), 5.12 (s, 2H, C(3)CH2O), 3.95 (s, 3H, CO2CH3), 3.46 (s, 3H, CH2OCH3), 2.43 (s, 3H, OC(O)CH3); 13C NMR (75 MHz, CDCl3) δ 168.5 (NOC(O)), 160.4 (CO2CH3), 136.9, 128.9, 127.1, 125.0, 123.7, 120.1, 118.2, 108.0 (Ar), 63.5 (CH2OCH3), 58.1 (C(3)CH2O), 52.4 (CO2CH3), 18.2 (OC(O)CH3); MS m/z 311 [M]+; HRMS (+ESI) calcd for C14H14ClNO5 [M]+ 311.0561, found 311.0559.
- Methyl 4-chloro-1-pivaloyloxy-3-[(methoxy)methyl]-1H-indole-2-carboxylate (1ay)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), methanol (9 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 1.5 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and pivaloyl chloride (28 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1ay (14.1 mg, 35%) as a pale-yellow solid. Mp 88–89 °C; Rf 0.47 (1:2 EtOAc/hexanes); HPLC tR 37.9 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.28–7.18 (m, 2H, Ar), 7.06 (dd, J = 7.8, 0.9 Hz, 1H, Ar), 5.13 (s, 2H, C(3)CH2O), 3.92 (s, 3H, CO2CH3), 3.44 (s, 3H, CH2OCH3), 1.47 (s, 9H, C(CH3)3); 13C NMR (75 MHz, CDCl3) δ 175.7 (NOC(O)), 160.2 (CO2CH3), 137.0, 128.9, 127.0, 125.7, 123.6, 120.3, 118.3, 107.8 (Ar), 63.5 (CH2OCH3), 58.0 (C(3)CH2O), 52.3 (CO2CH3), 38.8 (C(CH3)3), 27.4 (C(CH3)3); MS m/z 353 [M]+; HRMS (+ESI) calcd for C17H20ClNO5 [M]+ 353.1030, found 353.1028.
- Methyl 4-chloro-1-benzoyloxy-3-[(methoxy)methyl]-1H-indole-2-carboxylate (1az)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), methanol (9 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 1.5 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and benzoyl chloride (26 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1az (13.5 mg, 33%) as a pale-yellow solid. Mp 112–114 °C; Rf 0.39 (1:2 EtOAc/hexanes); HPLC tR 37.1 min; UV–Vis (CH3CN-H2O) λmax 236, 296 nm; 1H NMR (300 MHz, CDCl3) δ 8.22 (d, J = 7.6 Hz, 2H, Ar), 7.72 (t, J = 7.4 Hz, 1H, Ar), 7.57 (t, J = 7.7 Hz, 2H, Ar), 7.29–7.19 (m, 3H, Ar), 5.18 (s, 2H, C(3)CH2O), 3.83 (s, 3H, CO2CH3), 3.48 (s, 3H, CH2OCH3); 13C NMR (75 MHz, CDCl3) δ 164.6 (NOC(O)), 160.3 (CO2CH3), 137.4, 134.9, 130.6, 129.2, 128.9, 127.2, 126.4, 125.5, 123.8, 120.4, 118.8, 108.3 (Ar), 63.5 (CH2OCH3), 58.1 (C(3)CH2O), 52.4 (CO2CH3); MS m/z 373 [M]+; HRMS (+ESI) calcd for C19H16ClNO5 [M]+ 373.0717, found 373.0718.
- Methyl 4-chloro-1-acetoxy-3-[(n-butyloxy)methyl]-1H-indole-2-carboxylate (1bx)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-butanol (21 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 1.5 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and acetic anhydride (32 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1bx (9.7 mg, 25%) as a pale-yellow solid. Mp 48–50 °C; Rf 0.26 (1:4 EtOAc/hexanes); HPLC tR 27.9 min; UV–Vis (CH3CN-H2O) λmax 234, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.32–7.13 (m, 3H, Ar), 5.11 (s, 2H, C(3)CH2O), 3.94 (s, 3H, CO2CH3), 3.59 (t, J = 6.4 Hz, 2H, OCH2CH2), 2.43 (s, 3H, OC(O)CH3), 1.60 (quintet, J = 7.0 Hz, 2H, OCH2CH2), 1.39 (sextet, J = 7.4 Hz, 2H, O(CH2)2CH2), 0.89 (t, J = 7.2 Hz, 3H, O(CH2)3CH3); 13C NMR (75 MHz, CDCl3) δ 168.5 (NOC(O)), 160.4 (CO2CH3), 137.1, 129.0, 127.0, 125.0, 123.6, 120.3, 118.6, 108.0 (Ar), 70.4 (OCH2CH2), 62.0 (C(3)CH2O), 52.4 (CO2CH3), 32.1(OCH2CH2), 19.6 (O(CH2)2CH2), 18.2 (OC(O)CH3) 14.1 (O(CH2)3CH3); MS m/z 353 [M]+; HRMS (+ESI) calcd for C17H20ClNO5 [M]+ 353.1030, found 353.1031.
- Methyl 4-chloro-1-pivaloyloxy-3-[(n-butyloxy)methyl]-1H-indole-2-carboxylate (1by)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-butanol (21 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 1.5 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and pivaloyl chloride (28 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1by (12.0 mg, 28%) as a pale-yellow solid. Mp 66–68 °C; Rf 0.53 (1:4 EtOAc/hexanes); HPLC tR 33.3 min; UV–Vis (CH3CN-H2O) λmax 211, 230, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.27–7.17 (m, 2H, Ar), 7.04 (d, J = 7.8 Hz, 1H, Ar), 5.13 (s, 2H, C(3)CH2O), 3.91 (s, 3H, CO2CH3), 3.58 (t, J = 6.5 Hz, 2H, OCH2CH2), 1.68–1.32 (m, 4H, OCH2(CH2)2), 1.47 (s, 9H, C(CH3)3), 0.89 (t, J = 7.3 Hz, 3H, O(CH2)3CH3); 13C NMR (75 MHz, CDCl3) δ 175.7 (NOC(O)), 160.3 (CO2CH3), 137.2, 129.0, 126.9, 125.7, 123.5, 120.4, 118.8, 107.8 (Ar), 70.3 (OCH2CH2), 61.9 (C(3)CH2O), 52.3 (CO2CH3), 38.8 (C(CH3)3), 32.1 (OCH2CH2), 27.4 (C(CH3)3), 19.6 (O(CH2)2CH2), 14.1 (O(CH2)3CH3); MS m/z 395 [M]+; HRMS (+ESI) calcd for C20H26ClNO5 [M]+ 395.1500, found 395.1500.
