
Seeking Solvation: Exploring the Role of Protein Hydration in Silk Gelation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- Similarly, freezing the protein solution [9,16,20,21,22] produces a lower energy state for the water (i.e., as ice), thereby enticing it to leave the hydration shell. This example is particularly instructive, as it is the increased entropy of liquid water overcoming the stronger enthalpic component of ice crystals that defines its melting point.
- Most importantly, from the perspective of natural silk spinning, flow stress [8,9,10,20,23,24,25,26,27,28,29,30,31] causes the fibroin chain to deform away from an equilibrium shape. Under these conditions, the hydration shell becomes unstable through a further loss of entropy, or due to some peptide groups being forced to adopt conformations incompatible with the amount of H-bonding required to maintain stability.
- Firstly, the molecular size of B. mori fibroin was investigated in dilute solution, using small-angle X-ray scattering (SAXS) and dynamic light scattering (DLS), at room temperature and during heating to 80 °C.
- Infrared (IR) spectroscopy was used to probe thermally driven changes in the hydration of NSF and several amide-containing model compounds.
- Changes in hydration associated with gelation of NSF by freezing and thawing were also investigated using IR spectroscopy.
- Turbidity and protein aggregation measurements were used to investigate the solubility of diluted fibroin in salt solutions, where the free energy of the bulk aqueous phase can be determined.
2. Results
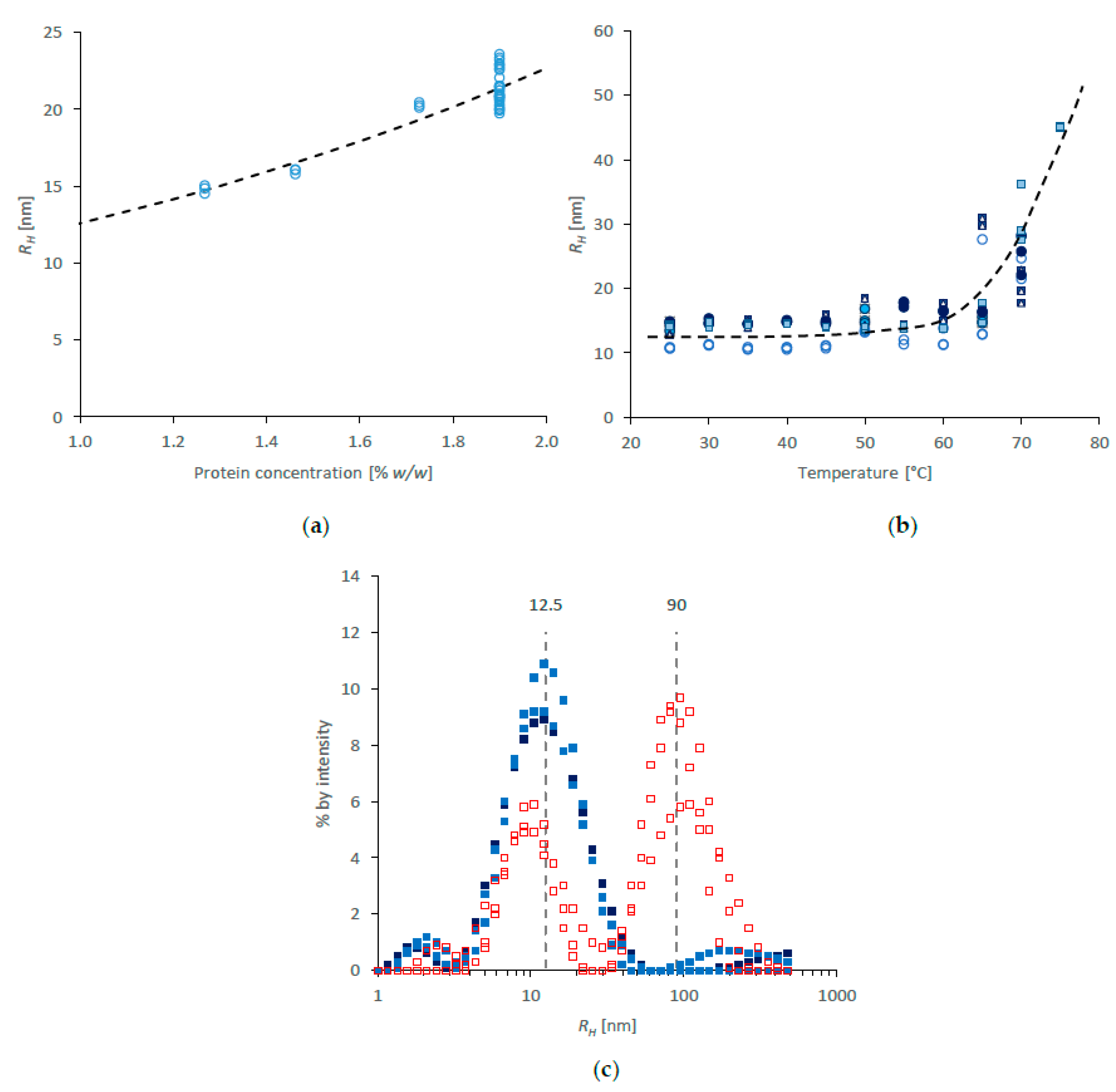
2.1. Protein Coil Size by SAXS
2.2. Protein Coil Size by DLS
2.3. Observing Silk Protein Hydration by Mid-IR Spectroscopy
- possibly also resonance between O-H stretching bands of water and the amide A and B bands.
2.4. Changes in Hydration during Heating
2.5. Observing Changes in Amide and Peptide Hydration by Near-IR Spectroscopy
2.6. Cloud-Point and Aggregation Measurements in Salt Solutions
3. Discussion
- (i)
- In dilute solution, the protein coil exhibits a three-dimensional Gaussian random walk configuration, typical of a polymer in solution under theta conditions. This implies an approximate equivalence between the strengths of monomer-monomer and monomer–solvent interactions. Moreover, while it was not possible to observe the coil geometry by SAXS (or SANS [60]) at higher concentration (the interpretation of scattering data required non-overlapping coils), the configuration determined at low concentration was consistent with previous NMR results [46,47,48,49] and rheology [9,99,178], which demonstrated that the protein in NSF behaved as a typical polymer in solution, albeit slightly modified by its natural propensity to form transient ‘sticky’ ionic interactions [66,67].
- (ii)
- (iii)
- The temperature at which the protein came out of solution (causing gelation of NSF or aggregation and turbidity in diluted solutions) appears to be independent of concentration. In terms of the Flory–Huggins theory [62,100], this implies that the configurational entropy of mixing is negligible, with solubility being dominated by the strength of monomer–solvent interactions (i.e., the interaction parameter, χ). It should be noted that, contrary to earlier descriptions, it is known that χ is not a constant describing purely enthalpic interactions, but varies with temperature and includes both entropic and enthalpic contributions [179,180,181,182].
- (iv)
- Coupling and resonance between vibrations of water and peptide groups affect much of the mid-range IR spectrum. Nevertheless, changes in amide band positions of fibroin in NSF and other amide models demonstrated that the strength of interaction between water and peptide groups decreases with increasing temperature. Moreover, this occurred as a precursor to—rather than a consequence of phase separation.
- (v)
- Subtle changes in hydration associated with gelation of NSF (by heating or freezing) were also demonstrated by NIR.
- (vi)
- Based on our hydration shell hypothesis, a thermodynamic model was developed that closely fitted the various data from gelation (of NSF) and aggregation or turbidity measurements (of diluted fibroin).
