
Multitargeted Molecular Docking and Dynamic Simulation Studies of Bioactive Compounds from Rosmarinus officinalis against Alzheimer’s Disease

 ,
,
Abstract
:1. Introduction
2. Results
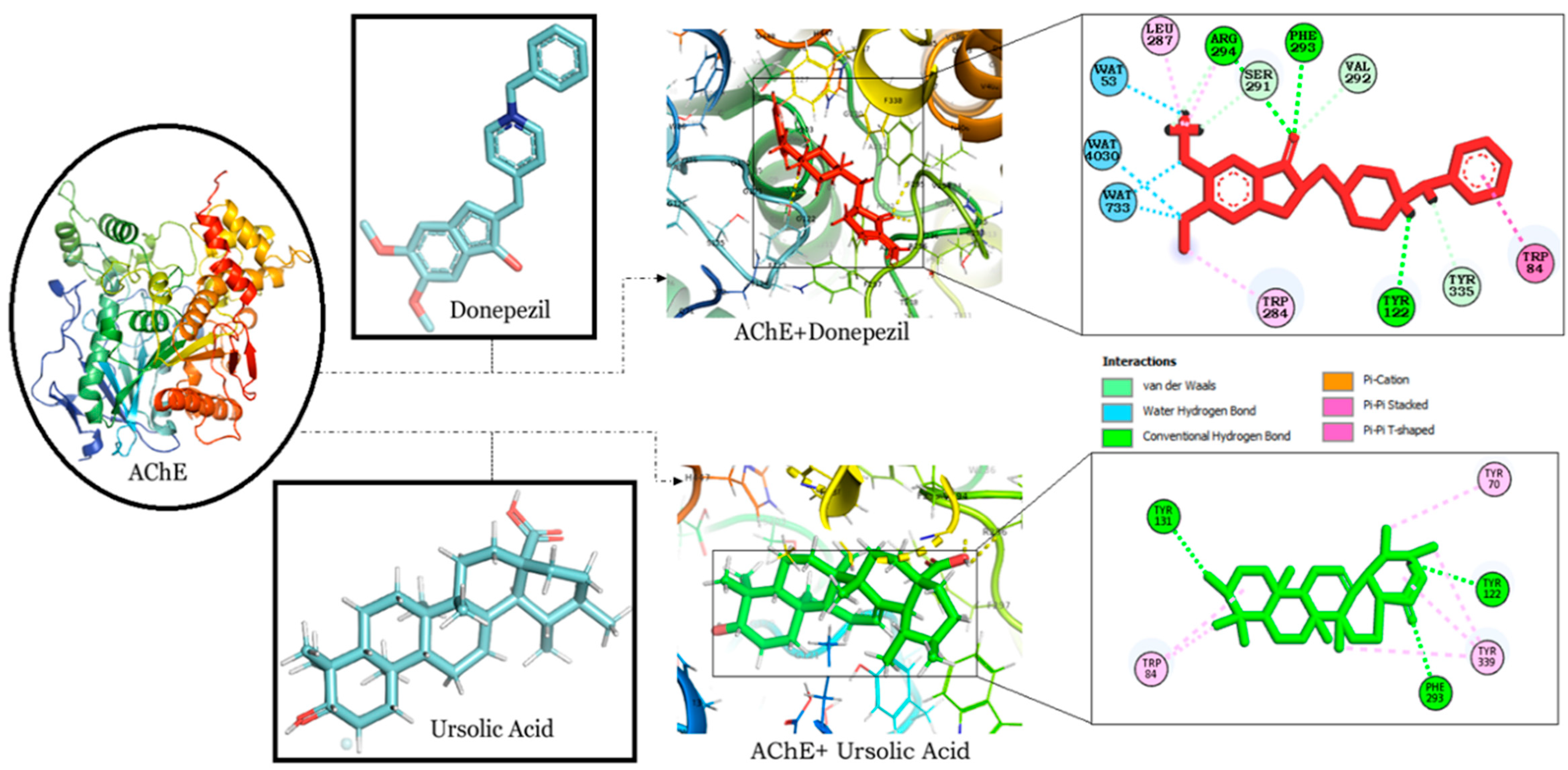
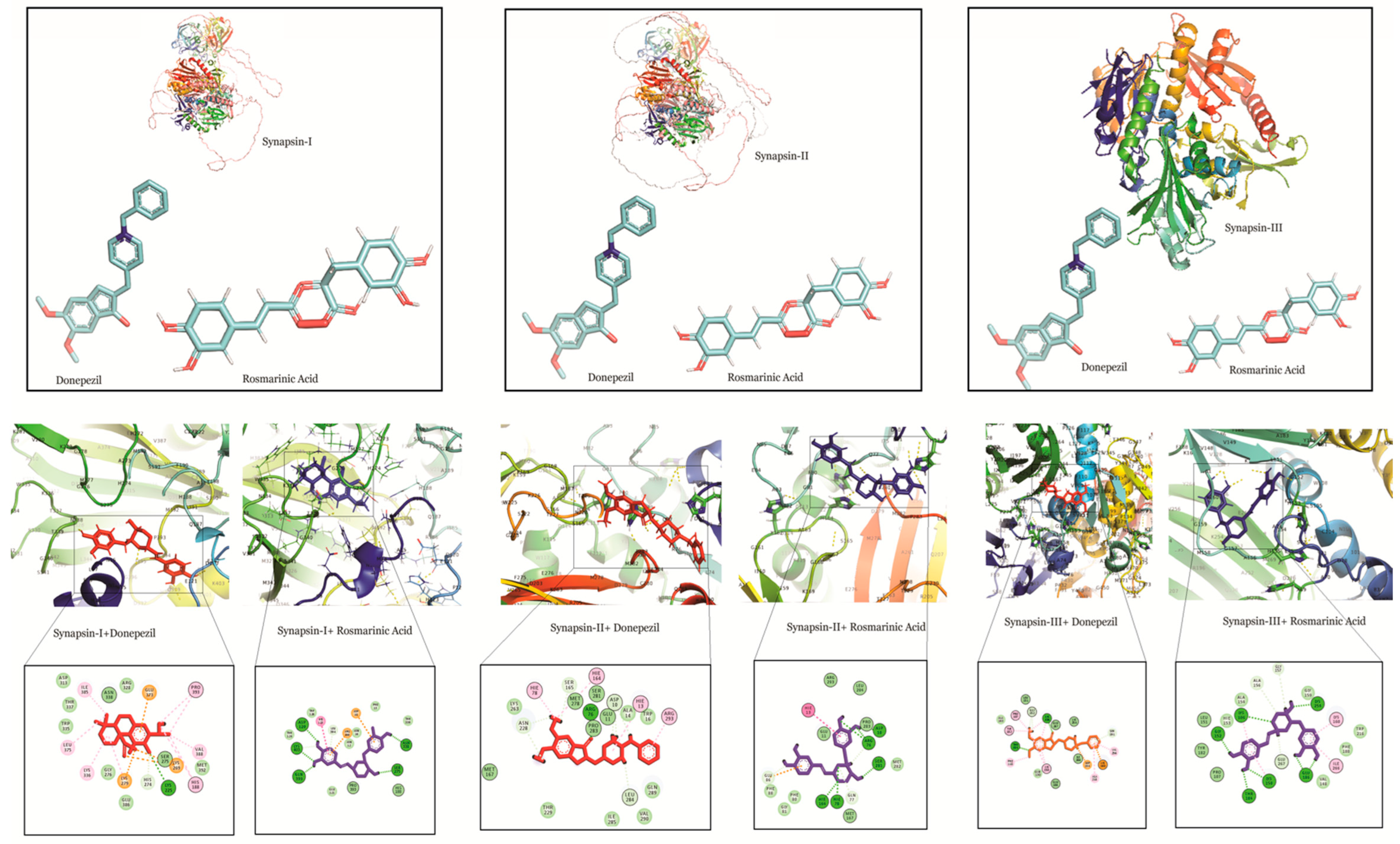
2.1. Molecular Docking Studies of Rosmarinic, Carnosic and Ursolic Acid on BACE1, AChE, Synapsin I, II and III
2.1.1. RA and UA Exhibit Binding Energy Comparable to Donepezil in Binding Interactions with AChE
2.1.2. RA Exhibits Strong Binding Interactions Strikingly Similar to Donepezil with BACE1
2.1.3. RA Exhibits Strong Binding Interactions with Synapsin I, II and III
2.2. Molecular Dynamic Simulation Studies of Rosmarinic, Carnosic and Ursolic Acid on BACE1, AChE, Synapsin I, II and III
2.3. Binding Free Energies of Interactions
2.4. Drug Likeness Analysis of CA, RA and UA
3. Discussion
4. Materials and Methods
4.1. Molecular Docking Simulations
4.2. Molecular Dynamics Simulation Analysis
4.3. Binding Free Energy Calculation Using MM/PBSA and MM/GBSA
4.4. Prediction of Drug Likeness of CA, RA and UA
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Ayllón, M.S.; Small, D.H.; Avila, J.; Sáez-Valero, J. Revisiting the role of acetylcholinesterase in Alzheimer’s disease: Cross-talk with P-tau and β-amyloid. Front. Mol. Neurosci. 2011, 4, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Fu, Y.; Yasvoina, M.; Shao, P.; Hitt, B.; O’Connor, T.; Logan, S.; Maus, E.; Citron, M.; Berry, R.; et al. β-Site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: Implications for Alzheimer’s disease pathogenesis. J. Neurosci. 2007, 27, 3639–3649. [Google Scholar] [CrossRef] [Green Version]
- Twilley, D.; Rademan, S.; Lall, N. A review on traditionally used South African medicinal plants, their secondary metabolites and their potential development into anticancer agents. J. Ethnopharmacol. 2020, 261, 113101. [Google Scholar] [CrossRef] [PubMed]
- Karthika, C.; Appu, A.P.; Akter, R.; Rahman, M.H.; Tagde, P.; Ashraf, G.M.; Abdel-Daim, M.M.; ul Hassan, S.S.; Abid, A.; Bungau, S. Potential innovation against Alzheimer’s disorder: A tricomponent combination of natural antioxidants (vitamin E, quercetin, and basil oil) and the development of its intranasal delivery. Environ. Sci. Pollut. Res. Int. 2022, 29, 10950–10965. [Google Scholar] [CrossRef]
- Tan, C.C.; Yu, J.T.; Wang, H.F.; Tan, M.S.; Meng, X.F.; Wang, C.; Jiang, T.; Zhu, X.C.; Tan, L. Efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimer’s Dis. 2014, 41, 615–631. [Google Scholar] [CrossRef]
- Haake, A.; Nguyen, K.; Friedman, L.; Chakkamparambil, B.; Grossberg, G.T. An update on the utility and safety of cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Drug. Saf. 2020, 19, 147–157. [Google Scholar] [CrossRef]
- Allegra, A.; Tonacci, A.; Pioggia, G.; Musolino, C.; Gangemi, S. Anticancer Activity of Rosmarinus officinalis L.: Mechanisms of Action and Therapeutic Potentials. Nutrients 2020, 12, 1739. [Google Scholar] [CrossRef]