- Methyl 4-chloro-1-benzoyloxy-3-[(n-butyloxy)methyl]-1H-indole-2-carboxylate (1bz)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-butanol (21 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 1.5 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and benzoyl chloride (26 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1bz (14.4 mg, 32%) as a pale-yellow solid. Mp 56–58 °C; Rf 0.38 (1:4 EtOAc/hexanes); HPLC tR 32.6 min; UV–Vis (CH3CN-H2O) λmax 229, 296 nm; 1H NMR (300 MHz, CDCl3) δ 8.22 (d, J = 7.3 Hz, 2H, Ar), 7.72 (t, J = 7.5 Hz, 1H, Ar), 7.57 (t, J = 7.8 Hz, 2H, Ar), 7.29–7.18 (m, 3H, Ar), 5.17 (s, 2H, C(3)CH2O), 3.83 (s, 3H, CO2CH3), 3.62 (t, J = 6.5 Hz, 2H, OCH2CH2), 1.63 (quintet, J = 6.5 Hz, 2H, OCH2CH2), 1.41 (sextet, J = 7.2 Hz, 2H, O(CH2)2CH2), 0.91 (t, J = 7.3 Hz, 3H, O(CH2)3CH3); 13C NMR (75 MHz, CDCl3) δ 164.6 (NOC(O)), 160.3 (CO2CH3), 137.5, 134.9, 130.6, 129.2, 129.0, 127.1, 126.3, 125.5, 123.7, 120.5, 119.2, 108.2 (Ar), 70.4 (OCH2CH2), 61.9 (C(3)CH2O), 52.3 (CO2CH3), 32.1 (OCH2CH2), 19.6 (O(CH2)2CH2), 14.1 (O(CH2)3CH3); MS m/z 415 [M]+; HRMS (+ESI) calcd for C22H22ClNO5 [M]+ 415.1187, found 415.1185.
- Methyl 4-chloro-1-acetoxy-3-[(n-hexyloxy)methyl]-1H-indole-2-carboxylate (1cx)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-hexanol (44 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and acetic anhydride (32 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1cx (11.3 mg, 27%) as a pale-yellow solid. Mp 51–53 °C; Rf 0.26 (1:4 EtOAc/hexanes); HPLC tR 31.4 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.29–7.19 (m, 2H, Ar), 7.14 (dd, J = 7.1, 0.9 Hz, 1H, Ar), 5.12 (s, 2H, C(3)CH2O), 3.94 (s, 3H, CO2CH3), 3.58 (t, J = 6.6 Hz, 2H, OCH2CH2), 2.42 (s, 3H, OC(O)CH3), 1.62 (quintet, J = 6.8 Hz, 2H, OCH2CH2), 1.40–1.25 (m, 6H, O(CH2)2(CH2)3), 0.86 (t, J = 6.5 Hz, 3H, O(CH2)5CH3); 13C NMR (75 MHz, CDCl3) δ 168.5 (NOC(O)), 160.4 (CO2CH3), 137.1, 129.0, 127.0, 125.0, 123.6, 120.3, 118.6, 108.0 (Ar), 70.7 (OCH2CH2), 62.0 (C(3)CH2O), 52.4 (CO2CH3), 31.9 (OCH2CH2), 30.0 (O(CH2)2CH2), 26.1 (O(CH2)3CH2), 22.8 (O(CH2)4CH2), 18.2 (OC(O)CH3), 14.3 (O(CH2)5CH3); MS m/z 381 [M]+; HRMS (+ESI) calcd for C19H24ClNO5 [M]+ 381.1343, found 381.1339.
- Methyl 4-chloro-1-pivaloyloxy-3-[(n-hexyloxy)methyl]-1H-indole-2-carboxylate (1cy)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-hexanol (44 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and pivaloyl chloride (28 μL, 0.22 mmol, 2.0 eq) for 1.5 h in general procedure afforded the title compound 1cy (13.7 mg, 29%) as a pale-yellow solid. Mp 57–58 °C; Rf 0.55 (1:4 EtOAc/hexanes); HPLC tR 36.5 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.27–7.18 (m, 2H, Ar), 7.05 (dd, J = 7.9, 0.9 Hz, 1H, Ar), 5.14 (s, 2H, C(3)CH2O), 3.92 (s, 3H, CO2CH3), 3.57 (t, J = 6.6 Hz, 2H, OCH2CH2), 1.68–1.25 (m, 8H, OCH2(CH2)4), 1.47 (s, 9H, C(CH3)3), 0.86 (t, J = 6.6 Hz, 3H, O(CH2)5CH3); 13C NMR (75 MHz, CDCl3) δ 175.6 (NOC(O)), 160.3 (CO2CH3), 137.2, 129.0, 126.9, 125.7, 123.5, 120.5, 118.8, 107.8 (Ar), 70.5 (OCH2CH2), 61.9 (C(3)CH2O), 52.2 (CO2CH3), 38.8 (C(CH3)3), 31.9 (OCH2CH2), 30.0 (O(CH2)2CH2), 27.4 (C(CH3)3), 26.1 (O(CH2)3CH2), 22.8 (O(CH2)4CH2), 14.2 (O(CH2)5CH3); MS m/z 423 [M]+; HRMS (+ESI) calcd for C22H30ClNO5 [M]+ 423.1813, found 423.1815.