4. Materials and Methods
4.1. Materials
4.2. Small-Angle X-ray Scattering (SAXS)
4.3. Dynamic Light Scattering (DLS)
4.4. Infrared Spectroscopy (IR)
4.5. Turbidity Measurements
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rudall, K.M.; Kenchington, W. Arthropod silks: The problem of fibrous protein in animal tissues. Annu. Rev. Entomol. 1971, 16, 73–96. [Google Scholar] [CrossRef]
- Kaplan, D.; Adams, W.W.; Farmer, B.; Viney, C. Silks: Biology, structure, properties and genetics. ACS Symp. Ser. 1994, 544, 2–16. [Google Scholar] [CrossRef] [Green Version]
- Craig, C.L. Evolution of arthropod silks. Annu. Rev. Entomol. 1997, 42, 231–267. [Google Scholar] [CrossRef]
- Sutherland, T.D.; Young, J.H.; Weisman, S.; Hayashi, C.Y.; Merritt, D.J. Insect silk: One name, many materials. Annu. Rev. Entomol. 2010, 55, 171–188. [Google Scholar] [CrossRef]
- Eisoldt, L.; Smith, A.; Scheibel, T. Decoding the secrets of spider silk. Mater. Today 2011, 14, 80–86. [Google Scholar] [CrossRef]
- Walker, A.A.; Holland, C.; Sutherland, T.D. More than one way to spin a crystallite: Multiple trajectories through liquid crystallinity to solid silk. Proc. R. Soc. B 2015, 282, 20150259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, M.; Johansson, J.; Rising, A. Silk spinning in spiders and silkworms. Int. J. Mol. Sci. 2016, 17, 1290. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, K.-Q. Silk fiber—Molecular formation mechanism, structure-property relationship and advanced applications. In Oligomerization of Chemical and Biological Compounds; Lesieur, C., Ed.; IntechOpen: London, UK, 2014. [Google Scholar] [CrossRef] [Green Version]
- Laity, P.R.; Holland, C. The rheology behind stress-induced solidification in native silk feedstocks. Int. J. Mol. Sci. 2016, 17, 1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foà, C. Die kolloiden Eigenschaften der natürlichen Seide (Properties of natural silk). Zeitschr. Chem. Ind. Kolloide 1912, 10, 7–12. [Google Scholar] [CrossRef]
- Magoshi, J.; Magoshi, Y.; Becker, M.A.; Kato, M.; Han, Z.; Tanaka, T.; Inoue, S.-I.; Nakamura, S. Crystallisation of silk fibroin from solution. Thermochim. Acta 2000, 352–353, 165–169. [Google Scholar] [CrossRef]
- Ochi, A.; Hossain, K.S.; Magoshi, J.; Nemoto, N. Rheology and dynamic light scattering of silk fibroin solution extracted from the middle division of Bombyx mori silkworm. Biomacromolecules 2002, 3, 1187–1196. [Google Scholar] [CrossRef]
- Tanaka, T.; Magoshi, J.; Magoshi, Y.; Inoue, S.; Kobayashi, M.; Tsuda, H.; Becker, M.A.; Nakamura, S. Thermal properties of Bombyx mori and several wild silkworm silks: Phase transition of liquid silk. J. Therm. Anal. Calorim. 2002, 70, 825–832. [Google Scholar] [CrossRef]
- Nagarkar, S.; Nicolai, T.; Chassenieux, C.; Lele, A. Structure and gelation mechanism of silk hydrogels. Phys. Chem. Chem. Phys. 2010, 12, 3834–3844. [Google Scholar] [CrossRef] [PubMed]
- Moriya, M.; Roschzttardtz, F.; Nakahara, Y.; Saito, H.; Masabuchi, Y.; Asakura, T. Rheological properties of native silk fibroins from domestic and wild silkworms, and flow analysis in each spinneret by a finite element method. Biomacromolecules 2009, 10, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Holland, C.; Hawkins, N.; Frydrych, M.; Laity, P.; Porter, D.; Vollrath, F. Differential scanning calorimetry of native silk feedstock. Macromol. Biosci. 2018, 19, 1800228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Shao, Z.; Marinkovic, N.S.; Miller, L.M.; Zhou, P.; Chance, M.R. Conformation transition kinetics of regenerated Bombyx mori silk fibroin membrane monitored by time-resolved FTIR spectroscopy. Biophys. Chem. 2001, 89, 25–34. [Google Scholar] [CrossRef]
- Kim, U.J.; Park, J.; Li, C.; Jin, H.-J.; Valluzzi, R.; Kaplan, D.L. Structure and properties of silk hydrogels. Biomacromolecules 2004, 5, 786–792. [Google Scholar] [CrossRef]
- Kasoju, N.; Hawkins, N.; Pop-Georgievski, O.; Kubies, D.; Vollrath, F. Silk fibroin gelation via non-solvent induced phase separation. Biomater. Sci. 2016, 4, 460. [Google Scholar] [CrossRef]
- Ramsden, W. Coagulation by shearing and by freezing. Nature 1938, 142, 1120–1121. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Luong, H.T.; Nguyen, H.D.; Tran, H.A.; Huynh, K.C.; Vo, T.V. Investigate the effect of thawing process on the self-assembly of silk protein for tissue applications. BioMed Res. Int. 2017, 2017, 4263762. [Google Scholar] [CrossRef]
- Kolahreez, D.; Morshed, M. Fabrication of porous three-dimensional fibroin structures through a freezing process. J. Appl. Polym. Sci. 2018, 135, 46537. [Google Scholar] [CrossRef]
- Iizuka, E. Mechanism of fiber formation by the silkworm, Bombyx mori, L. Biorheology 1966, 3, 141–152. [Google Scholar]
- Iizuka, E. The physico-chemical properties of silk fibers and the fiber spinning process. Experimentia 1983, 39, 449–454. [Google Scholar] [CrossRef]
- Iizuka, E. Silk thread: Mechanism of spinning and its mechanical properties. J. Appl. Polym. Sci. Polym. Symp. 1985, 41, 173–185. [Google Scholar]
- Magoshi, J.; Magoshi, Y.; Nakamura, S. Physical properties and structure of silk. 10: The mechanism of fibre formation from liquid silk of the silkworm Bombyx mori. Polym. Commun. 1985, 26, 309–311. [Google Scholar]
- Knight, D.P.; Knight, M.M.; Vollrath, F. Beta transition and stress-induced phase separation in the spinning of spider dragline silk. Int. J. Biol. Macromol. 2000, 27, 205–210. [Google Scholar] [CrossRef]
- Ohgo, K.; Bagusat, F.; Asakura, T.; Scheler, U. Investigation of structural transition of regenerated silk fibroin aqueous solution by rheo-NMR spectroscopy. J. Am. Chem. Soc. 2008, 130, 4182–4186. [Google Scholar] [CrossRef] [PubMed]
- Holland, C.; Urbach, J.S.; Blair, D.L. Direct visualization of shear dependent silk fibrillogenesis. Soft Matter 2012, 8, 2590. [Google Scholar] [CrossRef]
- Giesa, T.; Perry, C.C.; Buehler, M.J. Secondary structure transition and critical stress for a model of spider silk assembly. Biomacromolecules 2016, 17, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Sparkes, J.; Holland, C. The energy requirements for flow-induced solidification of silk. Macromol. Biosci. 2018, 1800229. [Google Scholar] [CrossRef] [PubMed]
- Ferscht, A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; WH. Freeman and Co.: New York, NY, USA, 1999. [Google Scholar]
- Whitford, D. Proteins: Structure and Function; John Wiley and Sons Ltd.: Chichester, UK, 2005. [Google Scholar]
- Raccosta, S.; Manno, M.; Bulone, D.; Giacomazza, D.; Militello, V.; Martorano, V.; San Biagio, P.L. Irreversible gelation of thermally unfolded proteins: Structural and mechanical properties of lysozyme aggregates. Eur. Biophys. J. 2010, 39, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.H.; Kavanagh, G.M.; Ross-Murphy, S.B. Globular protein gelation—theory and experiment. Food Hydrocoll. 2001, 15, 383–400. [Google Scholar] [CrossRef]
- Gosal, W.S.; Ross-Murphy, S.B. Globular protein gelation. Curr. Opin. Coll. Interface Sci. 2000, 5, 188–194. [Google Scholar] [CrossRef]
- Allain, A.-F.; Paquin, P.; Subirade, M. Relationships between conformation of β-lactoglobulin in solution and gel states as revealed by attenuated total reflection Fourier transform infrared spectroscopy. Int. J. Biol. Macromol. 1999, 26, 337–344. [Google Scholar] [CrossRef]
- Doi, E. Gels and gelling of globular proteins. Trends Food Sci. Technol. 1993, 4, 1–5. [Google Scholar] [CrossRef]
