
Synthesis and Application of New Salan Titanium Complexes in the Catalytic Reduction of Aldehydes


Abstract
:1. Introduction
2. Results and Discussion
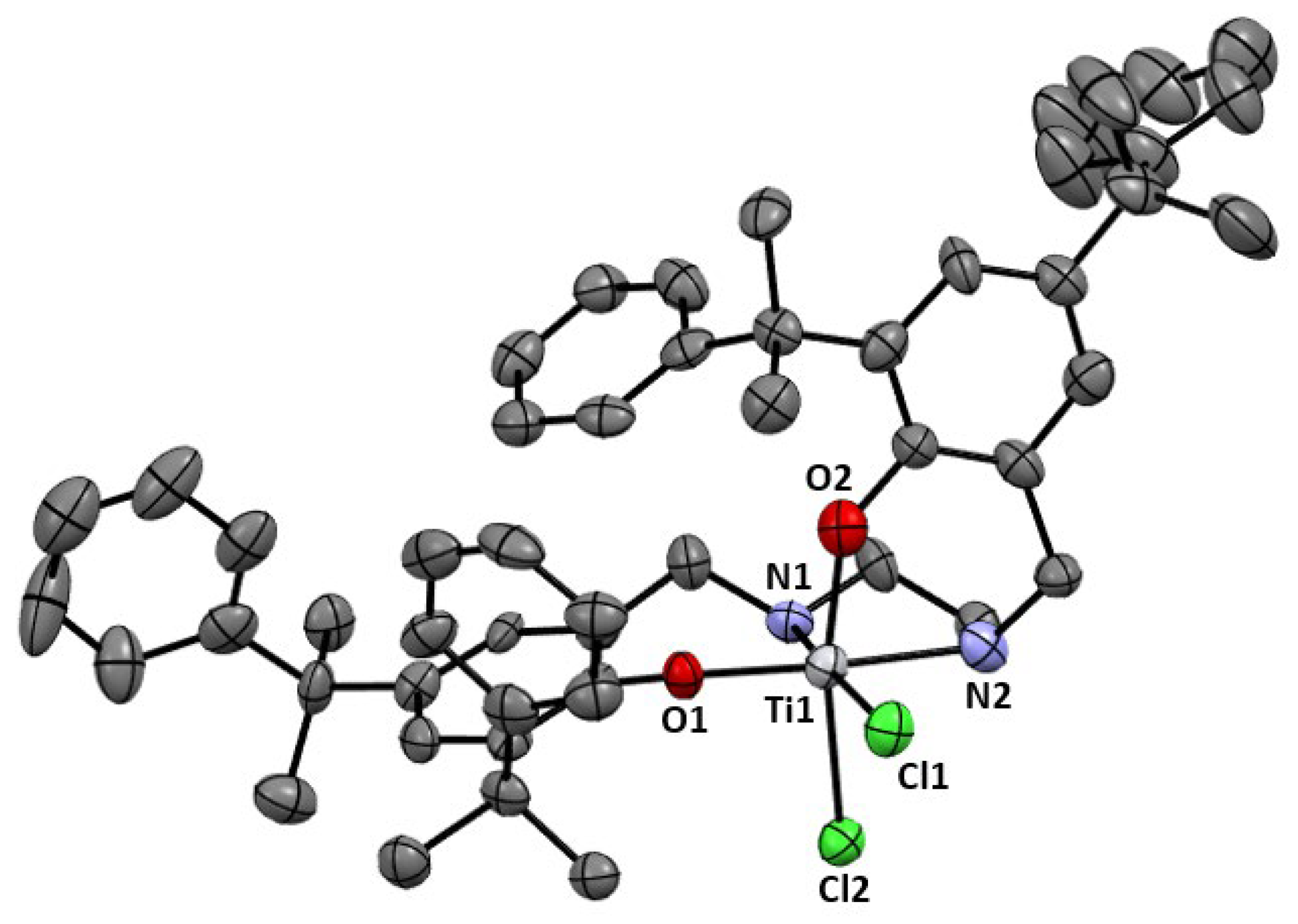
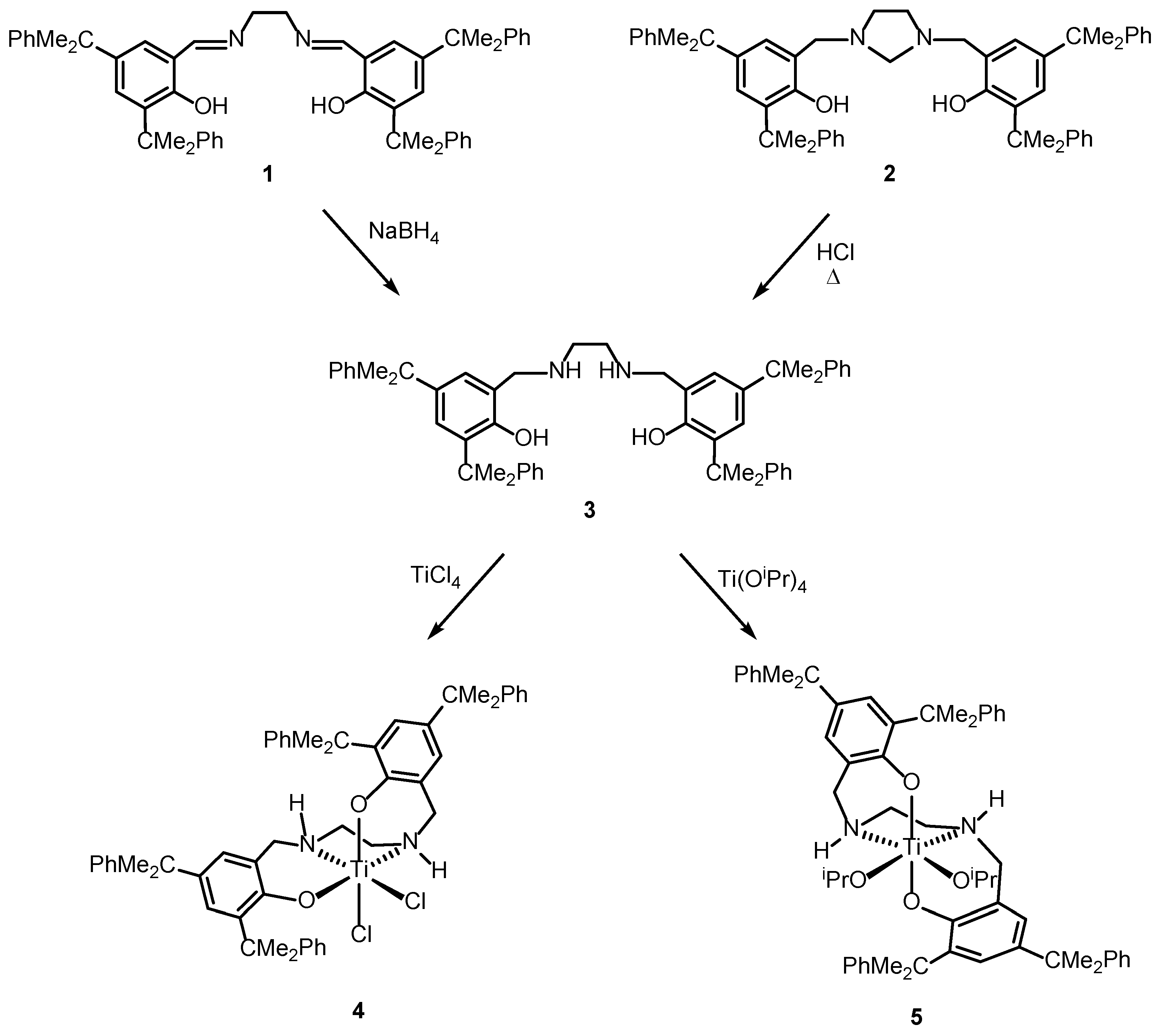
2.1. Synthesis and Characterization
2.2. Catalytic Studies
3. Materials and Methods
3.1. General Considerations
3.2. Synthesis and Characterization of the Compounds
3.2.1. 2-Hydroxy-3,5-Bis(2-Phenylpropan-2-yl)Benzaldehyde
3.2.2. 6,6′-((1E,1′E)-(Ethane-1,2-Diylbis(Azaneylylidene))Bis(methaneylylidene))Bis(2,4-Bis(2-Phenylpropan-2-yl)Phenol), 1
3.2.3. 6,6′-(Imidazolidine-1,3-Diylbis(Methylene))Bis(2,4-Bis(2-Phenylpropan-2-yl)Phenol), 2
3.2.4. 6,6′-((Ethane-1,2-Diylbis(Azanediyl))Bis(Methylene))Bis(2,4-Bis(2-Phenylpropan-2-yl)phenol), H2N2O2H2, 3
3.2.5. [(H2N2O2)TiCl2], 4
3.2.6. [(H2N2O2)Ti(OiPr)2], 5
3.2.7. H2N2O2Na2, 6
3.3. General Procedure for the Catalytic Reduction of Aldehydes
3.4. General Procedure for Single Crystal X-ray Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Xiong, H.; Li, L.; Liu, E.; Cheng, J.; Zhang, G. A chiral multidentate salan-supported heterobimetallic catalyst for asymmetric Friedel-Crafts reaction. Inorg. Chem. Commun. 2017, 84, 24–27. [Google Scholar] [CrossRef]
- Wang, Z.; He, J.; Mu, Y. Synthesis of chiral salan ligands with bulky substituents and their application in Cu-catalyzed asymmetric Henry reaction. J. Organomet. Chem. 2020, 928, 121546. [Google Scholar] [CrossRef]
- Chen, J.; Gu, H.; Zhu, X.; Nam, W.; Wang, B. Zirconium-Salan Catalyzed Enantioselective α-Hydroxylation of β-Keto Esters. Adv. Synth. Catal. 2020, 362, 2976–2983. [Google Scholar] [CrossRef]
- Bunda, S.; Udvardy, A.; Voronova, K.; Joó, F. Organic Solvent-Free, Pd(II)-Salan Complex-Catalyzed Synthesis of Biaryls via Suzuki-Miyaura Cross-Coupling in Water and Air. J. Org. Chem. 2018, 83, 15486–15492. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Kopilov, J.; Lamberti, M.; Venditto, V.; Kol, M. Same Ligand, Different Metals: Diiodo-Salan Complexes of the Group 4 Triad in Isospecific Polymerization of 1-Hexene and Propylene. Macromolecules 2010, 43, 1689–1691. [Google Scholar] [CrossRef]
