1. Introduction
With the gradual depletion of oil resources, the need to find new carbon sources becomes urgent and important [
1]. In addition, environmental issues such as climate warming and white pollution caused by the large-scale use of petroleum-based derivatives have made it challenging to continue to use traditional petroleum-based polymers [
2,
3]. Polylactic acid (PLA) is a biomass material whose raw material, lactic acid, can be fermented from natural biological materials such as corn and straw. And it can be degraded into CO
2 and H
2O in nature, so it is one of the most ideal biodegradable materials [
4]. Moreover, due to its favorable mechanical properties and processability, PLA is considered to be the most promising alternative to traditional petroleum-based polymers. PLA also has good spinnability, and can be made into monofilament or multifilament with round cross-sections, special-shaped fiber, fine denier fiber, crimped or non-crimped fiber, bicomponent fiber, spun-bonded nonwoven fabric, melt-blown nonwoven fabric, etc. Therefore, PLA fiber is expected to be used in apparel, household products and decoration, non-woven fabric, and in the hygiene and medical fields, etc. However, the textile dyeing and printing process needs to be carried out in a high-temperature and high-humidity environment. PLA products have a low softening point (60–70 °C) [
5], therefore easily deform under heating conditions. Moreover, PLA is prone to hydrolysis in a high-temperature and high-humidity environment, which greatly reduces the performance of PLA fiber products.
Since Ikada et al. [
6] first reported stereo-complexation (SC) formed by Poly(L-lactic acid)/poly(D-lactic acid) (PLLA/PDLA) blends in melt or solution, researchers have shown great interest in the crystallization behavior, crystal structure, and properties of SC [
7,
8,
9]. Compared with ordinary homocrystallization (HC) composed of enantiomerically neat PLLA or PDLA 10
3-helix chains, the enantiomers PLLA and PDLA chains can be arranged tightly side by side in a 3
1-helix conformation, resulting from the driving effect of the intermolecular hydrogen bond interaction [
10,
11]. Due to dense chain stacking and stronger intermolecular interactions, SC crystallization exhibited better physical properties, such as mechanical strength, hydrolytic stability, thermal stability, and heat resistance [
8,
9,
12,
13,
14]. In particular, the melting point (T
m) of SC crystallization is as high as 220–230 °C (about 50 °C higher than the T
m of HC), and the Vicat-softening temperature of PLA with almost complete SC structure can reach 200 °C [
5,
15]. Pan et al. [
16] proved that the fiber-softening temperature of pure PLA fiber was 72 °C, while that of PLA fiber with complete SC structure was up to 133 °C.
However, it proved difficult to extend to the existing industrial-scale production for the commonly used stereo-complex methods. Stereo-complex processes related to dissolution usually have high production costs and low efficiency, and require complex equipment to collect solvents, which may cause environmental problems due to solvent disposal [
17]. To achieve complete SC crystallization in PLLA/PDLA blends, additives were often used to promote nucleation (that is, the formation of spiral PLLA/PDLA pairs) and crystal growth, such as nucleating agents [
18,
19,
20], plasticizers [
21], and compatibilizer [
22,
23,
24]. However, the addition of these additives will bring processing and economic difficulties. The strong shear force induced by electrospinning at high voltage (10–20 kV) was also found to be conducive to the formation of SC crystallization [
25,
26,
27]. In addition, low-temperature sintering [
28,
29], oscillation shear injection molding (OSIM) [
5], the introduction of supercritical fluid [
17], and other methods have been used to prepare PLLA/PDLA blends with complete SC. However, the above-mentioned stereo-complex processes were complicated, uneconomical, and had low short-term industrialization potential. Since temperature and flow field are the key factors for SC formation, some researchers have prepared PLA fiber with a complete SC structure by melt-spinning on a laboratory scale [
16,
30,
31,
32]. The large-scale production of PLA products with complete SC structure from commercial high-molecular-weight PLA has recently become an issue worthy of attention.
In this work, commercially available high-molecular-weight PLLA and PDLA were used as raw materials and melt-spun into PLLA/PDLA fiber using a polyester fiber production line. The content of SC crystallites of PLLA/PDLA fiber was adjusted by high-temperature tension heat-setting. Thereby, the scalable preparation of complete stereo-complexation PLA fiber with favorable heat and hydrolysis resistances was achieved through a simple process.