- Dill, K.A. Dominant forces in protein folding. Biochemistry 1990, 29, 7133–7155. [Google Scholar] [CrossRef]
- Wuttke, R.; Hofmann, H.; Nettels, D.; Borgia, M.B.; Mittal, J.; Best, R.B.; Schuler, B. Temperature-dependent solvation modulates the dimensions of disordered proteins. Proc. Natl. Acad. Sci. USA 2014, 111, 5213–5218. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef]
- Tompa, P.; Fersht, A. Structure and Function of Intrinsically Disordered Proteins; Chapman and Hall/CRC: Boca Raton, FL, USA, 2009. [Google Scholar]
- Uversky, V.N. Intrinsically disordered proteins and their environment: Effects of strong denaturants, temperature, pH, counter ions, membranes, binding partners, osmolytes, and macromolecular crowding. Protein J. 2009, 28, 305–325. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Iizuka, E.; Yang, J.T. The disordered and β-conformations of silk fibroin in solution. Biochemistry 1968, 7, 2218–2228. [Google Scholar] [CrossRef]
- Asakura, T.; Suzuki, H.; Watanabe, Y. Conformational characterization of silk fibroin in intact Bombyx mori and Philosamia cynthia ricini silkworms by 13C NMR spectroscopy. Macromolecules 1983, 16, 1024–1026. [Google Scholar] [CrossRef]
- Asakura, T.; Watanabe, Y.; Uchida, A.; Minagawa, H. NMR of silk fibroin. 2. 13C NMR study of the chain dynamics and solution structure of Bombyx mori silk fibroin. Macromolecules 1984, 17, 1075–1081. [Google Scholar] [CrossRef]
- Asakura, T.; Okushita, K.; Williamson, M.P. Analysis of the structure of Bombyx mori silk fibroin by NMR. Macromolecules 2015, 48, 2345–2357. [Google Scholar] [CrossRef]
- Hijirida, D.H.; Do, K.G.; Michal, C.; Wong, S.; Zax, D.; Jelinski, L.W. 13C NMR of Nephila clavipes major ampullate silk gland. Biophys. J. 1996, 71, 3442–3447. [Google Scholar] [CrossRef] [Green Version]
- Dicko, C.; Vollrath, F.; Kenney, J.M. Spider silk protein refolding is controlled by changing pH. Biomacromolecules 2004, 5, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Dicko, C.; Knight, D.; Kenney, J.M.; Vollrath, F. Structural conformation of spidroin in solution: A synchrotron radiation circular dichroism study. Biomacromolecules 2004, 5, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, T.; Leclerc, J.; Rioux-Dubé, J.F.; Buffeteau, T.; Paquin, M.-C.; Rousseau, M.E.; Cloutier, I.; Auger, M.; Gagné, S.M.; Boudreault, S.; et al. Conformation of spider silk proteins in situ in the intact major ampullate gland and in solution. Biomacromolecules 2007, 8, 2342–2344. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre, T.; Boudreault, S.; Cloutier, C.; Pézolet, M. Conformational and orientational transformation of silk proteins in the major ampullate gland of Nephila clavipes spiders. Biomacromolecules 2008, 9, 2399–2407. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Yarger, J.L.; Holland, G.P. Exploring the backbone dynamics of native spider silk proteins in Black Widow silk glands with solution-state NMR spectroscopy. Polymer 2014, 55, 3879–3885. [Google Scholar] [CrossRef]
- Suzuki, Y. Structures of silk fibroin before and after spinning and biomedical applications. Polym. J. 2016, 48, 1039–1044. [Google Scholar] [CrossRef]
- Oktaviani, N.A.; Matsugami, A.; Malay, A.D.; Hayashi, F.; Kaplan, D.; Numata, K. Conformation and dynamics of soluble repetitive domain elucidates the initial β-sheet formation of spider silk. Nat. Commun. 2018, 9, 2121. [Google Scholar] [CrossRef]
- Greving, I.; Terry, E.A.; Holland, C.; Boulet-Audet, M.; Grillo, I.; Vollrath, F.; Dicko, C. Structural diversity of native major ampullate, minor ampullate, cylindriform, and flagelliform silk proteins in solution. Biomacromolecules 2020, 21, 3387–3393. [Google Scholar] [CrossRef]
- Asakura, T.; Suzuki, Y.; Nakazawa, Y.; Yazawa, K.; Holland, G.P. Silk structure studied with nuclear magnetic resonance. Prog. Nucl. Mag. Res. Spectr. 2013, 69, 23–68. [Google Scholar] [CrossRef]
- Miles, A.J.; Janes, R.W.; Wallace, B.A. Tools and methods for circular dichroism spectroscopy of proteins: A tutorial review. Chem. Soc. Rev. 2021, 50, 8400–8413. [Google Scholar] [CrossRef]
- Greving, I.; Dicko, C.; Terry, A.; Callow, P. Vollrath, Small angle neutron scattering of native and reconstituted silk fibroin. Soft Matter 2010, 6, 4389–4395. [Google Scholar] [CrossRef]
- Roe, R.-J. Methods of X-ray and Neutron Scattering in Polymer Science; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Gedde, U.W. Polymer Physics; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1999. [Google Scholar]
- Amino Acid Sequences Published on Genbank. Available online: https://www.ncbi.nlm.nih.gov/protein/ (accessed on 20 September 2020).
- Ainavarapu, S.R.K.; Brujić, J.; Huang, H.H.; Wiita, A.P.; Lu, H.; Li, L.; Walther, K.A.; Carrion-Vazquez, M.; Li, H.; Fernandez, J.M. Contour length and refolding rate of a small protein controlled by engineered disulfide bonds. Biophys. J. 2007, 92, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laity, P.R.; Baldwin, E.; Holland, C. Changes in silk feedstock rheology during cocoon construction: The role of calcium and potassium ions. Macromol. Biosci. 2018, 19, 1800188. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, C.; Laity, P.R.; Holland, C.; McLeish, T.C.B. Silk protein solution: A natural example of sticky reputation. Macromolecules 2020, 53, 2669–2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, C.; Laity, P.R.; Holland, C.; McLeish, T.C.B. Stretching of Bombyx mori silk protein in flow. Molecules 2021, 26, 1663. [Google Scholar] [CrossRef] [PubMed]
- Martel, A.; Burghammer, M.; Davies, R.; DiCola, E.; Panine, P.; Salmon, J.-B.; Riekel, C. A microfluidic cell for studying the formation of regenerated silk by synchrotron radiation small- and wide-angle X-ray scattering. Biomicrofluidics 2008, 2, 024104. [Google Scholar] [CrossRef] [Green Version]
- Hol, W.G.J. Effects of the α-helix dipole upon the functioning and structure of proteins and peptides. Adv. Biophys. 1985, 19, 133–165. [Google Scholar] [CrossRef]
- Sengupta, D.; Behera, R.N.; Smith, J.C.; Ullmann, G.M. The α-helix dipole: Screened out? Structure 2005, 13, 849–855. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar] [CrossRef]
- Damaschun, G.; Damaschun, H.; Gast, K.; Zirwer, D. Denatured states of yeast phosphoglycerate kinase. Biochemistry 1998, 63, 259–275. [Google Scholar]
- Zhou, H.-X. Dimensions of denatured protein chains from hydrodynamic data. J. Phys. Chem. B 2002, 106, 5769–5775. [Google Scholar] [CrossRef]
- Bernadó, P.; Blackledge, M. A self-consistent description of the conformational behavior of chemically denatured proteins from NMR and small angle scattering. Biophys. J. 2009, 97, 2839–2845. [Google Scholar] [CrossRef] [Green Version]
- Rawat, N.; Biswas, P. Size, shape, and flexibility of proteins and DNA. J. Chem. Phys. 2009, 131, 165104. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The alphabet of intrinsic disorder II. Various roles of glutamic acid in ordered and intrinsically disordered proteins. Proteins 2013, 1, e24684. [Google Scholar] [CrossRef] [Green Version]
- Machii, H. Varietal differences of nitrogen and amino acid contents in mulbery leaves. Acta Sericol. Entomol. 1989, 1, 51–61. [Google Scholar]
- Machii, H.; Koyama, A.; Yamanouchi, H. Mulberry breeding, cultivation and utilisation in Japan. In Proceedings of the FAO Electronic Conference on Mulberry for Animal Production, Online, May–August 2000; Available online: https://www.fao.org/3/X9895E/x9895e05.htm (accessed on 6 March 2021).