- De Oliveira, J.R.; Camargo SE, A.; de Oliveira, L.D. Rosmarinus officinalis L. (rosemary) as therapeutic and prophylactic agent. J. Biomed. Sci. 2019, 26, 5. [Google Scholar] [CrossRef]
- Andrade, J.M.; Faustino, C.; Garcia, C.; Ladeiras, D.; Reis, C.P.; Rijo, P. Rosmarinus officinalis L.: An update review of its phytochemistry and biological activity. Future Sci. OA 2018, 4, FSO283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borges, R.S.; Ortiz, B.L.S.; Pereira, A.C.M.; Keita, H.; Carvalho JC, T. Rosmarinus officinalis essential oil: A review of its phytochemistry, anti-inflammatory activity, and mechanisms of action involved. J. Ethnopharmacol. 2019, 229, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Rašković, A.; Milanović, I.; Pavlović, N.; Ćebović, T.; Vukmirović, S.; Mikov, M. Antioxidant activity of rosemary (Rosmarinus officinalis L.) essential oil and its hepatoprotective potential. BMC Complement. Altern. Med. 2014, 14, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahbardar, M.G.; Hosseinzadeh, H. Therapeutic effects of rosemary (Rosmarinus officinalis L.) and its active constituents on nervous system disorders. Iran J. Basic Med. Sci. 2020, 23, 1100. [Google Scholar]
- Amoah, S.K.; Sandjo, L.P.; Kratz, J.M.; Biavatti, M.W. Rosmarinic acid–pharmaceutical and clinical aspects. Planta Med. 2016, 82, 388–406. [Google Scholar] [CrossRef] [Green Version]
- Hase, T.; Shishido, S.; Yamamoto, S.; Yamashita, R.; Nukima, H.; Taira, S.; Kobayashi, S. Rosmarinic acid suppresses Alzheimer’s disease development by reducing amyloid β aggregation by increasing monoamine secretion. Sci. Rep. 2019, 9, 8711. [Google Scholar] [CrossRef] [Green Version]
- Kola, A.; Hecel, A.; Lamponi, S.; Valensin, D. Novel Perspective on Alzheimer ’s disease Treatment: Rosmarinic Acid Molecular Interplay with Copper (II) and Amyloid β. Life 2020, 10, 118. [Google Scholar] [CrossRef]
- Cornejo, A.; Aguilar Sandoval, F.; Caballero, L.; Machuca, L.; Muñoz, P.; Caballero, J.; Perry, J.; Ardiles, A.; Areche, C.; Melo, F. Rosmarinic acid prevents fibrillization and diminishes vibrational modes associated to β sheet in tau protein linked to Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2017, 32, 945–953. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Su, H.; Qu, Q.M. Carnosic acid prevents beta-amyloid-induced injury in human neuroblastoma sh-sy5y cells via the induction of autophagy. Neurochem. Res. 2016, 41, 2311–2323. [Google Scholar] [CrossRef]
- Rasoolijazi, H.; Azad, N.; Joghataei, M.T.; Kerdari, M.; Nikbakht, F.; Soleimani, M. The protective role of carnosic acid against beta-amyloid toxicity in rats. Sci. World J. 2013, 2013, 917082. [Google Scholar] [CrossRef] [Green Version]