3. Materials and Methods
3.1. Materials
Commercially available PLLA (trade mark Luminy® L130, with Mw = 17.3 × 104 g/mol, PDI = 1.95, and optical purity ≥ 99%) was supplied by Total Corbion (Amsterdam, The Netherlands). PDLA with Mw = 4.5 × 104 g/mol and PDI = 1.96 was purchased from Heilongjiang Xinda Enterprise Group Co., Ltd. (Harbin, China). The content of D-lactide in the PDLA is approximately 99%.
3.2. Sample Preparation and Stereo-Complexation
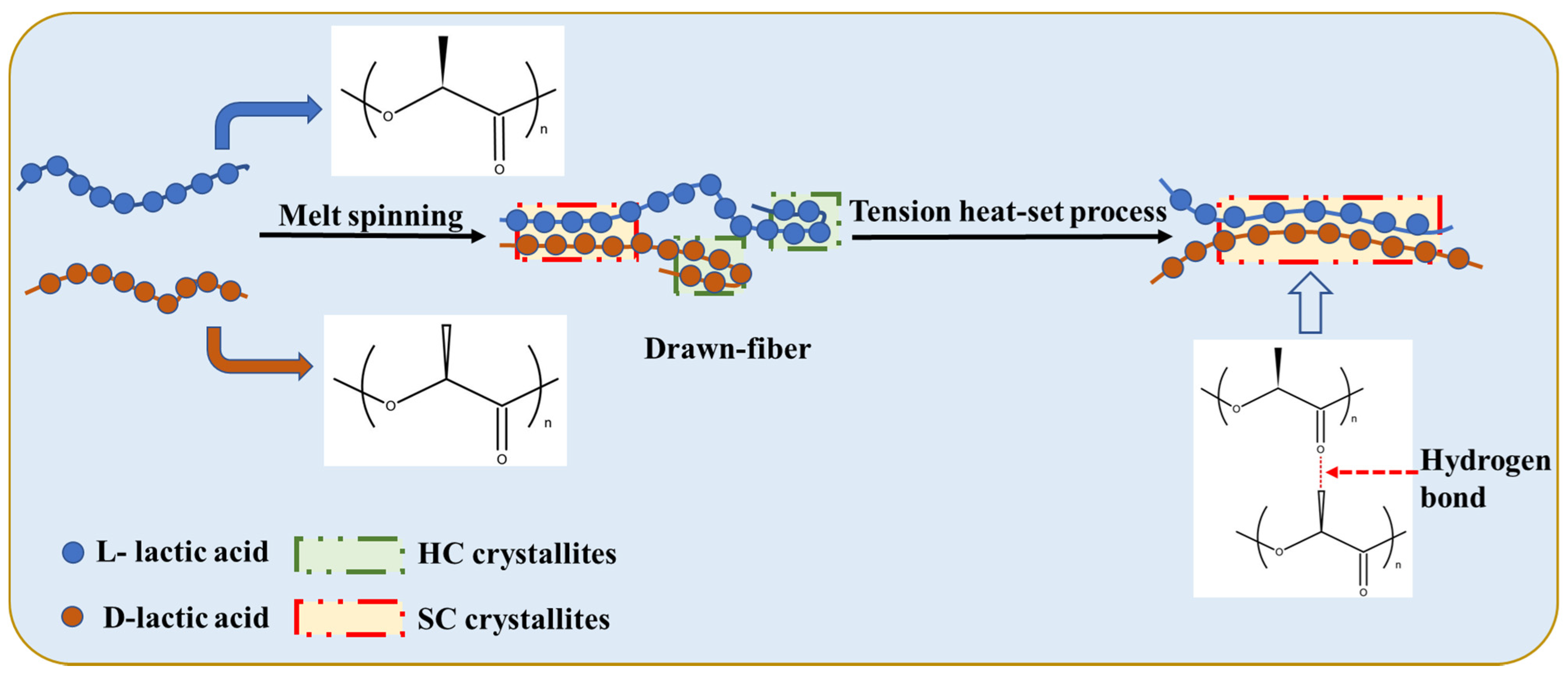
The production process, including pre-spinning and post-spinning, was performed on a polyester fiber production line. The pre-spinning process included drying slices, blended melt-spinning and winding shaping. PLLA and PDLA pellets (50:50, wt%) were dried in a vacuum drum. The drying process was as follows: feeding at drum temperature < 60 °C, cooling for 0.5 h after feeding, and then drying at 60 °C for 1 h, 70 °C for 4 h, 80 °C for 3 h, and 120 °C for 6.5 h. The total drying period was 15 h, and the vacuum was ≤−0.098 MPa. The pre-spinning temperatures are listed in
Table 8, and the spinning speed was 1020 m/min; metering pump frequency was 29.77 Hz. PLLA/PDLA as-spun fiber was obtained after pre-spinning.