- Hansen, J.C.; Lu, X.; Ross, E.D.; Woody, R.W. Intrinsic protein disorder, amino acid composition and histone terminal domains. J. Biol. Chem. 2006, 281, 1853–1856. [Google Scholar] [CrossRef] [Green Version]
- He, Y.-X.; Zhang, N.-N.; Li, W.-F.; Jia, N.; Chen, B.-Y.; Zhou, K.; Zhang, J.; Chen, Y.; Zhou, C.-Z. N-terminal domain of Bombyx mori fibroin mediates the assembly of silk in response to pH decrease. J. Mol. Biol. 2012, 418, 197–207. [Google Scholar] [CrossRef]
- Eisoldt, L.; Thamm, C.; Scheibel, T. Review: The role of terminal domains during storage and assembly of spider silk proteins. Biopolymers 2011, 97, 355–361. [Google Scholar] [CrossRef]
- Hagn, F.; Thamm, C.; Scheibel, T.; Kessler, H. pH-dependent dimerization and salt-dependent stabilization of the N-terminal domain of spider dragline silk—implications for fiber formation. Angew. Chem. Int. Ed. 2011, 50, 310–313. [Google Scholar] [CrossRef]
- Bauer, J.; Scheibel, T. Dimerization of the conserved N-terminal domain of a spider silk protein controls the self-assembly of the repetitive core domain. Biomacromolecules 2017, 18, 2521–2528. [Google Scholar] [CrossRef]
- Kronqvist, N.; Otikovs, M.; Chmyrov, V.; Chen, G.; Andersson, M.; Nordling, K.; Landreh, M.; Sarr, M.; Jörnvall, H.; Wennmalm, S.; et al. Sequential pH-driven dimerization and stabilization of the N-terminal domain enables rapid spider silk formation. Nat. Commun. 2014, 5, 3254. [Google Scholar] [CrossRef] [Green Version]
- Andersson, M.; Chen, G.; Otikovs, M.; Landreh, M.; Nordling, K.; Kronqvist, N.; Westermark, P.; Jörnvall, H.; Knight, S.; Ridderstråle, Y.; et al. Carbonic anhydrase generates CO2 and H+ that drive spider silk formation via opposite effects on the terminal domains. PLoS Biol. 2014, 12, e1001921. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.; Zwettler, F.U.; Johnson, C.M.; Neuweiler, H. The N-terminal domains of spider silk proteins assemble ultrafast and protected from charge screening. Nat. Commun. 2013, 4, 2815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrosa da Silva, F.L.; Pasquali, S.; Derreumaux, P.; Dias, L.G. Electrostatics analysis of the mutational and pH effects of the N-terminal domain self-association of the major ampullate spidroin. Soft Matter 2016, 12, 5600. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.; Leclerc, J.; Lefèvre, T.; Gagné, S.M.; Auger, M. Effect of pH on the structure of the recombinant C-terminal domain of Nephila clavipes dragline silk protein. Biomacromolecules 2014, 15, 4447–4454. [Google Scholar] [CrossRef] [PubMed]
- Gaines, W.A.; Sehorn, M.G.; Marcotte, W.R., Jr. Spidroin N-terminal domain promotes a pH-dependent association of silk proteins during self-assembly. J. Biol. Chem. 2010, 285, 40745–40753. [Google Scholar]
- Hossain, K.S.; Nemoto, N.; Magoshi, J. Dynamic and static light scattering of dilute aqueous solutions of silk fibroin collected from Bombyx mori silkworms. Langmuir 1999, 15, 4114–4119. [Google Scholar] [CrossRef]
- Jin, H.-J.; Kaplan, D.L. Mechanism of silk processing in insects and spiders. Nature 2003, 424, 1057–1061. [Google Scholar] [CrossRef]
- Lu, S.; Li, J.; Zhang, S.; Yin, Z.; Xing, T.; Kaplan, D. The influence of the hydrophilic–lipophilic environment on the structure of silk fibroin protein. J. Mater. Chem. B 2015, 3, 2599. [Google Scholar] [CrossRef]
- Malay, A.D.; Suzuki, T.; Katashima, T.; Kono, N.; Arakawa, K.; Numata, K. Spider silk self-assembly via modular liquid-liquid phase separation and nanofibrillation. Sci. Adv. 2020, 6, eabb6030. [Google Scholar] [CrossRef]
- Seib, F.P. Emerging silk material trends: Repurposing, phase separation and solution-based designs. Materials 2021, 14, 1160. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Makhatadze, G.I.; Privalov, P.L. Contribution of hydration to protein folding thermodynamics: I the enthalpy of hydration. J. Mol. Biol. 1993, 232, 639–659. [Google Scholar] [CrossRef] [PubMed]
- Privalov, P.L.; Makhatadze, G.I. Contribution of hydration to protein folding thermodynamics: II the entropy and Gibbs energy of hydration. J. Mol. Biol. 1993, 232, 660–679. [Google Scholar] [CrossRef] [PubMed]
- Laity, P.R.; Gilks, S.E.; Holland, C. Rheological behaviour of native silk feedstocks. Polymer 2015, 67, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Cowie, J.M.G. Polymers: Chemistry and Physics of Modern Materials, 2nd ed.; Chapmen and Hall: London, UK, 1997. [Google Scholar]
- Flippen, R.B. Photon correlation spectroscopy. In Modern Methods of Polymer Characterization; Barth, H.G., Mays, J.W., Eds.; John Wiley and Sons: New York, NY, USA, 1991; Chapter 10. [Google Scholar]
- Berry, G.C.; Cotts, P.M. Static and dynamic light scattering. In Modern Techniques for Polymer Characterisation; Pethrick, R.A., Dawkins, J.V., Eds.; John Wiley and Sons: Chichester, UK, 1999; Chapter 4. [Google Scholar]
- Pecora, R. Dynamic light scattering measurement of nanometer particles in liquids. J. Nanopart. Res. 2000, 2, 123–131. [Google Scholar] [CrossRef]
- Hossain, K.; Ochi, A.; Ooyama, E.; Magoshi, J.; Nemoto, N. Dynamic light scattering of native silk fibroin solution extracted from different parts of the middle division of the silk gland of the Bombyx mori silkworm. Biomacromolecules 2003, 4, 350–359. [Google Scholar] [CrossRef]
- Falk, M.; Ford, T.A. Infrared spectrum and structure of liquid water. Can. J. Chem. 1966, 44, 1699–1707. [Google Scholar] [CrossRef]
- Brubach, J.-B.; Mermet, A.; Filabozzi, A.; Gerschel, A.; Roy, P. Signatures of the hydrogen bonding in the infrared bands of water. J. Chem. Phys. 2005, 122, 184509. [Google Scholar] [CrossRef]
- Auer, B.M.; Skinner, J.L. IR and Raman spectra of liquid water: Theory and interpretation. J. Chem. Phys. 2008, 128, 224511. [Google Scholar] [CrossRef]
- Carpenter, W.B.; Fournier, J.A.; Biswas, R.; Voth, G.A.; Tokmakov, A. Delocalization and stretch-bend mixing of the HOH bend in liquid water. J. Chem. Phys. 2017, 147, 084503. [Google Scholar] [CrossRef] [PubMed]
- Kananenka, A.A.; Skinner, J.L. Fermi resonance in OH-stretch vibrational spectroscopy of liquid water and the water hexamer. J. Chem. Phys. 2018, 148, 244107. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.M.; Shakib, F.A.; Paesani, F. Disentangling coupling effects in the infrared spectra of liquid water. J. Phys. Chem. B 2018, 122, 10754–10761. [Google Scholar] [CrossRef] [PubMed]
- Grechko, M.; Hasegawa, T.; D’Angelo, F.; Ito, H.; Turchinovich, D.; Nagata, Y.; Bonn, M. Coupling between intra- and intermolecular motions in liquid water revealed by two dimensional terahertz-infrared-visible spectroscopy. Nat. Commun. 2018, 9, 885. [Google Scholar] [CrossRef]
- Zhang, B.; Yu, Y.; Zhang, Y.-Y.; Jiang, S.; Li, Q.; Hu, H.-S.; Li, G.; Zhao, Z.; Wang, C.; Xie, H.; et al. Infrared spectroscopy of neutral water clusters at finite temperature: Evidence for a noncyclic pentamer. Proc. Natl. Acad. Sci. USA 2020, 117, 15423–15428. [Google Scholar] [CrossRef] [PubMed]
- Barth, A.; Zscherp, C. What vibrations tell us about proteins. Quart. Rev. Biophys. 2020, 35, 369–430. [Google Scholar] [CrossRef]
- Barth, A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta 2007, 1767, 1073–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, H.; Mäntele, W. Infrared spectroscopies of proteins. In Handbook of Vibrational Spectroscopy; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2006. [Google Scholar] [CrossRef]