- Yi-Bin, W.; Xiang, L.; Bing, Y.; Qi, Z.; Fei-Tong, J.; Minghong, W.; Xiangxiang, Z.; Le, K.; Yan, L.; Ping, S.; et al. Inhibition of the CEBPβ-NFκB interaction by nanocarrier-packaged Carnosic acid ameliorates glia-mediated neuroinflammation and improves cognitive function in an Alzheimer’s disease model. Cell Death Dis. 2022, 13, 318. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Cui, D.; Li, Y.; Shi, J.; Xiang, L.; Bian, H.; Ma, Z.; Xia, W.; Wei, G. Carnosic Acid Reverses the Inhibition of ApoE4 on Cell Surface Level of ApoER2 and Reelin Signaling Pathway. J. Alzheimer’s Dis. 2020, 73, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Murakami, A.; Ohigashi, H. Ursolic acid: An anti-and pro-inflammatory triterpenoid. Mol. Nutr. Food Res. 2008, 52, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Gudoityte, E.; Arandarcikaite, O.; Mazeikiene, I.; Bendokas, V.; Liobikas, J. Ursolic and oleanolic acids: Plant metabolites with neuroprotective potential. Int. J. Mol. Sci. 2021, 22, 4599. [Google Scholar] [CrossRef] [PubMed]
- Loesche, A.; Köwitsch, A.; Lucas, S.D.; Al-Halabi, Z.; Sippl, W.; Al-Harrasi, A.; Csuk, R. Ursolic and oleanolic acid derivatives with cholinesterase inhibiting potential. Bioorg. Chem. 2019, 85, 23–32. [Google Scholar] [CrossRef]
- Liang, W.; Zhao, X.; Feng, J.; Song, F.; Pan, Y. Ursolic acid attenuates beta-amyloid-induced memory impairment in mice. Arq. Neuropsiquiatr. 2016, 74, 482–488. [Google Scholar] [CrossRef] [Green Version]
- Mirza, F.J.; Amber, S.; Hassan, D.; Ahmed, T.; Zahid, S. Rosmarinic acid and ursolic acid alleviate deficits in cognition, synaptic regulation and adult hippocampal neurogenesis in an Aβ1-42-induced mouse model of Alzheimer’s disease. Phytomedicine 2021, 83, 153490. [Google Scholar] [CrossRef]
- Shams Ul Hassan, S.; Abbas, S.Q.; Hassan, M.; Jin, H.Z. Computational Exploration of Anti-Cancer Potential of GUAIANE Dimers from Xylopia vielana by Targeting B-Raf Kinase Using Chemo-Informatics, Molecular Docking, and MD Simulation Studies. Anticancer Agents Med. Chem. 2022, 22, 731–746. [Google Scholar] [CrossRef]
- Chaniad, P.; Mungthin, M.; Payaka, A.; Viriyavejakul, P.; Punsawad, C. Antimalarial properties and molecular docking analysis of compounds from Dioscorea bulbifera L. as new antimalarial agent candidates. BMC Complement. Med. Ther. 2021, 21, 144. [Google Scholar] [CrossRef]
- Ouassaf, M.; Belaidi, S.; Chtita, S.; Lanez, T.; Abul Qais, F.; Md Amiruddin, H. Combined molecular docking and dynamics simulations studies of natural compounds as potent inhibitors against SARS-CoV-2 main protease. J. Biomol. Struct. Dyn. 2021, 27, 1–10. [Google Scholar] [CrossRef]
- Atali, S.; Dorandish, S.; Devos, J.; Williams, A.; Price, D.; Taylor, J.; Guthrie, J.; Heyl, D.; Evans, H.G. Interaction of amyloid beta with humanin and acetylcholinesterase is modulated by ATP. FEBS Open Bio 2020, 10, 2805–2823. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Mirza, F.J.; Zahid, S. The role of synapsins in neurological disorders. Neurosci. Bull. 2018, 34, 349–358. [Google Scholar] [CrossRef]
- Rong, X.; Jiang, L.; Qu, M.; Hassan, S.; Liu, Z. Enhancing Therapeutic Efficacy of Donepezil by Combined Therapy: A Comprehensive Review. Curr. Pharm. Des. 2021, 27, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Birks, J.S.; Harvey, R.J. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst. Rev. 2018, 6, 1–235. [Google Scholar] [CrossRef]