- Białek, M.; Pochwała, M.; Spaleniak, G. Olefin polymerization and copolymerization by complexes bearing [ONNO]-Type salan ligands: Effect of ligand structure and metal type (titanium, zirconium, and vanadium). J. Polym. Sci. A Polym. Chem. 2014, 52, 2111–2123. [Google Scholar] [CrossRef]
- Meppelder, G.-J.M.; Fan, H.-T.; Spaniol, T.P.; Okuda, J. Group 4 Metal Complexes Supported by [ONNO]-Type Bis(o-aminophenolato) Ligands: Synthesis, Structure, and α-Olefin Polymerization Activity. Organometallics 2009, 28, 5159–5165. [Google Scholar] [CrossRef]
- Ouyang, H.; Yuan, D.; Nie, K.; Zhang, Y.; Yao, Y.; Cui, D. Synthesis and Characterization of Dinuclear Salan Rare-Earth Metal Complexes and Their Application in the Homo- and Copolymerization of Cyclic Esters. Inorg. Chem. 2018, 57, 9028–9038. [Google Scholar] [CrossRef]
- Chmura, A.J.; Davidson, M.G.; Jones, M.D.; Lunn, M.D.; Mahon, M.F.; Johnson, A.F.; Khunkamchoo, P.; Roberts, S.L.; Wong, S.S.F. Group 4 Complexes with Aminebisphenolate Ligands and Their Application for the Ring Opening Polymerization of Cyclic Esters. Macromolecules 2006, 39, 7250–7257. [Google Scholar] [CrossRef]
- Sumrit, P.; Hormnirun, P. Aluminum Initiators Supported by Asymmetric [ONNO’]-Type Salan Ligands for the Ring-Opening Polymerization of rac-Lactide. Macromol. Chem. Phys. 2013, 214, 1845–1851. [Google Scholar] [CrossRef]
- Matsumoto, K.; Sawada, Y.; Katsuki, T. Asymmetric epoxidation of olefins catalyzed by Ti(salan) complexes using aqueous hydrogen peroxide as the oxidant. Pure Appl. Chem. 2008, 80, 1071–1077. [Google Scholar] [CrossRef]
- Jat, J.L.; De, S.R.; Kumar, G.; Adebesin, A.M.; Gandham, S.K.; Falck, J.R. Regio- and Enantioselective Catalytic Monoepoxidation of Conjugated Dienes: Synthesis of Chiral Allylic cis-Epoxides. Org. Lett. 2015, 17, 1058–1061. [Google Scholar] [CrossRef] [Green Version]
- Talsi, E.P.; Samsonenko, D.G.; Bryliakov, K.P. Titanium Salan Catalysts for the Asymmetric Epoxidation of Alkenes: Steric and Electronic Factors Governing the Activity and Enantioselectivity. Chem.-Eur. J. 2014, 20, 14329–14335. [Google Scholar] [CrossRef] [PubMed]