- Methyl 4-chloro-1-benzoyloxy-3-[(n-hexyloxy)methyl]-1H-indole-2-carboxylate (1cz)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), n-hexanol (44 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and benzoyl chloride (26 μL, 0.22 mmol, 2.0 eq) for 1 h in general procedure afforded the title compound 1cz (14.2 mg, 30%) as a pale-yellow solid. Mp 62–64 °C; Rf 0.44 (1:4 EtOAc/hexanes); HPLC tR 35.4 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 8.21 (d, J = 7.4 Hz, 2H, Ar), 7.72 (t, J = 7.4 Hz, 1H, Ar), 7.57 (t, J = 7.9 Hz, 2H, Ar), 7.28–7.17 (m, 3H, Ar), 5.17 (s, 2H, C(3)CH2O), 3.82 (s, 3H, CO2CH3), 3.60 (t, J = 6.6 Hz, 2H, OCH2CH2), 1.63 (quintet, J = 7.0 Hz, 2H, OCH2CH2), 1.41–1.22 (m, 6H, O(CH2)2(CH2)3), 0.86 (t, J = 6.6 Hz, 3H, O(CH2)5CH3); 13C NMR (75 MHz, CDCl3) δ 164.7 (NOC(O)), 160.4 (CO2CH3), 137.6, 134.9, 130.6, 129.2, 129.0, 127.1, 126.4, 126.0, 123.7, 120.5, 119.3, 108.3 (Ar), 70.7 (OCH2CH2), 61.9 (C(3)CH2O), 52.3 (CO2CH3), 31.9 (OCH2CH2), 30.0 (O(CH2)2CH2), 26.1 (O(CH2)3CH2), 22.9 (O(CH2)4CH2), 14.3 (O(CH2)5CH3); MS m/z 443 [M]+; HRMS (+ESI) calcd for C24H26ClNO5 [M]+ 443.1500, found 443.1500.
- Methyl 4-chloro-1-butanoyloxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1du)
- Use of SnCl2·2H2O (138 mg, 0.62 mmol, 3.3 eq), benzyl alcohol (38 μL, 0.37 mmol, 2.0 eq), and 2 (50 mg, 0.18 mmol, 1.0 eq) for 2.5 h at 40 °C, then use of DBU (388 μL, 2.60 mmol, 14.0 eq) and butanoyl chloride (38 μL, 0.37 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1du (20.4 mg, 26%) as a white solid. Mp 81–83 °C; Rf 0.56 (1:2 EtOAc/hexanes); HPLC tR 30.2 min; UV–Vis (CH3CN-H2O) λmax 214, 234, 297 nm; 1H NMR (300 MHz, CDCl3) δ 7.42–7.20 (m, 7H, Ar), 7.12 (d, J = 7.9 Hz, 1H, Ar), 5.20 (s, 2H, C(3)CH2O), 4.67 (s, 2H, OCH2Ph), 3.82 (s, 3H, CO2CH3), 2.68 (t, J = 7.4 Hz, 2H, OC(O)CH2), 1.86 (sextet, J = 7.4 Hz, 2H, OC(O)CH2CH2), 1.10 (t, J = 7.4 Hz, 3H, OC(O)(CH2)2CH3); 13C NMR (75 MHz, CDCl3) δ 171.2 (NOC(O)), 160.3 (CO2CH3), 138.7, 137.0, 128.9, 128.5, 128.2, 127.7, 127.0, 125.2, 123.6, 120.2, 117.9, 108.0 (Ar), 72.6 (OCH2Ph), 61.7 (C(3)CH2O), 52.3 (CO2CH3), 33.4 (OC(O)CH2), 18.3 (OC(O)CH2CH2), 13.9 (OC(O)(CH2)2CH3); MS m/z 415 [M]+; HRMS (+ESI) calcd for C22H22ClNO5 [M]+ 415.1187, found 415.1184.
- Methyl 4-chloro-1-hexanoyloxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1dv)
- Use of SnCl2·2H2O (138 mg, 0.61 mmol, 3.3 eq), benzyl alcohol (38 μL, 0.37 mmol, 2.0 eq), and 2 (50 mg, 0.18 mmol, 1.0 eq) for 2.5 h at 40 °C, then use of DBU (388 μL, 2.60 mmol, 14.0 eq) and hexanoyl chloride (52 μL, 0.37 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1dv (23.9 mg, 30%) as a pale-yellow solid. Mp 64–66 °C; Rf 0.62 (1:2 EtOAc/hexanes); HPLC tR 34.0 min; UV–Vis (CH3CN-H2O) λmax 211, 234, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.41–7.20 (m, 7H, Ar), 7.11 (dd, J = 7.9, 1.2 Hz, 1H, Ar), 5.20 (s, 2H, C(3)CH2O), 4.66 (s, 2H, OCH2Ph), 3.82 (s, 3H, CO2CH3), 2.68 (t, J = 7.5 Hz, 2H, OC(O)CH2), 1.83 (quintet, J = 7.4 Hz, 2H, OC(O)CH2CH2), 1.49–1.34 (m, 4H, OC(O)(CH2)2(CH2)2), 0.94 (t, J = 6.9 Hz, 3H, OC(O)(CH2)4CH3); 13C NMR (75 MHz, CDCl3) δ 171.4 (NOC(O)), 160.3 (CO2CH3), 138.7, 137.1, 129.0, 128.5, 128.2, 127.7, 127.0, 125.9, 123.6, 120.3, 118.0, 108.0 (Ar) 72.6 (OCH2Ph), 61.7 (C(3)CH2O), 52.2 (CO2CH3), 31.5 (OC(O)CH2), 31.4 (OC(O)CH2CH2), 24.4 (OC(O)(CH2)2CH2), 22.4 (OC(O)(CH2)3CH2), 14.1 (OC(O)(CH2)4CH3); MS m/z 443 [M]+; HRMS (+ESI) calcd for C24H26ClNO5 [M]+ 443.1500, found 443.1496.