- Sarno, T.A.; Talib, L.L.; Joaquim HP, G.; Bram JM, D.F.; Gattaz, W.F.; Forlenza, O.V. Protein expression of BACE1 is downregulated by donepezil in Alzheimer’s disease platelets. J. Alzheimer’s Dis. 2017, 55, 1445–1451. [Google Scholar] [CrossRef]
- Green, K.D.; Fosso, M.Y.; Garneau-Tsodikova, S. Multifunctional donepezil analogues as cholinesterase and BACE1 inhibitors. Molecules 2018, 23, 3252. [Google Scholar] [CrossRef] [Green Version]
- Halperin, I.; Ma, B.; Wolfson, H.; Nussinov, R. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins 2002, 47, 409–443. [Google Scholar] [CrossRef]
- Hassan, S.; Abbas, S.Q.; Ali, F.; Ishaq, M.; Bano, I.; Hassan, M.; Jin, H.-Z.; Bungau, S.G. A Comprehensive in Silico Exploration of Pharmacological Properties, Bioactivities, Molecular Docking, and Anticancer Potential of Vieloplain F from Xylopia vielana Targeting B-Raf Kinase. Molecules 2022, 27, 917. [Google Scholar] [CrossRef]
- Li, J.; Fu, A.; Zhang, L. An Overview of Scoring Functions Used for Protein-Ligand Interactions in Molecular Docking. Interdiscipl. Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef]
- Chen, Y.C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, T. Evaluation of AutoDock and AutoDock Vina on the CASF-2013 benchmark. J. Chem. Inf. Model 2018, 58, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Zev, S.; Raz, K.; Schwartz, R.; Tarabeh, R.; Gupta, P.K.; Major, D.T. Benchmarking the ability of common docking programs to correctly reproduce and score binding modes in SARS-CoV-2 protease Mpro. J. Chem. Inf. Model 2021, 61, 2957–2966. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model 2021, 61, 3891–3898. [Google Scholar]
- Zheng, L.; Meng, J.; Jiang, K.; Lan, H.; Wang, Z.; Lin, M.; Li, W.; Guo, H.; Wei, Y.; Mu, Y. Improving protein-ligand docking and screening accuracies by incorporating a scoring function correction term. Brief. Bioinform. 2022, 13, bbac051. [Google Scholar] [CrossRef]
- Pham, T.; Nguyen, T.H.; Tam, N.M.; YVu, T.; Pham, N.T.; Huy, N.T.; Mai, B.K.; Tung, N.T.; Pham, M.Q.; Vu, V.V.; et al. Improving ligand-ranking of AutoDock Vina by changing the empirical parameters. J. Comput. Chem. 2022, 43, 160–169. [Google Scholar] [CrossRef]
- Solis-Vasquez, L.; Tillack, A.F.; Santos-Martins, D.; Koch, A.; LeGrand, S.; Forli, S. Benchmarking the performance of irregular computations in AutoDock-GPU molecular docking. Parallel Comput. 2022, 109, 102861. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.; Nguyen, T.H.; Pham, T.; Huy, N.T.; Bay, M.V.; Pham, M.Q.; Nam, P.C.; Vu, V.V.; Ngo, S.T. Autodock Vina Adopts More Accurate Binding Poses but Autodock4 Forms Better Binding Affinity. J. Chem. Inf. Model 2020, 60, 204–211. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, M.; Dubey, R. Target enzyme in Alzheimer’s disease: Acetylcholinesterase inhibitors. Curr. Top. Med. Chem. 2019, 19, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Szeto, J.Y.; Lewis, S.J. Current treatment options for Alzheimer’s disease and Parkinson’s disease dementia. Curr. Neuropharmacol. 2016, 14, 326–338. [Google Scholar] [CrossRef]