- Adão, P.; Avecilla, F.; Bonchio, M.; Carraro, M.; Pessoa, J.C.; Correia, I. Titanium(IV)-Salan Catalysts for Asymmetric Sulfoxidation with Hydrogen Peroxide. Eur. J. Inorg. Chem. 2010, 2010, 5568–5578. [Google Scholar] [CrossRef]
- Talsi, E.P.; Bryliakov, K.P. Titanium-salan-catalyzed asymmetric sulfoxidations with H2O2: Design of more versatile catalysts. Appl. Organomet. Chem. 2013, 27, 239–244. [Google Scholar] [CrossRef]
- Talsi, E.P.; Bryliakov, K.P. Ti-Salan catalyzed asymmetric sulfoxidation of pyridylmethylthiobenzimidazoles to optically pure proton pump inhibitors. Cat. Today 2017, 279, 84–89. [Google Scholar] [CrossRef]
- Maru, M.S.; Barroso, S.; Adão, P.; Alves, L.G.; Martins, A.M. New salan and salen vanadium complexes: Synthesis and application in sulfoxidation catalysis. J. Organomet. Chem. 2018, 870, 136–144. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Zhu, C. Enantioselective Pinacol Coupling of Aryl Aldehydes Catalyzed by Chiral Salan-Mo(IV) Complexes. J. Org. Chem. 2007, 72, 10029–10034. [Google Scholar] [CrossRef]
- Sun, J.; Dai, Z.; Li, C.; Zhu, C. Enantioselective pinacol coupling reaction of aromatic aldehydes catalyzed by chiral vanadium complexes. J. Organomet. Chem. 2009, 694, 3219–3221. [Google Scholar] [CrossRef]
- Ramana, M.M.V.; Singh, B.K.D.; Parihar, J.A. Microwave-assisted coupling of carbonyl compound: An efficient synthesis of olefin. J. Chem. Res. 2004, 2004, 760–761. [Google Scholar] [CrossRef]
- Bravo, J.A.; Vila, J.L. Obtaining of alkenes by reductive coupling of carbonylic compounds; Synthesis of Z,E-6-dodecene, synthesis of flexibilene and isocaryophylene, mechanistic views. Boliv. J. Chem. 2018, 35, 73–84. [Google Scholar]
- Li, J.J. Wolff-Kishner Reaction. In Name Reactions, 6th ed.; Li, J.J., Ed.; Springer: Cham, Switzerland, 2021; pp. 583–585. [Google Scholar]
- Li, J.J. Clemmensen Reduction. In Name Reactions, 6th ed.; Li, J.J., Ed.; Springer: Cham, Switzerland, 2021; pp. 109–111. [Google Scholar]