- Methyl 4-chloro-1-hydrocinnamoyloxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1dw)
- Use of SnCl2·2H2O (138 mg, 0.61 mmol, 3.3 eq), benzyl alcohol (38 μL, 0.37 mmol, 2.0 eq), and 2 (50 mg, 0.18 mmol, 1.0 eq) for 3 h at 40 °C, then use of DBU (388 μL, 2.60 mmol, 14.0 eq) and hydrocinnamoyl chloride (55 μL, 0.37 mmol, 2.0 eq) for 3 h in general procedure afforded the title compound 1dw (28.6 mg, 32%) as a pale-yellow solid. Mp 103–104 °C; Rf 0.48 (1:2 EtOAc/hexanes); HPLC tR 32.7 min; UV–Vis (CH3CN-H2O) λmax 211, 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.41–7.14 (m, 13H, Ar), 5.19 (s, 2H, C(3)CH2O), 4.66 (s, 2H, OCH2Ph), 3.77 (s, 3H, CO2CH3), 3.14 (t, J = 7.1 Hz, 2H, (OC(O)CH2), 3.03 (t, J = 7.1 Hz, 2H, (OC(O)CH2CH2); 13C NMR (75 MHz, CDCl3) δ 170.6 (NOC(O)), 160.3 (CO2CH3), 139.7, 138.7, 137.0, 129.0, 128.8, 128.7, 128.5, 128.2, 127.7, 127.0, 126.9, 125.2, 123.6, 120.2, 118.1, 108.1 (Ar), 72.6 (OCH2Ph), 61.6 (C(3)CH2O), 52.2 (CO2CH3), 33.3 (OC(O)CH2), 30.7 (OC(O)CH2CH2); MS m/z 477 [M]+; HRMS (+ESI) calcd for C27H24ClNO5 [M]+ 477.1343, found 477.1345.
- Methyl 4-chloro-1-acetoxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1dx)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), benzyl alcohol (24 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and acetic anhydride (32 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1dx (13.0 mg, 30%) as a pale-yellow solid. Mp 68–69 °C; Rf 0.31 (1:2 EtOAc/hexanes); HPLC tR 26.6 min; UV–Vis (CH3CN-H2O) λmax 236, 299 nm; 1H NMR (300 MHz, CDCl3) δ 7.42–7.14 (m, 8H, Ar), 5.20 (s, 2H, C(3)CH2O), 4.67 (s, 2H, OCH2Ph), 3.82 (s, 3H, CO2CH3), 2.42 (s, 3H, OC(O)CH3); 13C NMR (75 MHz, CDCl3) δ 168.5 (NOC(O)), 160.4 (CO2CH3), 138.6, 137.0, 129.0, 128.5, 128.2, 127.7, 127.1, 125.0, 123.7, 120.2, 118.0, 108.0 (Ar), 72.6 (OCH2Ph), 61.7 (C(3)CH2O), 52.3 (CO2CH3), 18.2 (OC(O)CH3); MS m/z 387 [M]+; HRMS (+ESI) calcd for C20H18ClNO5 [M]+ 387.0874, found 387.0875.
- Methyl 4-chloro-1-pivaloyloxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1dy)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), benzyl alcohol (24 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and pivaloyl chloride (28 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1dy (15.7 mg, 33%) as a pale-yellow solid. Mp 80–82 °C; Rf 0.65 (1:2 EtOAc/hexanes); HPLC tR 31.6 min; UV–Vis (CH3CN-H2O) λmax 213, 234, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.41–7.19 (m, 7H, Ar), 7.06 (d, J = 7.7 Hz, 1H, Ar), 5.22 (s, 2H, C(3)CH2O), 4.66 (s, 2H, OCH2Ph), 3.82 (s, 3H, CO2CH3), 1.47 (s, 9H, C(CH3)3); 13C NMR (75 MHz, CDCl3) δ 175.7 (NOC(O)), 160.2 (CO2CH3), 138.8, 137.1, 129.0, 128.4, 128.2, 127.7, 126.9, 125.8, 123.6, 120.4, 118.2, 107.8 (Ar), 72.5 (OCH2Ph), 61.7 (C(3)CH2O), 52.2 (CO2CH3), 38.8 (OC(CH3)3), 27.4 (OC(CH3)3); MS m/z 429 [M]+; HRMS (+ESI) calcd for C23H24ClNO5 [M]+ 429.1343, found 429.1342.
- Methyl 4-chloro-1-benzoyloxy-3-[(benzyloxy)methyl]-1H-indole-2-carboxylate (1dz)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), benzyl alcohol (24 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and benzoyl chloride (26 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1dz (16.5 mg, 33%) as a pale-yellow solid. Mp 112–114 °C; Rf 0.50 (1:2 EtOAc/hexanes); HPLC tR 31.0 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 8.20 (d, J = 7.2 Hz, 2H, Ar), 7.70 (t, J = 7.5 Hz, 1H, Ar), 7.55 (t, J = 7.9 Hz, 2H, Ar), 7.42–7.17 (m, 8H, Ar), 5.25 (s, 2H, C(3)CH2O), 4.68 (s, 2H, OCH2Ph), 3.72 (s, 3H, CO2CH3); 13C NMR (75 MHz, CDCl3) δ 164.6 (NOC(O)), 160.3 (CO2CH3), 138.7, 137.5, 134.9, 130.6, 129.2, 129.0, 128.2, 128.1, 127.7, 127.1, 126.7, 125.6, 123.7, 120.5, 118.6, 108.3 (Ar), 72.6 (OCH2Ph), 61.7 (C(3)CH2O), 52.3 (CO2CH3); MS m/z 449 [M]+; HRMS (+ESI) calcd for C25H20ClNO5 [M]+ 449.1030, found 449.1028.