- Silva, M.A.; Kiametis, A.S.; Treptow, W. Donepezil inhibits acetylcholinesterase via multiple binding modes at room temperature. J. Chem. Inf. Model 2020, 60, 3463–3471. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Youn, K.; Ho, C.T.; Karwe, M.V.; Jeong, W.S.; Jun, M. p-Coumaric acid and ursolic acid from Corni fructus attenuated β-amyloid(25-35)-induced toxicity through regulation of the NF-κB signaling pathway in PC12 cells. J. Agric. Food Chem. 2014, 62, 4911–4916. [Google Scholar] [CrossRef]
- Das, B.; Yan, R. Role of BACE1 in Alzheimer’s synaptic function. Transl. Neurodegener. 2017, 6, 23. [Google Scholar] [CrossRef]
- Ahmed, R.R.; Holler, C.J.; Webb, R.L.; Li, F.; Beckett, T.L.; Murphy, M.P. BACE1 and BACE2 enzymatic activities in Alzheimer’s disease. J. Neurochem. 2010, 112, 1045–1053. [Google Scholar] [CrossRef] [Green Version]
- Benjannet, S.; Elagoz, A.; Wickham, L.; Mamarbachi, M.; Munzer, J.S.; Basak, A.; Lazure, C.; Cromlish, J.A.; Sisodia, S.; Checler, F.; et al. Post-translational processing of β-secretase (β-amyloid-converting enzyme) and its ectodomain shedding: The pro-and transmembrane/cytosolic domains affect its cellular activity and amyloid-β production. J. Biol. Chem. 2001, 276, 10879–10887. [Google Scholar] [CrossRef] [Green Version]
- Haniu, M.; Denis, P.; Young, Y.; Mendiaz, E.A.; Fuller, J.; Hui, J.O.; Bennett, B.D.; Kahn, S.; Ross, S.; Burgess, T.; et al. Characterization of Alzheimer’s beta-secretase protein BACE. A pepsin family member with unusual properties. J. Biol. Chem. 2000, 275, 21099–21106. [Google Scholar] [CrossRef] [Green Version]
- Ermolieff, J.; Loy, J.A.; Koelsch, G.; Tang, J. Proteolytic activation of recombinant pro-memapsin 2 (pro-β-secretase) studied with new fluorogenic substrates. Biochemistry 2000, 39, 12450–12456. [Google Scholar] [CrossRef]
- Huse, J.T.; Pijak, D.S.; Leslie, G.J.; Lee VM, Y.; Doms, R.W. Maturation and endosomal targeting of β-site amyloid precursor protein-cleaving enzyme: The Alzheimer’s disease β-secretase. J. Biol. Chem. 2000, 275, 33729–33737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, L.; Tang, J. Flap position of free memapsin 2 (β-secretase), a model for flap opening in aspartic protease catalysis. Biochemistry 2004, 43, 4689–4695. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Osswald, H.L. BACE1 (β-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6765–6813. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, R.; Hatayama, K.; Takahashi, T.; Hayashi, T.; Sato, Y.; Sato, D.; Ohta, K.; Nakano, H.; Seki, C.; Endo, Y.; et al. Structure-activity relations of rosmarinic acid derivatives for the amyloid β aggregation inhibition and antioxidant properties. Eur. J. Med. Chem. 2017, 138, 1066–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gürbüz, P.; Dokumacı, A.H.; Gündüz, M.G.; Perez, C.; Göger, F.; Paksoy, M.Y.; Yerer, M.B.; Demirezer, L.Ö. In vitro biological activity of Salvia fruticose Mill. infusion against amyloid β-peptide-induced toxicity and inhibition of GSK-3β, CK-1δ, and BACE-1 enzymes relevant to Alzheimer’s disease. Saudi Pharm. J. 2021, 29, 236–243. [Google Scholar] [CrossRef]