- Wolfrom, M.L.; Karabinos, J.V. Carbonyl Reduction by Thioacetal Hydrogenolysis. J. Am. Chem. Soc. 1944, 66, 909–911. [Google Scholar] [CrossRef]
- Sousa, S.C.A.; Fernandes, T.A.; Fernandes, A.C. Highly Efficient Deoxygenation of Aryl Ketones to Arylalkanes Catalyzed by Dioxidomolybdenum Complexes. Eur. J. Org. Chem. 2016, 2016, 3109–3112. [Google Scholar] [CrossRef]
- Argouarch, G. Mild and efficient rhodium-catalyzed deoxygenation of ketones to alkanes. New. J. Chem. 2019, 43, 11041–11044. [Google Scholar] [CrossRef]
- Kalutharage, N.; Yi, C.S. Scope and Mechanistic Analysis for Chemoselective Hydrogenolysis of Carbonyl Compounds Catalyzed by a Cationic Ruthenium Hydride Complex with a Tunable Phenol Ligand. J. Am. Chem. Soc 2015, 137, 11105–11114. [Google Scholar] [CrossRef] [Green Version]
- Hancock, S.L.; Mahon, M.F.; Jones, M.D. Monomeric Ti(IV) homopiperazine complexes and their exploitation for the ring opening polymerization of rac-lactide. Chem. Cent. J. 2013, 7, 135. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.; Tshuva, E.Y. Synthesis of Pure Enantiomers of Titanium(IV) Complexes with Chiral Diaminobis(Phenolato) Ligands and Their Biological Reactivity. Sci. Rep. 2018, 8, 9705. [Google Scholar] [CrossRef] [Green Version]
- Yeori, A.; Groysman, S.; Goldberg, I.; Kol, M. Diastereoisomerically Selective Enantiomerically Pure Titanium Complexes of Salan Ligands: Synthesis, Structure, and Preliminary Activity Studies. Inorg. Chem. 2005, 44, 4466–4468. [Google Scholar] [CrossRef]
- Jones, M.D.; Davidson, M.G.; Kociok-Kohn, G. New titanium and zirconium initiatores for the production of polylactide. Polyhedron 2010, 29, 697–700. [Google Scholar] [CrossRef]
- MacMillan, S.N.; Jung, C.F.; Shalumova, T.; Tanski, J.M. Chiral-at-metal tetrahydrosalen complexes of resolved titanium(IV) sec-butoxides: Ligand wrapping and multiple asymmetric catalytic induction. Inorg. Chim. Acta 2009, 362, 3134–3146. [Google Scholar] [CrossRef]
- Manna, C.M.; Tshuva, E.Y. Markedly different cytotoxicity of the two enantiomers of C2-symmetrical Ti(IV) penolato complexes; mechanistic implications. Dalton Trans. 2010, 39, 1182–1184. [Google Scholar] [CrossRef] [PubMed]