- Methyl 4-chloro-1-acetoxy-3-[(phenylethyloxy)methyl]-1H-indole-2-carboxylate (1ex)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), 2-phenylethyl alcohol (27 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and acetic anhydride (32 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1ex (11.5 mg, 26%) as a pale-yellow solid. Mp 70–71 °C; Rf 0.21 (1:4 EtOAc/hexanes); HPLC tR 27.6 min; UV–Vis (CH3CN-H2O) λmax 211, 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.29–7.13 (m, 8H, Ar), 5.17 (s, 2H, C(3)CH2O), 3.89 (s, 3H, CO2CH3), 3.80 (t, J = 7.2 Hz, 2H, OCH2CH2), 2.94 (t, J = 7.4 Hz, 2H, OCH2CH2), 2.42 (s, 3H, OC(O)CH3); 13C NMR (75 MHz, CDCl3) δ 168.5 (NOC(O)), 160.4 (CO2CH3), 139.2, 137.0, 129.1, 129.0, 128.4, 127.0, 126.2, 125.0, 123.7, 120.2, 118.3, 108.0 (Ar), 71.4 (OCH2CH2), 62.1 (C(3)CH2O), 52.4 (CO2CH3), 36.5 (OCH2CH2), 18.2 (OC(O)CH3); MS m/z 401 [M]+; HRMS (+ESI) calcd for C21H20ClNO5 [M]+ 401.1030, found 401.1029.
- Methyl 4-chloro-1-pivaloyloxy-3-[(phenylethyloxy)methyl]-1H-indole-2-carboxylate (1ey)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), 2-phenylethyl alcohol (27 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and pivaloyl chloride (28 μL, 0.22 mmol, 2.0 eq) for 1.5 h in general procedure afforded the title compound 1ey (13.1 mg, 27%) as a pale-yellow solid. Mp 83–84 °C; Rf 0.59 (1:4 EtOAc/hexanes); HPLC tR 33.1 min; UV–Vis (CH3CN-H2O) λmax 212, 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.32–7.18 (m, 7H, Ar), 7.05 (d, J = 8.0 Hz, 1H, Ar), 5.20 (s, 2H, C(3)CH2O), 3.88 (s, 3H, CO2CH3), 3.80 (t, J = 7.3 Hz, 2H, OCH2CH2), 2.94 (t, J = 7.4 Hz, 2H, OCH2CH2), 1.47 (s, 9H, C(CH3)3); 13C NMR (75 MHz, CDCl3) δ 175.7 (NOC(O)), 160.3 (CO2CH3), 139.3, 137.2, 129.2, 129.0, 128.5, 127.0, 126.4, 125.9, 123.6, 120.4, 118.6, 108.1 (Ar), 71.3 (OCH2CH2), 62.1 (C(3)CH2O), 52.3 (CO2CH3), 38.8 (OC(CH3)3), 36.6 (OCH2CH2), 27.4 (OC(CH3)3); MS m/z 443 [M]+; HRMS (+ESI) calcd for C24H26ClNO5 [M]+ 443.1500, found 443.1503.
- Methyl 4-chloro-1-benzoyloxy-3-[(phenylethyloxy)methyl]-1H-indole-2-carboxylate (1ez)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), 2-phenylethyl alcohol (27 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 1.5 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and benzoyl chloride (26 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1ez (16.3 mg, 32%) as a pale-yellow solid. Mp 112–113 °C; Rf 0.44 (1:4 EtOAc/hexanes); HPLC tR 32.0 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 8.22 (d, J = 7.2 Hz, 2H, Ar), 7.73 (t, J = 7.4 Hz, 1H, Ar), 7.58 (t, J = 7.7 Hz, 2H, Ar), 7.31–7.18 (m, 8H, Ar), 5.24 (s, 2H, C(3)CH2O), 3.84 (t, J = 7.4Hz, 2H, OCH2CH2), 3.79 (s, 3H, CO2CH3), 2.97 (t, J = 7.4 Hz, 2H, OCH2CH2); 13C NMR (75 MHz, CDCl3) δ 164.6 (NOC(O)), 160.3 (CO2CH3), 139.3, 137.5, 134.9, 130.6, 129.2, 129.1, 129.0, 128.4, 127.1, 126.4, 126.2, 125.5, 123.7, 120.4, 119.0, 108.3 (Ar), 71.4 (OCH2CH2), 62.0 (C(3)CH2O), 52.3 (CO2CH3), 36.5 (OCH2CH2); MS m/z 463 [M]+; HRMS (+ESI) calcd for C26H22ClNO5 [M]+ 463.1187, found 463.1188.
- Methyl 4-chloro-1-acetoxy-3-[(cyclohexyloxy)methyl]-1H-indole-2-carboxylate (1fx)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), cyclohexanol (23 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and acetic anhydride (32 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1fx (10.2 mg, 24%) as a pale-yellow solid. Mp 80–82 °C; Rf 0.42 (1:2 EtOAc/hexanes); HPLC tR 24.9 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.29–7.13 (m, 3H, Ar), 5.14 (s, 2H, C(3)CH2O), 3.94 (s, 3H, CO2CH3), 3.51–3.44 (m, 1H, OCH), 2.43 (s, 3H, OC(O)CH3), 2.06–1.25 (m, 10H, (CH2)5); 13C NMR (75 MHz, CDCl3) δ 168.6 (NOC(O)), 160.4 (CO2CH3), 137.1, 129.0, 127.0, 125.0, 123.6, 120.2, 119.0, 108.0 (Ar), 77.9 (OCH), 59.4 (C(3)CH2O), 52.3 (CO2CH3), 32.6, 26.1, 24.6 (OCHCH2CH2CH2), 18.2 (N(1)OC(O)CH3); MS m/z 379 [M]+; HRMS (+ESI) calcd for C19H22ClNO5 [M]+ 379.1187, found 379.1183.