- Harrick, N.J. Surface chemistry from spectral analysis of totally internally reflected radiation. J. Phys. Chem. 1960, 64, 1110–1114. [Google Scholar] [CrossRef]
- Ekgasit, S.; Padermshoke, A. Optical contact in ATR/FT-IR spectroscopy. Appl. Spectr. 2001, 55, 1352–1359. [Google Scholar] [CrossRef]
- Milosevic, M. Internal reflection and ATR spectroscopy. Appl. Spectr. Rev. 2004, 39, 365–384. [Google Scholar] [CrossRef]
- Boulet-Audet, M.; Buffeteau, T.; Boudreault, S.; Daugey, N.; Pézolet, M. Quantitative determination of band distortions in diamond attenuated total reflectance infrared spectra. J. Phys. Chem. B. 2010, 114, 8255–8261. [Google Scholar] [CrossRef] [PubMed]
- Corujo, M.P.; Sklepari, M.; And, D.L.; Millichip, M.; Reason, A.; Goodchild, S.C.; Wormell, P.; Amarasinghe, D.P.; Lindo, V.; Chmel, N.P.; et al. Infrared absorbance spectroscopy of aqueous proteins: Comparison of transmission and ATR data collection and analysis of structure fitting. Chirality 2018, 30, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.G.; Schweitzer-Stenner, R.; Krimm, S.; Mirkin, N.G.; Asher, S.A. N-methylacetamide and its hydrogen bonded water molecules are vibrationally coupled. J. Am. Chem. Soc. 1994, 116, 11141–11142. [Google Scholar] [CrossRef]
- Cazade, P.-A.; Hédin, F.; Xu, Z.-H.; Meuwly, M. Vibrational relaxation and energy migration of N-methylacetamide in water: The role of nonbonded interactions. J. Phys. Chem. B. 2015, 119, 3112–3122. [Google Scholar] [CrossRef] [PubMed]
- Panuszko, A.; Gojlo, E.; Zielkiewicz, J.; Śmiechowski, M.; Krakowiak, J.; Stangret, J. Hydration of simple amides. FTIR spectra of HDO and theoretical studies. J. Phys. Chem. B. 2008, 112, 2483–2493. [Google Scholar] [CrossRef]
- Panuszko, A.; Wojciechowski, M.; Bruździak, P.; Rakowska, P.W.; Stangrat, J. Characteristics of hydration water around hen egg lysozyme as the protein model in aqueous solution. FTIR spectroscopy and molecular dynamics simulation. Phys. Chem. Chem. Phys. 2012, 14, 15765–15773. [Google Scholar] [CrossRef]
- Panuszko, A.; Nowak, M.G.; Bruździak, P.; Stasiulewicz, M.; Stangrat, J. Amides as models to study the hydration of proteins and peptides—Spectroscopic and theoretical approach on hydration in various temperatures. J. Mol. Liq. 2019, 278, 706–715. [Google Scholar] [CrossRef]
- Iwamoto, R. Infrared and near-infrared study of the interaction of amide C=O with water in ideally inert medium. J. Phys. Chem. A 2010, 114, 7398–7407. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.K.; Bates, S.P.; Crain, J.; Martyna, G.J. Solution structure of the aqueous model peptide N-methylacetamide. J. Phys. Chem. B 2006, 110, 21319–21326. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Zhang, J.; Li, C.; Luo, Y.; Ye, S. Ultrafast energy relaxation dynamics of amide I vibrations coupled with protein-bound water molecules. Nat. Commun. 2019, 10, 1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dannenberg, J.J. Enthalpies of hydration of N-methylacetamide by one, two, and three waters and the effect upon the C=O stretching frequency, an ab initio DFT study. J. Phys. Chem. A 2006, 110, 5798–5802. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.A.; Dobbs, K.D.; Valentini, J.J. Amide-water and amide-amide hydrogen bond strengths. J. Phys. Chem. 1994, 98, 13435–13439. [Google Scholar] [CrossRef]
- Xiao, X.; Tan, Y.; Zhu, L.; Guo, Y.; Wen, Z.; Li, M.; Pu, X.; Tian, A. Effects of the position and manner of hydration on the stability of solvated N-methylacetamides and the strength of binding between N-methylacetamide and water clusters: A computational study. J. Mol. Model. 2012, 18, 1389–1399. [Google Scholar] [CrossRef]
- Cai, K.; Su, T.; Lin, S.; Zheng, R. Molecular mechanics force field-based general map for the solvation effect on amide I probe of peptide in different micro-environments. Spectrochim. Acta A Mol. Biomol. Spectr. 2014, 117, 548–556. [Google Scholar] [CrossRef]
- Yadav, V.K.; Chandra, A. First-principles simulation study of vibrational spectral diffusion and hydrogen bond fluctuations in aqueous solution of N-methylacetamide. J. Phys. Chem. B. 2015, 119, 9858–9867. [Google Scholar] [CrossRef]
- Amunson, K.; Kubelka, J. On the temperature dependence of amide I frequencies of peptides in solution. J. Phys. Chem. B. 2007, 111, 9993–9998. [Google Scholar] [CrossRef]
- Ingrosso, F.; Monard, G.; Farag, M.H.; Bastida, A.; Ruiz-López, M.F. Importance of polarisation and charge transfer effects to model the infrared spectra of peptides in solution. J. Chem. Theory Comput. 2011, 7, 1840–1849. [Google Scholar] [CrossRef]
- Donati, G.; Petrone, A.; Rega, N. Multiresolution continuous wavelet transform for studying coupled solute–solvent vibrations via ab initio molecular dynamics. Phys. Chem. Chem. Phys. 2020, 22, 22645. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Swenson, C.J. Optimized intermolecular potential functions for amides and peptides. Hydration of amides. J. Am. Chem. Soc. 1985, 107, 1489–1496. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Gao, J. Cis-trans energy difference for the peptide bond in the gas phase and aqueous solution. J. Am. Chem. Soc. 1988, 110, 4212–4216. [Google Scholar] [CrossRef]
- Gaigeot, M.P.; Vuilleumier, R.; Sprik, M.; Borgis, D. Infrared spectroscopy of N-methylacetamide revisited by ab initio molecular dynamics simulations. J. Chem. Theory Comput. 2005, 1, 772–789. [Google Scholar] [CrossRef]
- Deetz, M.J.; Fahey, J.E.; Smith, B.D. NMR studies of hydrogen bonding interactions with secondary amide and urea groups. J. Phys. Org. Chem. 2001, 14, 463–467. [Google Scholar] [CrossRef]
- Di Gioacchino, M.; Bruni, F.; Ricci, M.A. N-methylacetamide aqueous solutions: A neutron diffraction study. J. Phys. Chem. B 2019, 123, 1808–1814. [Google Scholar] [CrossRef]
- Kang, Y.K.; Park, H.S. Internal rotation about the C-N bond of amides. J. Mol. Struct. 2004, 676, 171–176. [Google Scholar] [CrossRef]
- Mantz, Y.A.; Branduardi, D.; Bussi, G.; Parrinello, M. Ensemble of transition state structures for the cis-trans isomerization of N-methylacetamide. J. Phys. Chem. B. 2009, 113, 12521–12529. [Google Scholar] [CrossRef]
- Stark, E.; Luchter, K.; Margoshes, M. Near-infrared analysis (NIRA): A technology for quantitative and qualitative analysis. Appl. Spectr. Revs. 1986, 22, 335–339. [Google Scholar] [CrossRef]
- Bokobza, L. Near infrared spectroscopy. J. Near Infrared Anal. 1998, 6, 3–17. [Google Scholar] [CrossRef]
- Pasquini, C. Near infrared spectroscopy: Fundamentals, practical aspects and analytical applications. J. Braz. Chem. Soc. 2003, 14, 198–219. [Google Scholar] [CrossRef] [Green Version]
- Burns, D.A.; Ciurczak, E.W. Handbook of Near-Infrared Analysis, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Vandermuelen, D.L.; Ressler, N. A near-infrared analysis of water-macromolecule interactions: Hydration and the spectra of aqueous solutions of intact proteins. Arch. Biochem. Biophys. 1980, 199, 197–205. [Google Scholar] [CrossRef]
- Lamanna, R.; Delmelle, M.; Cannistraro, S. A near-infrared study of hydrogen bonding in human albumin aqueous solutions. Chem. Phys. Lett. 1990, 172, 312–316. [Google Scholar] [CrossRef]
- Liu, Y.; Czarnecki, M.A.; Ozaki, Y. Fourier-transform near-infrared study of dissociation and thermodynamic properties of N-methylacetamide in a carbon tetrachloride solution. Bull. Inst. Chem. Res. Kyoto Univ. 1993, 71, 133–139. Available online: https://hdl.handle.net/2433/77510 (accessed on 6 March 2021).