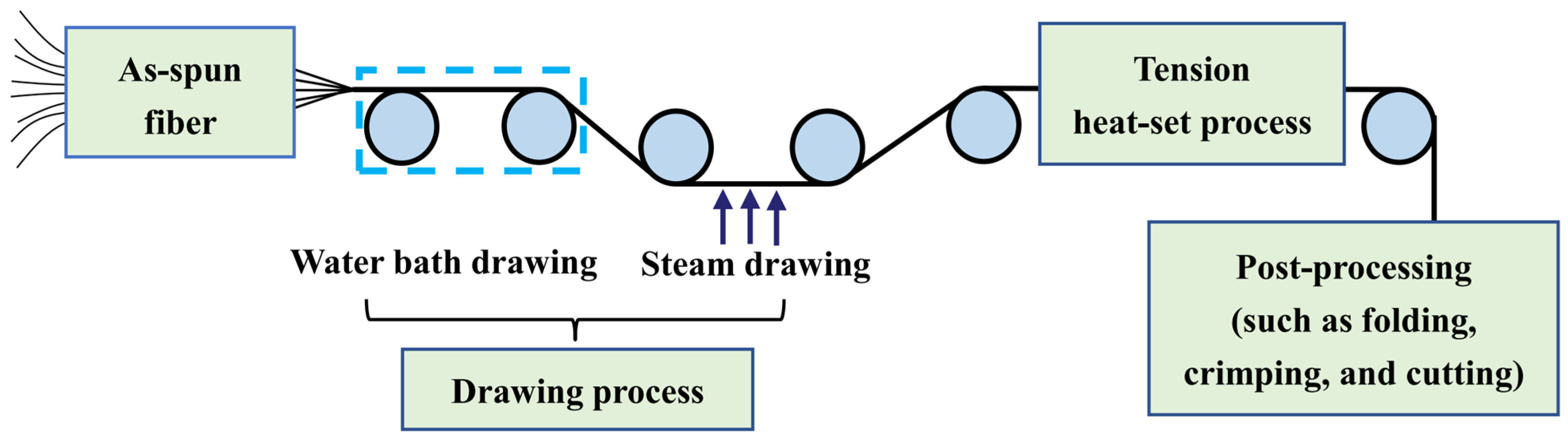
The post-spinning process of the PLLA/PDLA fiber is shown in
Figure 10. The drawing process consisted of two steps: water bath drawing at 62 ± 2 °C and steam drawing. The total drawing ratio was up to 3 times. To obtain PLLA/PDLA fiber with different stereo-complex rate, a high-temperature tension heat-setting process at 160 °C, 170 °C, 180 °C, and 190 °C was adopted respectively.
The PLLA/PDLA fiber after the drawing process was named drawn−fiber. The drawn−fiber after high-temperature tension heat-setting treatment was named SC−PLA fiber, and the fiber corresponding to 160 °C, 170 °C, 180 °C, and 190 °C tension heat-setting treatment was named SC−160, SC−170, SC−180, and SC−190, respectively.