- Zheng, K.; Hu, F.; Zhou, Y.; Zhang, J.; Zheng, J.; Lai, C.; Xiong, W.; Cui, K.; Hu, Y.-Z.; Han, Z.-T.; et al. miR-135a-5p mediates memory and synaptic impairments via the Rock2/Adducin1 signaling pathway in a mouse model of Alzheimer’s disease. Nat. Comm. 2021, 12, 1903. [Google Scholar] [CrossRef]
- Marsh, J.; Bagol, S.H.; Williams, R.S.; Dickson, G.; Alifragis, P. Synapsin I phosphorylation is dysregulated by beta-amyloid oligomers and restored by valproic acid. Neurobiol. Dis. 2017, 106, 63–75. [Google Scholar] [CrossRef]
- Maesako, M.; Zoltowska, K.M.; Berezovska, O. Synapsin 1 promotes Aβ generation via BACE1 modulation. PLoS ONE 2019, 14, e0226368. [Google Scholar] [CrossRef] [Green Version]
- Okimoto, N.; Futatsugi, N.; Fuji, H.; Suenaga, A.; Morimoto, G.; Yanai, R.; Ohno, Y.; Narumi, T.; Taiji, M. High-performance drug discovery: Computational screening by combining docking and molecular dynamics simulations. PLoS Comput. Biol. 2009, 5, e1000528. [Google Scholar] [CrossRef]
- Santos, L.H.; Ferreira, R.S.; Caffarena, E.R. Integrating molecular docking and molecular dynamics simulations. In Docking Screens for Drug Discovery; Humana: New York, NY, USA, 2019; pp. 13–34. [Google Scholar]
- Maia, E.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef]
- Cerón-Carrasco, J.P. When virtual screening yields inactive drugs: Dealing with false theoretical friends. Chem. Med. Chem. 2022, 17, e202200278. [Google Scholar] [CrossRef]
- Jinhua, W.; Ying, Z.; Yuhua, L. PXR-ABC drug transporters/CYP-mediated ursolic acid transport and metabolism in vitro and vivo. Arch. Pharm. 2020, 353, e2000082. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Huang, M.; Liu, D.; Li, Y.; Luo, Q.; Pham, K.; Wang, M.; Zhang, J.; Zhang, R.; Peng, Z.; et al. Absorption and Transport Characteristics and Mechanisms of Carnosic Acid. Biology 2021, 10, 1278. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Bitencourt-Ferreira, G.; de Azevedo, W.F. Molecular Docking Simulations with ArgusLab. In Docking Screens for Drug Discovery; Humana: New York, NY, USA, 2019; pp. 203–220. [Google Scholar]
- Systèmes, D. Biovia, Discovery Studio Modeling Environment; Dassault Systèmes Biovia: San Diego, CA, USA, 2016. [Google Scholar]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L.; DeLano, W. PyMOL. The PyMOL Molecular Graphics System, Version 2; Schrödinger, LLC.: New York, NY, USA, 2020. [Google Scholar]
- Turner, P.J. XMGRACE, Version 5.1. 19. Center for Coastal and Land-Margin Research; Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005; Volume 2. [Google Scholar]
- Chen, J.; Zhang, S.; Wang, W.; Sun, H.; Zhang, Q.; Liu, X. Binding of Inhibitors to BACE1 Affected by pH-Dependent Protonation: An Exploration from Multiple Replica Gaussian Accelerated Molecular Dynamics and MM-GBSA Calculations. ACS Chem. Neurosci. 2021, 12, 2591–2607. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Ligand | Binding Energy | Interacting Residues |
|---|---|---|---|
| AChE | Rosmarinic acid | −9.56 | H Bonds: Phe 293, Phe 336, Tyr 335, Asn 85, Glu 200 Pi-Pi interaction: Phe 295, Trp 84 Pi-Sigma: Tyr 339 |