- Meker, S.; Manna, C.M.; Peri, D.; Tshuva, E.Y. Major impact of N-methylation on cytotoxic and hydrolysis of salan Ti(IV) complexes: Sterics and electronics are intertwined. Dalton Trans. 2011, 40, 9802–9809. [Google Scholar] [CrossRef] [PubMed]
- SAINT, Version 7.03A; Bruker AXS Inc.: Madison, WI, USA, 1997–2003.
- Sheldrick, G.M.; SADABS. Software for Empirical Absorption Corrections; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Cryst. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Crystallogr. 2005, 38, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure and refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Cryst. C Struct. Chem. 2015, C71, 9–18. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst | Solvent | T (°C) | t (h) | Conv. (%) | Alkane (%) | Alcohol (%) | Diol (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 4 | THF | 30 | 24 | 93 | 75 | 9 | 4 |
| 2 | 4 | THF | 55 | 4 | 82 | 78 | 0 | 2 |
| 3 c | 4 | THF | 55 | 4 | 55 | 3 | 0 | 0 |
| 4 | 4 | THF | 55 | 7 | 91 | 89 | 0 | 1 |
| 5 | 4 | THF | 55 | 24 | 94 | 92 | 0 | 1 |
| 6 | 4 | EtOH | 55 | 4 | 47 | 46 | 1 | 0 |
| 7 | 4 | n-Bu2O/H2O | 55 | 24 | 44 | 40 | 4 | 0 |
| 8 | 5 | THF | 30 | 24 | 75 | 69 | 5 | 0 |
| 9 | - | THF | 55 | 4 | 29 | 2 | 4 | 0 |
| 10 | - | EtOH | 55 | 4 | 3 | 3 | 0 | 0 |

| Entry | Substrate | t (h) | Conv. (%) | Alkane (%) | Alcohol (%) |
|---|---|---|---|---|---|
| 1 | PhCH2C(O)H | 24 | 99 | 87 | 0 |
| 2 | PhCH2CH2C(O)H | 24 | 99 | 84 | 0 |
| Compound | 42.(C4H10O)3 | 5 | 7 |
|---|---|---|---|
| Empirical formula | C116 H146 Cl4 N4 O7 Ti2 | C58 H72 N2 O4 Ti | C58 H76 N2 O4 Si2 Ti |
| Formula weight | 1945.91 | 909.05 | 969.26 |
| Temperature (K) | 150 (2) | 150 (2) | 294 (2) |
| Crystal system, space group | Tetragonal, I41/a | Triclinic, P-1 | Triclinic, P-1 |
| a (Å) | 25.4100 (1) | 11.664 (3) | 10.315 (2) |
| b (Å) | 25.4100 (1) | 15.050 (6) | 14.434 (4) |
| c (Å) | 33.3570 (2) | 16.062 (5) | 19.035 (5) |
| α(º) | 90 | 73.87 (2) | 79.30 (1) |
| β(º) | 90 | 86.84 (1) | 86.65 (1) |
| γ(º) | 90 | 67.29 (1) | 88.76 (2) |