3.3. Accelerated Hydrolysis Tests
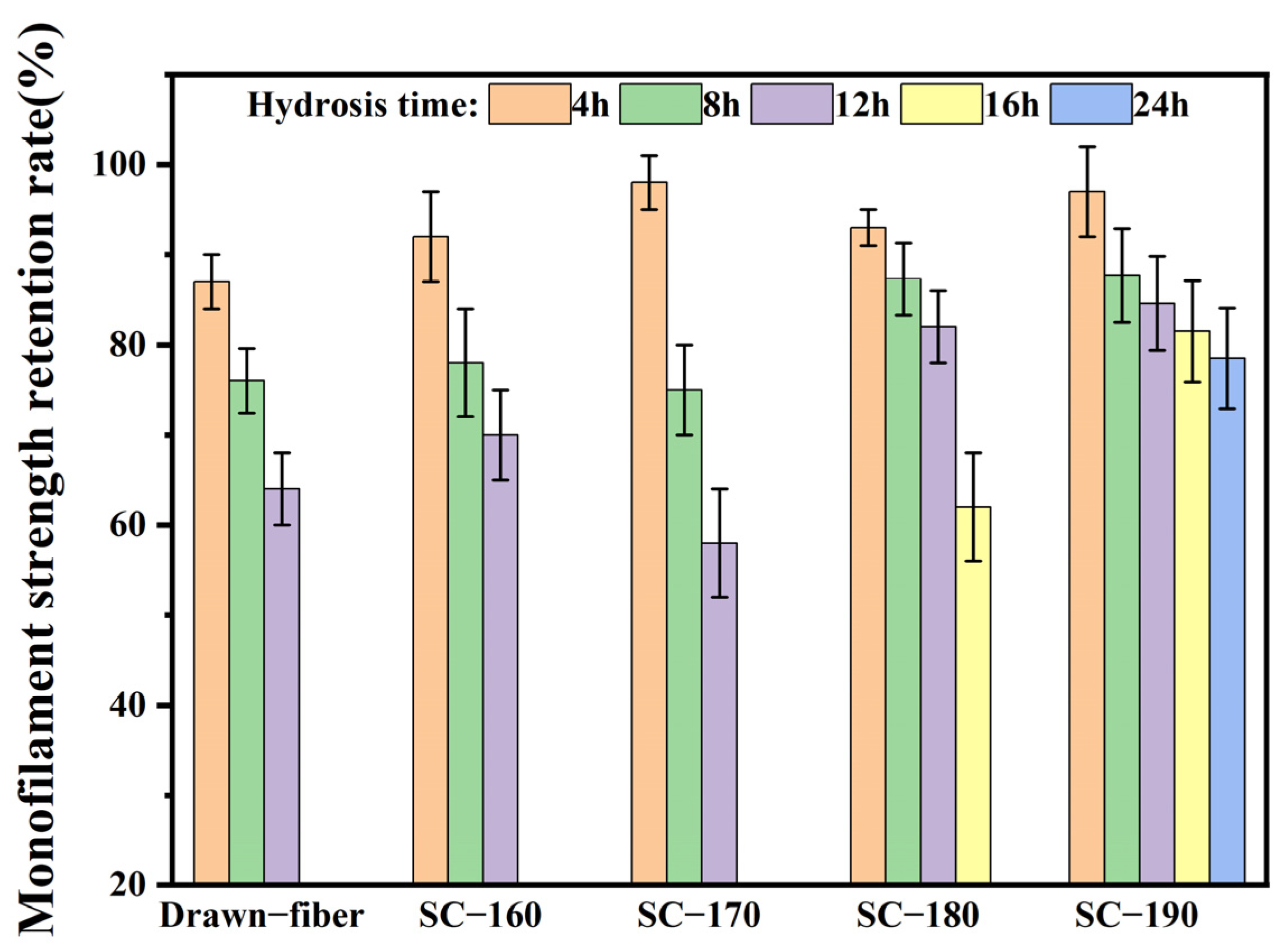
Since textile dyeing and printing processes are often performed in a high-temperature and high-humidity environment, accelerated hydrolysis at 95 °C was used to characterize the hydrolysis resistance of drawn−fiber and SC−PLA fiber. The fiber samples including drawn−fiber, SC−160, SC−170, SC−180, and SC−190 were put into a 95 °C constant-temperature water bath for different times (4 h, 8 h, 12 h, 16 h, 24 h, or 32 h), and then rinsed with distilled water. The residual water in the hydrolyzed fiber samples was absorbed by long-fiber paper towels, and then the samples were dried at room temperature and transferred to a desiccator. Before the characterization, all fiber samples were placed in a standard test environment (65% relative humidity, 21 °C) for 24 h.