- Methyl 4-chloro-1-pivaloyloxy-3-[(cyclohexyloxy)methyl]-1H-indole-2-carboxylate (1fy)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), cyclohexanol (23 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and pivaloyl chloride (28 μL, 0.22 mmol, 2.0 eq) for 1.5 h in general procedure afforded the title compound 1fy (13.9 mg, 30%) as a pale-yellow solid. Mp 98–100 °C; Rf 0.63 (1:2 EtOAc/hexanes); HPLC tR 34.7 min; UV–Vis (CH3CN-H2O) λmax 212, 234, 296 nm; 1H NMR (300 MHz, CDCl3) δ 7.24–7.15 (m, 2H, Ar), 7.01 (d, J = 7.9 Hz, 1H, Ar), 5.13 (s, 2H, C(3)CH2O), 3.89 (s, 3H, CO2CH3), 3.48–3.41 (m, 1H, OCH), 1.44 (s, 9H, (CH3)3), 2.10–1.23 (m, 10H, (CH2)5); 13C NMR (75 MHz, CDCl3) δ 175.7 (NOC(O)), 160.3 (CO2CH3), 137.2, 129.0, 126.9, 125.7, 123.5, 120.5, 119.3, 107.8 (Ar), 77.7 (OCH), 59.4 (C(3)CH2O), 52.2 (CO2CH3), 38.8 (OC(CH3)3), 32.6, 26.1, 24.6 (OCHCH2CH2CH2), 27.4 (OC(CH3)3); MS m/z 421 [M]+; HRMS (+ESI) calcd for C22H28ClNO5 [M]+ 421.1656, found 421.1655.
- Methyl 4-chloro-1-benzoyloxy-3-[(cyclohexyloxy)methyl]-1H-indole-2-carboxylate (1fz)
- Use of SnCl2·2H2O (82.8 mg, 0.37 mmol, 3.3 eq), cyclohexanol (23 μL, 0.22 mmol, 2.0 eq), and 2 (30 mg, 0.11 mmol, 1.0 eq) for 2 h at 40 °C, then use of DBU (233 μL, 1.56 mmol, 14.0 eq) and benzoyl chloride (26 μL, 0.22 mmol, 2.0 eq) for 2 h in general procedure afforded the title compound 1fz (13.1 mg, 27%) as a pale-yellow solid. Mp 96–97 °C; Rf 0.52 (1:2 EtOAc/hexanes); HPLC tR 33.8 min; UV–Vis (CH3CN-H2O) λmax 235, 296 nm; 1H NMR (300 MHz, CDCl3) δ 8.22 (d, J = 7.2 Hz, 2H, Ar), 7.73 (t, J = 7.4 Hz, 1H, Ar), 7.57 (t, J = 7.9 Hz, 2H, Ar), 7.29–7.17 (m, 8H, Ar), 5.20 (s, 2H, C(3)CH2O), 3.83 (s, 3H, CO2CH3), 3.58–3.47 (m, 1H, OCH), 2.08–1.22 (m, 10H, (CH2)5); 13C NMR (75 MHz, CDCl3) δ 164.7 (NOC(O)), 160.4 (CO2CH3), 137.6, 134.9, 130.6, 129.2, 129.0, 127.1, 126.4, 125.5, 123.7, 120.5, 119.7, 108.3 (Ar), 77.9 (OCH), 59.4 (C(3)CH2O), 52.3 (CO2CH3), 32.6, 26.1, 24.6 (OCHCH2CH2CH2); MS m/z 441 [M]+; HRMS (+ESI) calcd for C24H24ClNO5 [M]+ 441.1343, found 441.1340.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Acheson, R.M.; Bolton, R.G.; Hunter, I. 1-Hydroxyindoles, and Products from the Reaction of 2-Nitrophenylsuccinic Anhydride with Fluorosulphonic Acid. J. Chem. Soc. C 1970, 1067–1070. [Google Scholar] [CrossRef]
- Acheson, R.M.; Littlewood, D.M.; Rosenberg, H.E. Synthesis of 1-Methoxyindoles. J. Chem. Soc. Chem. Commun. 1974, 671a. [Google Scholar] [CrossRef]
- Bartsch, A.; Bross, M.; Spiteller, P.; Spiteller, M.; Steglich, W. Birnbaumin A and B: Two Unusual 1-Hydroxyindole Pigments from the “Flower Pot Parasol” Leucocoprinus Birnbaumii. Angew. Chem. Int. Ed. 2005, 44, 2957–2959. [Google Scholar] [CrossRef]
- Wang, J.; Pearce, A.N.; Chan, S.T.S.; Taylor, R.B.; Page, M.J.; Valentin, A.; Bourguet-Kondracki, M.; Dalton, J.P.; Wiles, S.; Copp, B.R. Biologically Active Acetylenic Amino Alcohol and N-Hydroxylated 1,2,3,4-Tetrahydro-β-carboline Constituents of the New Zealand Ascidian Pseudodistoma opacum. J. Nat. Prod. 2016, 79, 607–610. [Google Scholar] [CrossRef]
- Escolano CStephacidin, B. The Avrainvillamide Dimer: A Formidable Synthetic Challenge. Angew. Chem. Int. Ed. 2005, 44, 7670–7673. [Google Scholar] [CrossRef] [PubMed]
- Gmelin, R.; Virtanen, A.I. Neoglucobrassicin, ein zweiter SCN--Precursor vom Indoltyp in Brassica-Arten. Acta. Chem. Scand. 1962, 16, 1378–1384. [Google Scholar] [CrossRef]
- Johns, S.R.; Lamberton, J.A.; Occolowitz, J.L. 1,5-Dimethoxy-3-(dimethylaminomethyl)indole, the major alkaloid from Gymnacranthera paniculata (A. DC.) Warb. var. zippeliana (Miq.) J. Sinclair (family Myristicaceae). Aust. J. Chem. 1967, 20, 1737–1742. [Google Scholar] [CrossRef]
- Acheson, R.M.; Nwankwo, J.O. The metabolism of some 1-hydroxylated Indoles in the rat. Xenobiotica 1984, 14, 877–883. [Google Scholar] [CrossRef]
- Somei, M. 1-Hydroxyindoles. Heterocycles 1999, 50, 1157–1211. [Google Scholar] [CrossRef]