- Wang, J.; Sowa, M.G.; Ahmed, K.; Mantsch, H.H. Photoacoustic near-infrared investigation of homo-polypeptides. J. Phys. Chem. 1994, 98, 4748–4755. [Google Scholar] [CrossRef]
- Czarnecki, M.A.; Haufa, K.Z. Effect of temperature and concentration on the structure of N-methylacetamide-water complexes: Near-infrared spectroscopic study. J. Phys. Chem. A. 2005, 109, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Izutsu, K.; Fujimaki, Y.; Kuwabara, A.; Hiyama, Y.; Yomota, C.; Aoyagi, N. Near-infrared analysis of protein secondary structure in aqueous solutions and freeze-dried solids. J. Pharm. Sci. 2006, 95, 781–789. [Google Scholar] [CrossRef]
- Ma, L.; Cui, X.; Cai, W.; Shao, X. Understanding the function of water during the gelation of globular proteins by temperature dependent near infrared spectroscopy. Phys. Chem. Chem. Phys. 2018, 20, 20132–20140. [Google Scholar] [CrossRef]
- Mo, C.; Wu, P.; Chen, X.; Shao, Z. Near-infrared characterization on the secondary structure of regenerated Bombyx mori silk fibroin. Appl. Spectr. 2006, 60, 1438–1441. [Google Scholar] [CrossRef]
- Mo, C.; Wu, P.; Chen, X.; Shao, Z. The effect of water on the conformation transition of Bombyx mori silk fibroin. Vibr. Spectr. 2009, 51, 105–109. [Google Scholar] [CrossRef]
- Mapelli, M.; Greco, F.; Gussoni, M.; Consonni, R.; Zetta, L. Application of NMR microscopy to the morphological study of the silkworm Bombyx mori during its metamorphosis. Mag. Res. Imag. 1997, 15, 639–700. [Google Scholar] [CrossRef]
- Baldwin, R.L. How Hofmeister ion interactions affect protein stability. Biophys. J. 1996, 71, 2056–2063. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cremer, P.S. Interactions between macromolecules and ions: The Hofmeister series. Curr. Opin. Chem. Biol. 2006, 10, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Sedlák, E.; Stagg, L.; Wittung-Stafshede, P. Effect of Hofmeister ions on protein thermal stability: Roles of ionhydration and peptide groups? Arch. Biochem. Biophys. 2008, 479, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Salis, A.; Ninham, B.W. Models and mechanisms of Hofmeister effects in electrolyte solutions, and colloid and protein systems revisited. Chem. Soc. Rev. 2014, 43, 7358. [Google Scholar] [CrossRef] [Green Version]
- Senske, M.; Constantinescu-Aruxandei, D.; Havenith, M.; Herrmann, C.; Weingärtner, H.; Ebbinghaus, S. The temperature dependence of the Hofmeister series: Thermodynamic fingerprints of cosolute–protein interactions. Phys. Chem. Chem. Phys. 2016, 18, 29698. [Google Scholar] [CrossRef] [Green Version]
- Debye, P. Light scattering in solutions. J. Appl. Phys. 1944, 15, 338–342. [Google Scholar] [CrossRef]
- Doty, P.; Steiner, R.F. Light scattering and spectrophotometry of colloidal solutions. J. Chem. Phys. 1950, 18, 1211–1220. [Google Scholar] [CrossRef]
- Parker, T.G.; Dalgleish, D.G. The use of light scattering and turbidity measurements to study the kinetics of extensively aggregated proteins: αs-casein. Biopolymers 1977, 16, 2533–2547. [Google Scholar] [CrossRef]
- Camerini-Otero, R.D.; Day, L.A. The wavelength dependence of the turbidity of solutions of macromolecules. Biopolymers 1978, 17, 2241–2249. [Google Scholar] [CrossRef]
- Hall, D.; Minton, A.P. Turbidity as a probe of tubulin polymerisation kinetics: A theoretical and experimental re-examination. Anal. Biochem. 2005, 345, 198–213. [Google Scholar] [CrossRef]
- Grollman, A. The vapour pressure of aqueous solutions with special reference to the problem of the state of water in biological fluids. J. Gen. Physiol. 1931, 14, 661–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, R.A. The vapour pressure of solutions of potassium chloride and sodium chloride. Trans. R. Soc. N. Z. 1945, 75, 203–217. Available online: https://paperspast.natlib.govt.nz/periodicals/TPRSNZ1945-75.2.4.23 (accessed on 6 March 2021).
- Robinson, R.A. Activity coefficients of sodium chloride and potassium chloride in mixed aqueous solutions at 25°. J. Phys. Chem. 1961, 65, 662–667. [Google Scholar] [CrossRef]
- Robinson, R.A.; Bower, V.E. An additivity rule for the vapour pressure lowering of aqueous solutions. J. Res. Nat. Bureau Stand. A Phys. Chem. 1965, 69A, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, R.; Ziamajidi, F. Water activities of ternary mixtures of poly(ethylene glycol), NaCl and water over the temperature range of 293.15 K to 313.15 K. J. Chem. Thermodyn. 2006, 38, 1335–1343. [Google Scholar] [CrossRef]
- Wagner, W.; Pruß, A. The IAPWS formulation 1995 for the thermodynamic properties of ordinary water substance for general and scientific use. J. Phys. Chem. Ref. Data 2002, 31, 387–535. [Google Scholar] [CrossRef] [Green Version]
- Feistel, R.; Wagner, W. A new equation of state for H2O ice Ih. J. Phys. Chem. Ref. Data 2006, 35, 1021–1047. [Google Scholar] [CrossRef] [Green Version]
- Reiss, H.; Wilson, I.B. The effect of surface on melting point. J. Colloid Sci. 1948, 3, 551–561. [Google Scholar] [CrossRef]
- Still, R.C.; Skapski, A.S. Method for the determination of the surface tension of solids from their melting points in thin wedges. J. Chem. Phys. 1956, 24, 644–651. [Google Scholar] [CrossRef]
- Hay, J.N.; Laity, P.R. Observations of water migration during thermoporometry studies of cellulose films. Polymer 2000, 41, 6171–6180. [Google Scholar] [CrossRef]
- Laity, P.R.; Holland, C. Thermo-rheological behaviour of native silk feedstocks. Eur. Polym. J. 2017, 87, 519–534. [Google Scholar] [CrossRef] [Green Version]
- Russell, T.P.; Hjelm, R.P.; Seeger, P.A. Temperature dependence of the interaction parameter of polystyrene and poly(methyl methacrylate). Macromolecules 1990, 23, 890–893. [Google Scholar] [CrossRef]
- Etxabarren, C.; Iriarte, M.; Uriarte, C.; Etxeberrie, A.; Iruin, J.J. Polymer-solvent interaction parameter in polymer solutions at high polymer concentrations. J. Chromatog. 2002, 969, 245–254. [Google Scholar] [CrossRef]
- Dudowicz, J.; Freed, K.F.; Douglas, J.F. Solvation of polymers as mutual association. I. General theory. J. Chem. Phys. 2013, 138, 164901. [Google Scholar] [CrossRef]
- Dudowicz, J.; Freed, K.F.; Douglas, J.F. Solvation of polymers as mutual association. II. Basic thermodynamic properties. J. Chem. Phys. 2013, 138, 164902. [Google Scholar] [CrossRef]
- Mills, E.A.; Plotkin, S.S. Protein transfer free energy obeys entropy-enthalpy compensation. J. Phys. Chem. B 2015, 119, 14130–14144. [Google Scholar] [CrossRef]
- Pan, A.; Kar, T.; Rakshit, A.; Moulik, S.P. Enthalpy–entropy compensation (eec) effect: Decisive role of free energy. J. Phys. Chem. B. 2016, 120, 10531–10539. [Google Scholar] [CrossRef]
- Bigman, L.S.; Levy, Y. Entropy-enthalpy compensation in conjugated proteins. Chem. Phys. 2018, 514, 95–105. [Google Scholar] [CrossRef]
- Khrapunov, S. The enthalpy-entropy compensation phenomenon. Limitations for the use of some basic thermodynamic equations. Curr. Protein Pept. Sci. 2018, 19, 1088–1091. [Google Scholar] [CrossRef]
- Moulik, S.P.; Naskar, B.; Rakshit, A.K. Current status of enthalpy-entropy compensation phenomenon. Curr. Sci. 2019, 117, 1286–1291. [Google Scholar] [CrossRef]