3.4. Accelerated Hydrolysis Tests
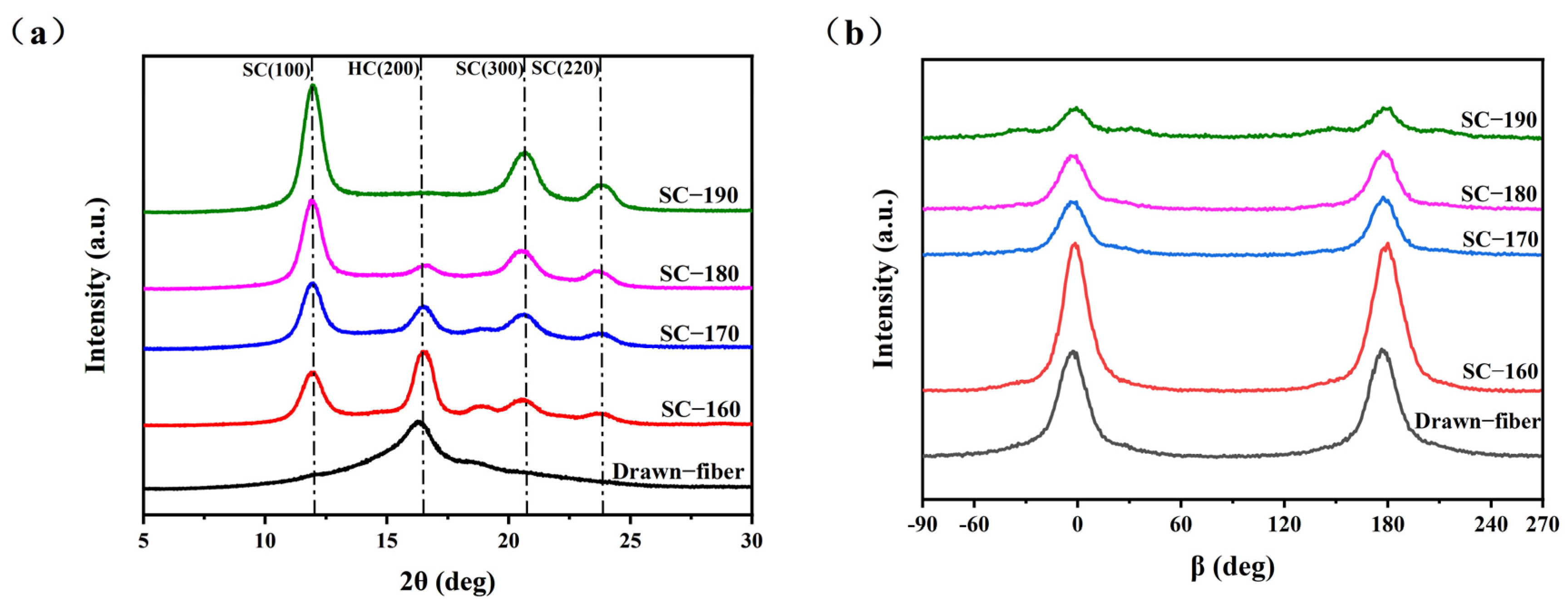
3.4.1. Wide-angle X-ray Diffraction (WAXD)
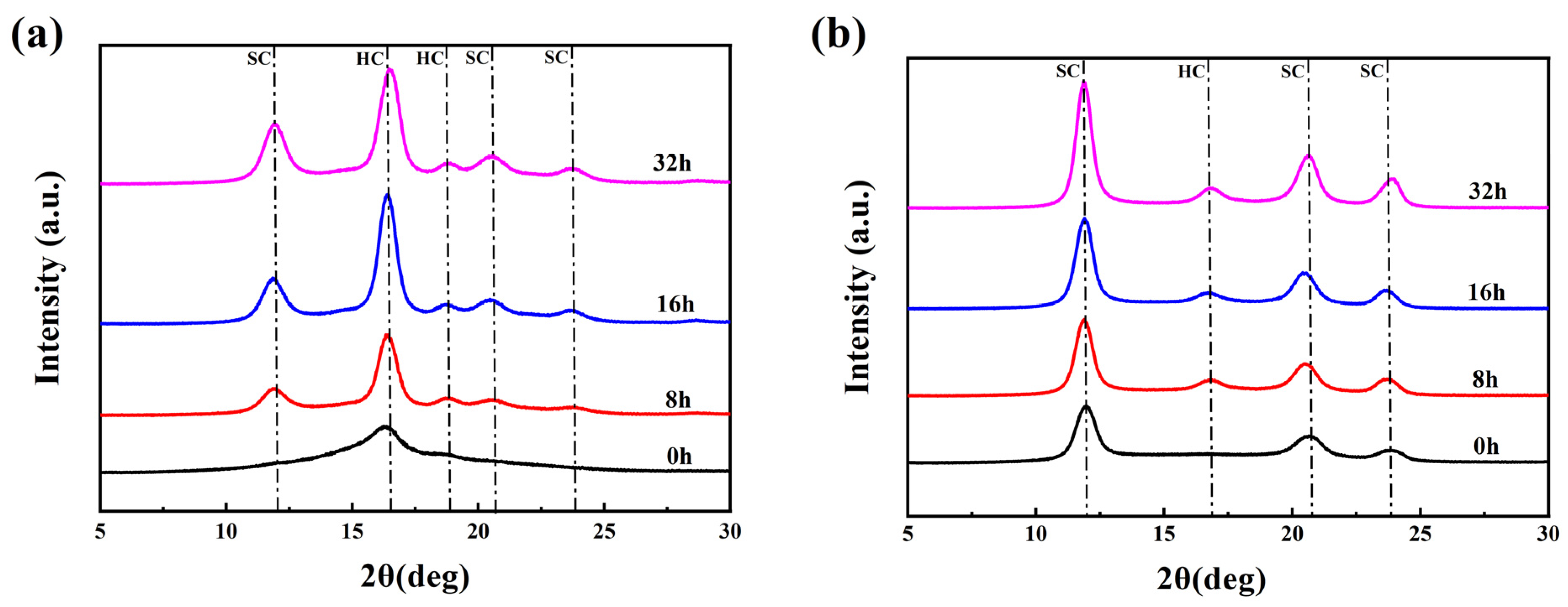
The crystal structure was characterized using an X-ray diffractometer (Bruker D8 Advance, Bremen, Germany) equipped with a Ni-filtered Cu Kα radiation source (λ = 0.154 nm). The measurements were carried out at 40 kV and 150 mA with scan angles from 5° to 60° at a scan rate of 2°/min.
The obtained patterns were split and then the crystallinity and other related parameters of the fiber samples were calculated. The percentages of the diffraction peak areas of HC crystallites and SC crystallites to the total area of the diffraction pattern were used to represent the HC crystallinity (
, the absolute content of HC crystallites) and the SC crystallites crystallinity (
, the absolute content of SC crystallites) respectively.
represents the total crystallinity of the fiber samples.
represents the stereo-complex rate, which is the percentage of SC crystallites in the total crystals. The specific calculation formulas were as follows:
where S
sc, S
hc, and S
a are the integral area of the SC crystallites, HC and the amorphous region, respectively.
The grain size was calculated via the Scherer formula:
where D is the grain size (Å); K is the Scherrer constant, about 0.89; B is the half-height width of the diffraction peak of the measured sample; θ is the diffraction angle (°); and γ is the X-ray wavelength, about 0.154 nm.
The orientation structure of the crystal region was tested using a D/max-2550VB/PC (RigaKu, Tokyo, Japan). The measurements were carried out at 40 kV and 150 mA with scan angles from −90° to 270° and a scan rate of 5°/min. The orientation factor (
) of the crystal region was calculated by the following formula.
where
is the ith half-width of the peak in the azimuthal scan from WAXD determination.