- Somei, M. Recent Advances in the Chemistry of 1-Hydroxyindoles, 1-Hydroxytryptophans, and 1-Hydroxytryptamines. Adv. Heterocycl. Chem. 2002, 82, 101–155. [Google Scholar]
- Jump, S.M.; Kung, J.; Staub, R.; Kinseth, M.A.; Cram, E.J.; Yudina, L.N.; Preobrazhenskaya, M.N.; Bjeldanes, L.F.; Firestone, G.L. N-Alkoxy derivatization of indole-3-carbinol Increases the efficacy of the G1 cell cycle arrest and of I3C-specific regulation of cell cycle gene transcription and activity in human breast cancer cells. Biochem. Pharmacol. 2008, 75, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Roy, S.; Giacomelli, C.; Macchia, M.; Tuccinardi, T.; Martinelli, A.; Lanza, M.; Betti, L.; Giannaccini, G.; Lucacchini, A.; et al. Discovery of N-Hydroxyindole-Based Inhibitors of Human Lactate Dehydrogenase Isoform A (LDH-A) as Starvation Agents against Cancer Cells. J. Med. Chem. 2011, 54, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Calvaresi, E.C.; Tuccinardi, T.; Paterni, I.; Macchia, M.; Martinelli, A.; Hergenrother, P.J.; Minutolo, F. Assessing the differential action on cancer cells of LDH-A inhibitors based on the N-hydroxyindole-2-carboxylate (NHI) and malonic (Mal) scaffolds. Org. Biomol. Chem. 2013, 11, 6588–6596. [Google Scholar] [CrossRef] [Green Version]
- Bagley, M.C.; Dale, J.W.; Merritt, E.A.; Xiong, X. Thiopeptide Antibiotics. Chem. Rev. 2005, 105, 685–714. [Google Scholar] [CrossRef] [PubMed]
- Somei, M.; Yamada, K.; Hasegawa, M.; Tabata, M.; Nagahama, Y.; Morikawa, H.; Yamada, F. Preparations of 1-Hydroxyindole Derivatives and Their Potent Inhibitory Activities on Platelet Aggregation. Heterocycles 1996, 43, 1855–1858. [Google Scholar] [CrossRef]
- Fiume, L. Inhibition of Aerobic Glycolysis in Yoshida Ascites Hepatoma by Tartronic Acid. Nature 1960, 187, 792–793. [Google Scholar] [CrossRef] [PubMed]
- Munshi, K.L.; Kohl, H.; de Souza, N.J. Synthesis of 2-Amino-3-carbethoxy-1-hydroxyindoles. J. Heterocycl. Chem. 1977, 14, 1145–1146. [Google Scholar] [CrossRef]
- Belley, M.; Beaudoin, D.; Duspara, P.; Sauer, E.; St-Pierre, G.; Trimble, L.A. Synthesis and Reactivity of N-Hydroxy-2-Amino-3-Arylindoles. Synlett 2007, 19, 2991–2994. [Google Scholar] [CrossRef]
- Somei, M.; Kawasaki, T. A New and Simple Synthesis of 1-Hydroxyindole Derivatives. Heterocycles 1989, 29, 1251–1254. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kodama, A.; Nishida, T.; Shimizu, K.; Somei, M. Preparation of 1-Hydroxyindole Derivatives and a New Route to 2-Substituted Indoles. Heterocycles 1991, 32, 221–227. [Google Scholar]
- Chirkova, Z.V.; Kabanova, M.V.; Sergeev, S.S.; Filimonov, S.I.; Abramov, I.G.; Samet, A.V.; Suponitsky, K.Y. Synthesis of 3-acyl-1-hydroxy-1H-indole-5,6-dicarbonitriles. Mendeleev Commun. 2015, 25, 315–317. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Lee, S.H.; Estrada, A.A.; Zak, M. Construction of Substituted N-Hydroxyindoles: Synthesis of a Nocathiacin I Model System. Angew. Chem. 2005, 117, 3802–3806. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Estrada, A.A.; Freestone, G.C.; Lee, S.H.; Alvarez-Mico, X. New synthetic technology for the construction of N-hydroxyindoles and synthesis of nocathiacin I Model Systems. Tetrahedron 2007, 63, 6088–6114. [Google Scholar] [CrossRef]
- Wong, A.; Kuethe, J.T.; Davies, I.W. A General Synthesis of N-Hydroxyindoles. J. Org. Chem. 2003, 68, 9865–9866. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Cho, H.; Lim, Y.J.; Kim, C.; Lee, S.H. One-Pot Synthesis of Novel Multisubstituted 1-Alkoxyindoles. Molecules 2021, 26, 1466. [Google Scholar] [CrossRef] [PubMed]
- Yun, Z.; Cheng, R.; Sun, J.; Zhang-Negrerie, D.; Du, Y. Iodobenzene Dichloride/Zinc Chloride-Mediated Synthesis of N-Alkoxyindole-3-carbonitriles from 3-Alkoxyimino-2-arylalkylnitriles via Intramolecular Heterocyclization. Adv. Synth. Catal. 2018, 360, 250–254. [Google Scholar] [CrossRef]
- Acheson, R.M.; Hunt, P.G.; Littlewood, D.M.; Murrer, B.A.; Rosenberg, H.E. The Synthesis, Reactions, and Spectra of 1-Acetoxy-, 1-Hydroxy-, and 1-Methoxy-Indoles. J. Chem. Soc. Perkin Trans. 1 1978, 1, 1117–1125. [Google Scholar] [CrossRef]