- Wang, X.; Yang, X.; Chen, H.; Yang, X.; Xu, Z. Entropy–enthalpy compensation in peptide adsorption on solid surfaces: Dependence on surface hydration. Langmuir 2020, 36, 10822–10829. [Google Scholar] [CrossRef]
- Dunitz, J.D. Win some, lose some: Enthalpy-entropy compensation in weak intermolecular interactions. Chem. Biol. 1995, 2, 709–712. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Kaplan, D.; Cebe, P. Dynamic protein-water relationships during β-sheet formation. Macromolecules 2008, 41, 3939–3948. [Google Scholar] [CrossRef]
- Michnik, A. Thermal stability of bovine serum albumin: DSC study. J. Thermal Anal. Calorim. 2003, 71, 509–519. [Google Scholar] [CrossRef]
- Michnik, A.; Michalik, K.; Kluczewska, A.; Drzazga, Z. Comparative dsc study of human and bovine serum albumin. J. Therm. Anal. Calorim. 2006, 84, 113–117. [Google Scholar] [CrossRef]
- Weijers, M.; Barneveld, P.A.; Cohen-Stuart, M.A.; Visschers, R.W. Heat-induced denaturation and aggregation of ovalbumin at neutral pH described by irreversible first-order kinetics. Protein Sci. 2003, 12, 2693–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Gehoh, M.; Yazuriha, K. Crystal structure of silk (Bombyx mori). J. Polym. Sci. B Polym. Phys. 1991, 29, 889–891. [Google Scholar] [CrossRef]
- Marsh, R.E.; Corey, R.B.; Pauling, L. An investigation of the structure of silk fibroin. Biochim. Biophys. Acta 1955, 16, 1–34. [Google Scholar] [CrossRef]
- Warwicker, J.O. The crystal structure of silk fibroin. Acta Cryst. 1954, 7, 565–573. [Google Scholar] [CrossRef]
- Öjelund, G.; Sköld, R.; Wadsö, I. Thermochemistry of solutions of biochemical model compounds, 5. Transfer of N-alkylamides from water to non-aqueous media. J. Chem. Thermodyn. 1976, 8, 45–54. [Google Scholar] [CrossRef]
- Della Gatta, G.; Barone, G.; Elia, V. Enthalpies of solvation for N-alkylamides in water and carbon tetrachloride at 25 °C. J. Solut. Chem. 1986, 15, 157–167. [Google Scholar] [CrossRef]
- Spencer, J.N.; Berger, S.K.; Powell, C.R.; Henning, B.D.; Furman, G.S.; Loffredo, W.M.; Rydberg, E.M.; Neubert, R.A.; Shoop, C.E.; Blauch, D.N. Amide interactions in aqueous and organic medium. J. Phys. Chem. 1981, 85, 1236–1241. [Google Scholar] [CrossRef]
- Akiyama, M. Study on hydration enthalpy of N-methylacetamide in water. Spectrochim. Acta A 2002, 58, 1943–1950. [Google Scholar] [CrossRef]
- Kreis, R.W.; Wood, R.H. Enthalpy of fusion and cryoscopic constant of N-methylacetamide. J. Chem. Thermodyn. 1969, 1, 523–526. [Google Scholar] [CrossRef]
- Ahlers, J.; Lohmann, J.; Gmehling, J. Binary solid-liquid equilibria of organic systems containing different amides and sulfolane. J. Chem. Eng. Data 1999, 44, 727–730. [Google Scholar] [CrossRef]
- Specific Heat of Some Liquids and Fluids, The Engineering Toolbox. Available online: https://www.engineeringtoolbox.com/specific-heat-fluids-d_151.html (accessed on 1 October 2021).
- Lide, D.R. (Ed.) Handbook of Chemistry and Physics, 82nd ed.; CRC Press: Boca Raton, FL, USA, 2001. [Google Scholar]
- Bondi, A. Estimation of the heat capacity of liquids. Ind. Eng. Chem. Fundam. 1966, 5, 442–449. [Google Scholar] [CrossRef]
- Shaw, R. Heat capacities of liquids. Estimation of heat capacity at constant pressure and 25 °C, using additivity rules. J. Chem. Eng. Data 1969, 14, 461–465. [Google Scholar] [CrossRef]
- Cerdeiriña, C.A.; Gonzáles-Salgado, D.; Romani, L.; del Carmen Dalgado, M.; Torres, L.A.; Costas, M. Towards an understanding of the heat capacity of liquids. A simple two-state model for molecular association. J. Chem. Phys. 2004, 120, 6648. [Google Scholar] [CrossRef]
- Al-Shorachi, H.N.; Hashim, E.T. Estimation of heat capacity for liquids. Pet. Sci. Technol. 2007, 25, 1513–1517. [Google Scholar] [CrossRef]
- Bolmatov, D.; Brazhkin, V.V.; Trachenko, K. The phonon theory of liquid thermodynamics. Sci. Rep. 2012, 2, 421. [Google Scholar] [CrossRef]
- Schliesser, J.M.; Woodfield, B.F. Development of a Debye heat capacity model for vibrational modes with a gap in the density of states. J. Phys. Condens. Matter 2015, 27, 285402. [Google Scholar] [CrossRef] [Green Version]
- Naef, R. Calculations of the isobaric heat capacities of the liquid and solid phase of organic compounds at and around 298.15 K based on their ‘true’ molecular volume. Molecules 2019, 24, 1626. [Google Scholar] [CrossRef] [Green Version]
- Fomin, Y.D. Anomalously high heat capacity of liquids, relation to structural properties. Mol. Phys. 2019, 117, 2786–2792. [Google Scholar] [CrossRef]
- Benson, S.W. Heat capacity and structure in liquids. Application to the structure of water. J. Am. Chem. Soc. 1978, 100, 5640–5644. [Google Scholar] [CrossRef]
- Benson, S.W.; Siebert, E.D. A simple two-structure model for liquid water. J. Am. Chem. Soc. 1992, 114, 4269–4276. [Google Scholar] [CrossRef]
- Sceats, M.G.; Rice, S.A. The enthalpy and heat capacity of liquid water and the ice polymorphs from a random network model. J. Chem Phys. 1980, 72, 3248–3259. [Google Scholar] [CrossRef]
- Vidler, M.; Tennyson, J. Accurate partition function and thermodynamic data for water. J. Chem. Phys. 2000, 113, 9766–9771. [Google Scholar] [CrossRef] [Green Version]
- Abe, H. Estimation of heat capacity and properties of water by spectrum decomposition of the second overtone band of OH stretching vibration. J. Near Infrared Spectrosc. 2004, 12, 45–54. [Google Scholar] [CrossRef]
- Lishchuk, S.V.; Malomuzh, N.P.; Makhlaichuk, P.V. Contribution of H-bond vibrations to heat capacity of water. Phys. Lett. A 2011, 375, 2656–2660. [Google Scholar] [CrossRef]
- Makhlaichuk, V.N.; Malomuzh, N.P. Manifestation of cluster excitations in dielectric properties of water vapor and liquid water as well as their heat capacity. J. Mol. Liq. 2018, 253, 83–90. [Google Scholar] [CrossRef]
- Chen, H.; Hansen, K. Low temperature heat capacity of water clusters. Chem. Phys. Lett. 2014, 610–611, 369–374. [Google Scholar] [CrossRef]
- Putintsev, N.M.; Putintsev, D.N. Heat capacity and thermal expansion of water and helium. J. Therm. Sci. 2017, 26, 125–131. [Google Scholar] [CrossRef]
- Marshall, B.D. A resummed thermodynamic perturbation theory for positive and negative hydrogen bond cooperativity in water. J. Phys. Condens. Matter 2019, 31, 184001. [Google Scholar] [CrossRef]
- Höhne, G.W.H.; Hemminger, W.F.; Flammersheim, H.-J. Differential Scanning Calorimetry; Springer: Berlin, Germany, 2003. [Google Scholar]
- Spink, C.H. Differential scanning calorimetry. Methods Cell Biol. 2008, 84, 115–141. [Google Scholar] [CrossRef]
- Privalov, P.L. Microcalorimetry of macromolecules: The physical basis of biological structures. J. Solut. Chem. 2015, 44, 1141–1161. [Google Scholar] [CrossRef]
- Sochava, I.V.; Smirnova, O.I. Heat capacity of hydrated and dehydrated globular proteins: Denaturation increment of heat capacity. Food Hydrocoll. 1993, 6, 513–524. [Google Scholar] [CrossRef]
- Myers, J.K.; Pace, C.N.; Scholtz, J.M. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995, 4, 2138–2148. [Google Scholar] [CrossRef] [Green Version]