| AChE | Carnosic acid | −7.91 | H Bonds: Ser 123, Tyr 335 Alkyl Bonds: Val 71, Pro 86 Pi-Sigma: Trp 84 Carbon-Hydrogen bond: Gly 119 |
| AChE | Ursolic acid | −9.17 | H Bonds: Tyr 131, Tyr 122, Phe 293 Pi- Alkyl Bonds: Trp 84, Tyr 70, Tyr 339 |
| AChE | Donepezil | −9.17 | H Bonds: Tyr 122, Phe 293, Arg 294 Alkyl Bonds: Leu 287, Trp 284 Pi-pi stacking: Trp 84 Carbon-hydrogen bond: Ser 291, Val 292, Tyr 335 |
| BACE1 | Rosmarinic acid | −7.45 | H Bonds: Tyr 76, Gly 235, Arg 240 |
| BACE1 | Carnosic acid | −5.85 | H Bonds: Asp 37, Asp 233 Alkyl Bond: Val 337, Tyr 203, Tyr 76, Phe 113 Carbon-hydrogen bond: Gly 39 |
| BACE1 | Ursolic acid | −5.48 | H Bonds: Phe 113 Alkyl Bonds: Tyr 76 |
| BACE1 | Donepezil | −8.27 | H Bonds: Tyr 71, Gly 34, Arg 235 Van der waals: Asp 228, Thr 329 |
| Synapsin I | Rosmarinic acid | −8.49 | H Bonds: Asp 120, Lys 403, Gln 399, Ser 275, Asn 338 Pi-anion: Asp 10, Arg 186 Alkyl Bond: His 123 |
| Synapsin I | Carnosic acid | −7.13 | H Bonds: Phe 222 Alkyl Bonds: Val 219 Pi-pi interaction: His 188 |
| Synapsin I | Ursolic acid | −5.69 | H Bonds: His 123, Ser 275 Alkyl Bonds: Ala 193, His 188, Pro 393 |
| Synapsin I | Donepezil | −6.5 | H Bonds: Lys 225 Alkyl Bonds: His 188, Val 388, Pro 393, Lys 336, Leu 375, Ile 385 Pi-Anion: Glu 373, Lys 269, Lys 279 |
| Synapsin II | Rosmarinic acid | −7.02 | H Bonds: Arg 76, Ser 281, Asp 10, His 78, His 164 Carbon-hydrogen bonds: Gln 77, Glu 86 |
| Synapsin II | Carnosic acid | −5.08 | H Bonds: Asp 120 Alkyl Bonds: Ala 124, Leu 394, His 188 Salt bridge: Arg 186 Carbon hydrogen bond: His 123, Pro 393, His 188, His 274 |
| Synapsin II | Ursolic acid | −6.02 | H Bonds: Gln 399 Carbon-hydrogen bond: Leu 394 Alkyl Bonds: Ala 124, His 123, Pro 393 |
| Synapsin II | Donepezil | −6.50 | H Bonds: Arg 76 Alkyl Bonds: Arg 293, His 78, His 164, His 13 Carbon-hydrogen Bond: Asp 10, Leu 284, Asp 228, Ser 165 |
| Synapsin III | Rosmarinic acid | −8.05 | H Bonds: Lys 106, Lys 254, Lys 150, Thr 184, Gly 152, Glu 186 Alkyl Bonds: Lys 160, Ile 266 Carbon-hydrogen bond: Ala 156, Gly 157, Glu 267, |
| Synapsin III | Carnosic acid | −4.59 | H Bonds: Ala 316, Asp 292, Asn 317 Alkyl Bonds: Lys 315, Val 354, Phe 286, Ile 364, Val 246, Ile 287, Lys 258 Salt bridge: Lys 352, Arg 307 Pi-pi interaction: Trp 314 |
| Synapsin III | Ursolic acid | −5.73 | H Bonds: Gly 312, Asn 313 Alkyl Bonds: Ala 316, Phe 286 Carbon-hydrogen Bond: Ser 309 |
| Synapsin III | Donepezil | −7.28 | H Bonds: Lys 106, Thr 184 Alkyl Bonds: Ala 218, Val 256, Lys 150, Phe 110, Tyr 182, Pro 107 Carbon-hydrogen bond: Gly 152, Ser 191 Pi anion: Asp 194, Lys 160 |
| AChE | ΔGcomplex | ΔGreceptor | ΔGligand | ΔGbind |
|---|---|---|---|---|
| Rosmarinic acid | −46,014.79 | −45,849.86 | −124.2334 | −40.6927 |
| Carnosic acid | −9927.076 | −12,735.73 | 2850.116 | −41.4654 |
| Ursolic acid | −12,566.42 | −12,652.49 | 129.8068 | −43.7367 |
| Donepezil | −12,641.44 | −12,680.03 | 81.1693 | −42.5805 |
| BACE | ||||
| Rosmarinic acid | −34,885.23 | −34,728.67 | −132.9516 | −23.6095 |