- Somei, M.; Shoda, T. A Facile Route to 1-Acetoxy- and 1-Methoxyindols. Heterocycles 1981, 16, 1523–1525. [Google Scholar] [CrossRef]
- Nagayoshi, T.; Saeki, S.; Hamana, M. Studies on Tertiary Amine Oxides. LXXIX. Reactions of 2-Ethoxycarbonyl-1-hydroxyindole in the Presence of Acylating Agents. Chem. Pharm. Bull. 1984, 32, 3678–3682. [Google Scholar] [CrossRef] [Green Version]
- Chirkova, Z.V.; Kabanova, M.V.; Filimonov, S.I.; Abramov, I.G.; Samet, A.V.; Stashina, G.A.; Suponitsky, K.Y. The C-3 acylation of 1-hydroxyindoles. Tetrahedron Lett. 2017, 58, 755–757. [Google Scholar] [CrossRef]
- Belley, M.; Sauer, E.; Beaudoin, D.; Duspara, P.; Trimble, L.A.; Dubé, P. Synthesis and reactivity of N-hydroxy-2-aminoindoles. Tetrahedron Lett. 2006, 47, 159–162. [Google Scholar] [CrossRef]
- Park, Y.K.; Kim, H.; Kim, D.S.; Cho, H.; Moon, A.; Jeong, C.; Yoon, H.; Lee, S.H. Synthesis of New 2,3-Disubstituted 4-Chloro-1-Hydroxyindoles. Bull. Korean Chem. Soc. 2015, 36, 2095–2100. [Google Scholar] [CrossRef]
- Park, Y.K.; Kim, H.; Lee, S.H. Synthesis of New Highly Substituted and Hindered 1-Hydroxyindole-2-Carboxylates. Bull. Korean Chem. Soc. 2016, 37, 82–90. [Google Scholar] [CrossRef]
- Park, Y.K.; Lee, S.H. Synthesis of New 1-Hydroxyindole-2-Carboxylates and Mechanistic Studies on Reaction Pathways. J. Heterocycl. Chem. 2017, 54, 1995–2002. [Google Scholar] [CrossRef]
- Bellamy, F.D.; Ou, K. Selective reduction of aromatic nitro compounds with stannous chloride in non acidic and non aqueous medium. Tetrahedron Lett. 1984, 25, 839–842. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | 8d Formation | Acylation | Yield (%) | ||
| SnCl2∙2H2O (eq) | BnOH (eq) | DBU (eq) | Pivaloyl Chloride (eq) | ||
| 1 | 2.5 | 2.0 | 10.6 | 2.0 | 21 |
| 2 | 2.9 | 2.0 | 12.3 | 2.0 | 27 |
| 3 | 3.3 | 1.5 | 14.0 | 1.5 | 25 |
| 4 | 3.3 | 1.5 | 14.0 | 3.0 | 26 |
| 5 | 3.3 | 2.0 | 14.0 | 2.0 | 35 |
| 6 | 3.3 | 3.0 | 14.0 | 3.0 | 35 |
| 7 | 3.7 | 2.0 | 15.7 | 2.0 | 22 |
 | ||||
|---|---|---|---|---|
| Entry | ROH | RCOX | Product | Yield (%) |
| 1 | MeOH | acetic anhydride | 1ax | 28 |
| 2 | MeOH | pivaloyl chloride | 1ay | 35 |
| 3 | MeOH | benzoyl chloride | 1az | 33 |
| 4 | n-BuOH | acetic anhydride | 1bx | 25 |
| 5 | n-BuOH | pivaloyl chloride | 1by | 28 |
| 6 | n-BuOH | benzoyl chloride | 1bz | 32 |
| 7 | n-HexOH | acetic anhydride | 1cx | 27 |
| 8 | n-HexOH | pivaloyl chloride | 1cy | 29 |
| 9 | n-HexOH | benzoyl chloride | 1cz | 30 |
| 10 | BnOH | butanoyl chloride | 1du | 26 |
| 11 | BnOH | hexanoyl chloride | 1dv | 30 |
| 12 | BnOH | hydrocinnamoyl chloride | 1dw | 32 |
| 13 | BnOH | acetic anhydride | 1dx | 30 |
| 14 | BnOH | pivaloyl chloride | 1dy | 33 |
| 15 | BnOH | benzoyl chloride | 1dz | 33 |
| 16 | PhCH2CH2OH | acetic anhydride | 1ex | 26 |
| 17 | PhCH2CH2OH | pivaloyl chloride | 1ey | 27 |
| 18 | PhCH2CH2OH | benzoyl chloride | 1ez | 32 |
| 19 | c-HxOH | acetic anhydride | 1fx | 24 |
| 20 | c-HxOH | pivaloyl chloride | 1fy | 30 |
| 21 | c-HxOH | benzoyl chloride | 1fz | 27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.E.; Lim, Y.J.; Kim, C.; Jeong, Y.R.; Cho, H.; Lee, S.H. Syntheses of New Multisubstituted 1-Acyloxyindole Compounds. Molecules 2022, 27, 6769. https://doi.org/10.3390/molecules27196769
Kim YE, Lim YJ, Kim C, Jeong YR, Cho H, Lee SH. Syntheses of New Multisubstituted 1-Acyloxyindole Compounds. Molecules. 2022; 27(19):6769. https://doi.org/10.3390/molecules27196769
Chicago/Turabian StyleKim, Ye Eun, Yoo Jin Lim, Chorong Kim, Yu Ra Jeong, Hyunsung Cho, and Sang Hyup Lee. 2022. "Syntheses of New Multisubstituted 1-Acyloxyindole Compounds" Molecules 27, no. 19: 6769. https://doi.org/10.3390/molecules27196769
APA StyleKim, Y. E., Lim, Y. J., Kim, C., Jeong, Y. R., Cho, H., & Lee, S. H. (2022). Syntheses of New Multisubstituted 1-Acyloxyindole Compounds. Molecules, 27(19), 6769. https://doi.org/10.3390/molecules27196769