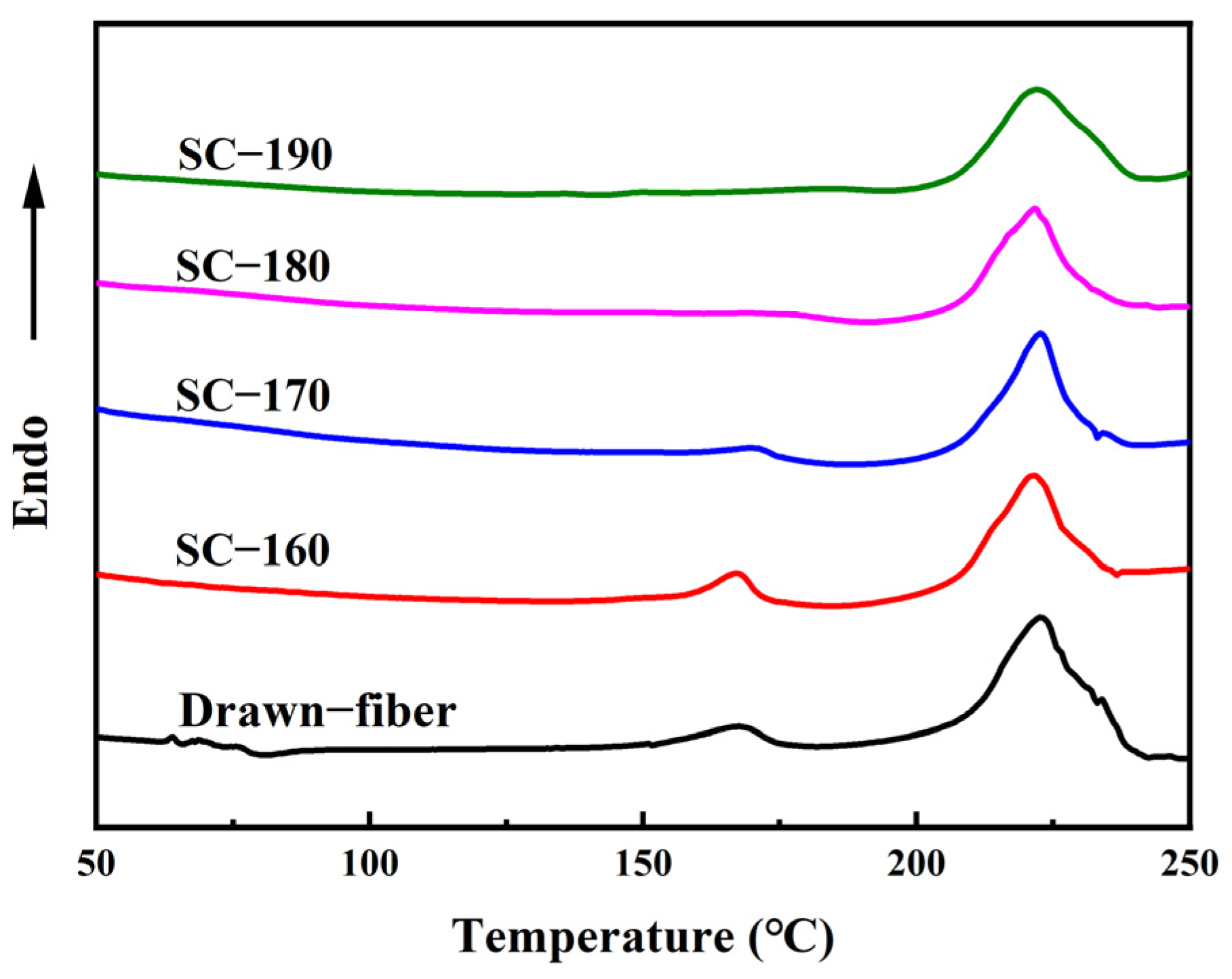
3.4.2. Differential Scanning Calorimetry (DSC)
DSC measurements of fiber samples were performed on a DSC8500 (PerkinElmer Inc., Waltham, MA, USA) under a nitrogen atmosphere at a flow rate of 50 mL/min. The temperature range was between 50 °C and 250 °C and the heating rate was set at 10 °C/min.