- Makhatadze, G.I. Heat capacities of amino acids, peptides and proteins. Biophys. Chem. 1998, 71, 133–156. [Google Scholar] [CrossRef]
- Mazurenko, S.; Kunka, A.; Beerens, K.; Johnson, C.M.; Damborsky, J.; Prokop, Z. Exploration of protein unfolding by modelling calorimetry data from reheating. Sci. Rep. 2017, 7, 16321. [Google Scholar] [CrossRef] [Green Version]
- Li-Blatter, X.; Seelig, J. Thermal and chemical unfolding of lysozyme. multistate Zimm−Bragg theory versus two-state model. J. Phys. Chem. B 2019, 123, 10181–10191. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Ruggeri, F.S.; Vigolo, D.; Kamada, A.; Qamar, S.; Levin, A.; Iserman, C.; Albert, S.; St. George-Hyslop, P.; Knowles, T.P.J. Biomolecular condensates undergo a generic shear-mediated liquid-to-solid transition. Nat. Nanotech. 2020, 15, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, V.Y.; Burova, T.V.; Grinberg, N.V.; Moskalets, A.P.; Dubovik, A.S.; Plashchina, I.G.; Khokhlov, A.R. Energetics and mechanisms of poly(n-isopropylacrylamide) phase transitions in water−methanol solutions. Macromolecules 2020, 53, 10765–10772. [Google Scholar] [CrossRef]
- Kametani, S.; Sekine, S.; Ohkubo, T.; Hirano, T.; Ute, K.; Cheng, H.N.; Asakura, T. NMR studies of water dynamics during sol-to-gel transition of poly(N-isopropylacrylamide) in concentrated aqueous solution. Polymer 2017, 109, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Halle, B. Protein hydration dynamics in solution: A critical survey. Philos. Trans. Biol. Sci. 2004, 359, 1207–1224. [Google Scholar] [CrossRef]
- Ebbinghaus, S.; Kim, S.J.; Heyden, M.; Yu, X.; Heugen, U.; Gruebele, M.; Leitner, D.M.; Havenith, M. An extended dynamical hydration shell around proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 20749–20752. Available online: https://pnas.org/cgi/doi/10.1073/pnas.0709207104 (accessed on 6 March 2021). [CrossRef] [Green Version]
- Ebbinghaus, S.; Kim, S.J.; Heyden, M.; Yu, X.; Heugen, U.; Gruebele, M.; Leitner, D.M.; Havenith, M. Protein sequence and pH-dependent hydration probed by Terahertz spectroscopy. J. Am. Chem. Soc. 2008, 130, 2374–2375. [Google Scholar] [CrossRef]
- Russo, D.; Ollivier, J.; Teixera, J. Water hydrogen bond analysis on hydrophilic and hydrophobic biomolecule sites. Phys. Chem. Chem. Phys. 2008, 10, 4968–4974. [Google Scholar] [CrossRef]
- Grebenkov, D.S.; Goddard, Y.A.; Diakova, G.; Korb, J.-P.; Bryant, R.G. Dimensionality of diffusive exploration at the protein interface in solution. J. Phys. Chem. B. 2009, 113, 13347–13356. [Google Scholar] [CrossRef] [Green Version]
- Perticaroli, S.; Comez, L.; Paolantoni, M.; Sassi, P.; Morresi, A.; Fioretto, D. Extended frequency range depolarised light scattering study of N-acetyl-leucine-methylamide–water solutions. J. Am. Chem. Soc. 2011, 133, 12063–12068. [Google Scholar] [CrossRef]
- Gallat, F.-X.; Laganowsky, A.; Wood, K.; Gabel, F.; van Eijck, L.; Wuttke, J.; Moulin, M.; Härtlein, M.; Eisenberg, D.; Colletier, J.-P.; et al. Dynamical coupling of intrinsically disordered proteins and their hydration water: Comparison with folded soluble and membrane proteins. Biophys. J. 2012, 103, 129–136. [Google Scholar] [CrossRef] [Green Version]
- King, J.T.; Kubarych, K.J. Site-specific coupling of hydration water and protein flexibility studied in solution with ultrafast 2D-IR spectroscopy. J. Am. Chem. Soc. 2012, 134, 18705–18712. [Google Scholar] [CrossRef]
- Nickels, J.D.; O’Neill, H.; Hong, L.; Tyagi, M.; Ehlers, G.; Weiss, K.L.; Zhang, Q.; Yi, Z.; Mamontov, E.; Smith, J.C.; et al. Dynamics of protein and its hydration water: Neutron scattering studies in fully deuterated GFP. Biophys. J. 2012, 103, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Conti-Nibali, V.; Havenith, M. New insights into the role of water in biological function: Studying solvated biomolecules using terahertz absorption spectroscopy in conjunction with molecular dynamics simulations. J. Am. Chem. Soc. 2014, 136, 12800–12807. [Google Scholar] [CrossRef]
- Xu, Y.; Havenith, M. Perspective: Watching low-frequency vibrations of water in biomolecular recognition by THz spectroscopy. J. Chem. Phys. 2015, 143, 170901. [Google Scholar] [CrossRef] [Green Version]
- Jose, J.C.; Khatua, P.; Bansal, N.; Sengupta, N.; Bandyopadhyay, S. Microscopic hydration properties of the Aβ1-42 peptide monomer and the globular protein ubiquitin: A comparative molecular dynamics study. J. Phys. Chem. B. 2014, 118, 11591–11604. [Google Scholar] [CrossRef] [PubMed]
- Sushko, O.; Dubrovka, R.; Donnan, R.S. Sub-terahertz spectroscopy reveals that proteins influence the properties of water at greater distance than previously detected. J. Chem. Phys. 2015, 142, 055101. [Google Scholar] [CrossRef]
- Bellissent-Funel, M.-C.; Hassanali, A.; Havenith, M.; Henchman, R.; Pohl, P.; Sterpone, F.; van der Spoel, D.; Yu, Y.; Garcia, A.E. Water determines the structure and dynamics of proteins. Chem. Rev. 2016, 116, 7673–7697. [Google Scholar] [CrossRef]
- Serin, G.; Nguyen, H.H.; Marty, J.-D.; Micheau, J.-C.; Gernigon, V.; Mingotaud, A.-F.; Bajon, D.; Soulet, T.; Massenot, S.; Coudret, C. Terahertz time-domain spectroscopy of thermoresponsive polymers in aqueous solution. J. Phys. Chem. B 2016, 120, 9778–9787. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.; Schmollngruber, M.; Steinhauser, O. Rotational dynamics of water molecules near biological surfaces with implications for nuclear quadrupole relaxation. Phys. Chem. Chem. Phys. 2016, 18, 24620–24630. [Google Scholar] [CrossRef]
- Gavrilov, Y.; Leuchter, J.D.; Levy, J. On the coupling between the dynamics of protein and water. Phys. Chem. Chem. Phys. 2017, 19, 8243–8257. [Google Scholar] [CrossRef]
- Yamamoto, N.; Ito, S.; Nakanishi, M.; Chatani, E.; Inoue, K.; Kandori, H.; Tominaga, K. Effect of temperature and hydration level on purple membrane dynamics studied using broadband dielectric spectroscopy from sub-GHz to THz regions. J. Phys. Chem. B 2018, 122, 1367–1377. [Google Scholar] [CrossRef]
- Batys, P.; Zhang, Y.; Lutkenhaus, J.L.; Sammalkorpi, M. Hydration and temperature response of water mobility in poly(diallyldimethylammonium)–poly(sodium 4-styrenesulfonate) complexes. Macromolecules 2018, 51, 8268–8277. [Google Scholar] [CrossRef] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laity, P.R.; Holland, C. Seeking Solvation: Exploring the Role of Protein Hydration in Silk Gelation. Molecules 2022, 27, 551. https://doi.org/10.3390/molecules27020551
Laity PR, Holland C. Seeking Solvation: Exploring the Role of Protein Hydration in Silk Gelation. Molecules. 2022; 27(2):551. https://doi.org/10.3390/molecules27020551
Chicago/Turabian StyleLaity, Peter R., and Chris Holland. 2022. "Seeking Solvation: Exploring the Role of Protein Hydration in Silk Gelation" Molecules 27, no. 2: 551. https://doi.org/10.3390/molecules27020551
APA StyleLaity, P. R., & Holland, C. (2022). Seeking Solvation: Exploring the Role of Protein Hydration in Silk Gelation. Molecules, 27(2), 551. https://doi.org/10.3390/molecules27020551