| Volume (Å3) | 21537.6 (2) | 2494.3 (15) | 2779.9 (12) |
| Z | 8 | 2 | 2 |
| Calculated density (g m−3) | 1.200 | 1.210 | 1.158 |
| Absorption coefficient (mm−1) | 0.303 | 0.220 | 0.242 |
| F (000) | 8304 | 976 | 1040 |
| Crystal size (mm) | 0.20 × 0.26 × 0.34 | 0.10 × 0.16 × 0.22 | 0.08 × 0.08 × 0.14 |
| θ range for data collection (°) | 1.007–25.576 | 3.163–27.046 | 1.978–26.558 |
| Limiting indices | −30 ≤ h ≤ 30, −30 ≤ k ≤ 30, −37 ≤ l ≤ 40 | −14 ≤ h ≤ 11, −18 ≤ k ≤ 14, −20 ≤ l ≤ 20 | −12 ≤ h ≤ 12, −18 ≤ k ≤ 18, −23 ≤ l ≤ 23 |
| Reflections collected/unique | 117534/10101 [Rint = 0.2329] | 26502/10473 [Rint = 0.0730] | 62151/10227 [Rint = 0.3149] |
| Completeness to θ = 25.242 | 99.9 | 99.5 | 88.8 |
| Data/restraints/parameters | 10029/0/612 | 10473/0/606 | 10227/0/626 |
| Goodness-of-fit on F2 | 1.519 | 0.961 | 0.842 |
| Final R indices [I > 2σ(I)] a | R1 = 0.1579, wR2 = 0.4442 | R1 = 0.0558, wR2 = 0.1186 | R1 = 0.0897, wR2 = 0.2013 |
| R indices (all data) a | R1 = 0.2840, wR2 = 0.4777 | R1 = 0.1105, wR2 = 0.1365 | R1 = 0.2545, wR2 = 0.2425 |
| Largest diff. peak/hole (e Å−3) | 1.176 and −0.592 | 0.379 and −0.720 | 0.748 and −0.469 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hipólito, J.; Martins, A.M.; Alves, L.G. Synthesis and Application of New Salan Titanium Complexes in the Catalytic Reduction of Aldehydes. Molecules 2022, 27, 6821. https://doi.org/10.3390/molecules27206821
Hipólito J, Martins AM, Alves LG. Synthesis and Application of New Salan Titanium Complexes in the Catalytic Reduction of Aldehydes. Molecules. 2022; 27(20):6821. https://doi.org/10.3390/molecules27206821
Chicago/Turabian StyleHipólito, Joana, Ana M. Martins, and Luis G. Alves. 2022. "Synthesis and Application of New Salan Titanium Complexes in the Catalytic Reduction of Aldehydes" Molecules 27, no. 20: 6821. https://doi.org/10.3390/molecules27206821
APA StyleHipólito, J., Martins, A. M., & Alves, L. G. (2022). Synthesis and Application of New Salan Titanium Complexes in the Catalytic Reduction of Aldehydes. Molecules, 27(20), 6821. https://doi.org/10.3390/molecules27206821