| Carnosic acid | −5294.862 | −5588.338 | 2835.457 | −2541.98 |
| Ursolic acid | −8016.414 | −5540.046 | 134.2603 | −2610.629 |
| Donepezil | −8008.447 | −5585.226 | 115.4342 | −2538.656 |
| Synapsin-I | ||||
| Rosmarinic acid | −28,807.57 | −28,635.99 | −121.7649 | −49.8122 |
| Carnosic acid | −3588.86 | −6284.642 | 2750.101 | −54.3182 |
| Ursolic acid | −6185.364 | −6295.104 | 132.3759 | −22.6355 |
| Donepezil | −28,678.94 | −28,607.5 | −48.6825 | −22.7567 |
| Synapsin-II | ||||
| Rosmarinic acid | −29,054.36 | −28,900.69 | −118.5544 | −35.1146 |
| Carnosic acid | −29,031.46 | −28,950.41 | −57.2692 | −23.7774 |
| Ursolic acid | −6071.435 | −6187.76 | 134.8061 | −18.4811 |
| Donepezil | −6133.151 | −6183.642 | 68.3527 | −17.8623 |
| Synapsin-III | ||||
| Rosmarinic acid | −27,429.88 | −27,273.57 | −123.8107 | −32.4908 |
| Carnosic acid | −2686.096 | −5320.382 | 2641.009 | −6.723 |
| Ursolic acid | −5793.038 | −5918.352 | 130.956 | −5.6419 |
| Donepezil | −5851.622 | −5905.256 | 82.103 | −28.4686 |
| Carnosic Acid | Rosmarinic Acid | Ursolic Acid | |
|---|---|---|---|
| Lipinski Rule of Five | |||
| Molecular Mass | 332.44 | 360.32 | 456.71 |
| Hydrogen Bond Donors | 1 | 5 | 2 |
| Hydrogen Bond Acceptors | 4 | 8 | 3 |
| Log P | 4.18 | 1.53 | 7.005 |
| Molar Refractivity | 98.60 | 81.95 | 152.11 |
| ADMET analysis | |||
| Human intestinal absorption | + | + | + |
| Caco2 permeability | + | - | + |
| Subcellular localization | Mitochondria | Mitochondria | Mitochondria |
| OATP1B1 & OATP1B3 inhibitor | + | + | + |
| CYP inhibitory promiscuity | - | - | - |
| Carcinogenicity | - | - | - |
| Ames mutagenesis | - | - | - |
| Estrogen receptor binding | + | + | + |
| Androgen receptor binding | - | + | + |
| Thyroid receptor binding | + | + | + |
| Glucocorticoid receptor binding | + | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirza, F.J.; Zahid, S.; Amber, S.; Sumera; Jabeen, H.; Asim, N.; Ali Shah, S.A. Multitargeted Molecular Docking and Dynamic Simulation Studies of Bioactive Compounds from Rosmarinus officinalis against Alzheimer’s Disease. Molecules 2022, 27, 7241. https://doi.org/10.3390/molecules27217241
Mirza FJ, Zahid S, Amber S, Sumera, Jabeen H, Asim N, Ali Shah SA. Multitargeted Molecular Docking and Dynamic Simulation Studies of Bioactive Compounds from Rosmarinus officinalis against Alzheimer’s Disease. Molecules. 2022; 27(21):7241. https://doi.org/10.3390/molecules27217241
Chicago/Turabian StyleMirza, Fatima Javed, Saadia Zahid, Sanila Amber, Sumera, Hira Jabeen, Noreen Asim, and Syed Adnan Ali Shah. 2022. "Multitargeted Molecular Docking and Dynamic Simulation Studies of Bioactive Compounds from Rosmarinus officinalis against Alzheimer’s Disease" Molecules 27, no. 21: 7241. https://doi.org/10.3390/molecules27217241
APA StyleMirza, F. J., Zahid, S., Amber, S., Sumera, Jabeen, H., Asim, N., & Ali Shah, S. A. (2022). Multitargeted Molecular Docking and Dynamic Simulation Studies of Bioactive Compounds from Rosmarinus officinalis against Alzheimer’s Disease. Molecules, 27(21), 7241. https://doi.org/10.3390/molecules27217241