The ratio of the sum of melting enthalpy of HC crystallites and SC crystallites to the melting enthalpy of complete crystallization was used to express the degree of crystallinity (
). The melting enthalpy of SC crystallites as a percentage of the melting enthalpy of HC crystallites and SC crystallites is expressed as the content of SC crystallites (
). Since the enthalpy of complete crystallization of HC crystallites and SC crystallites are different, following formulas were established to calculate
and
.
where ΔH
m−H and ΔH
m−S are the melting enthalpy of HC crystallites and SC crystallites, respectively, ΔH
om−H is the melting enthalpy of complete crystallization of HC, which is 93.7 J/g, and ΔH
om−S is the melting enthalpy of complete crystallization of SC crystallites, which is 142.0 J/g [
12].
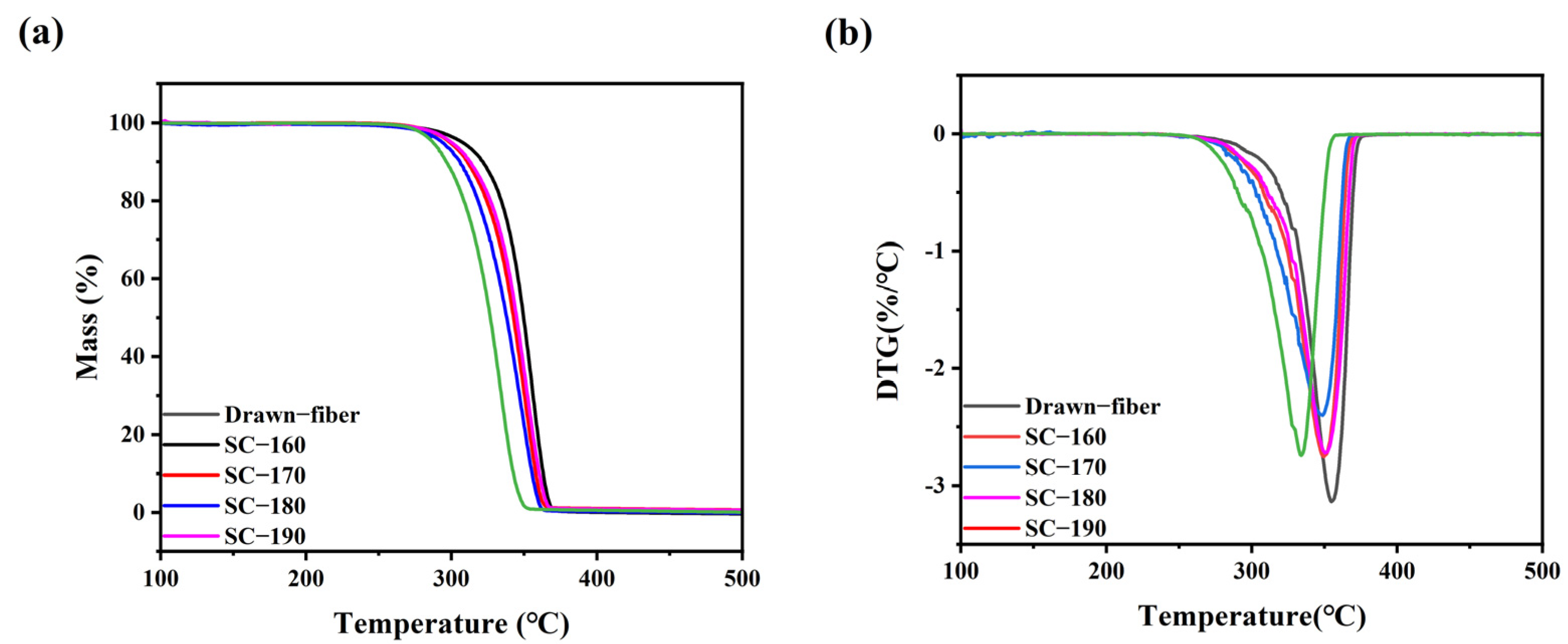
3.4.3. Thermogravimetric Analysis (TGA)
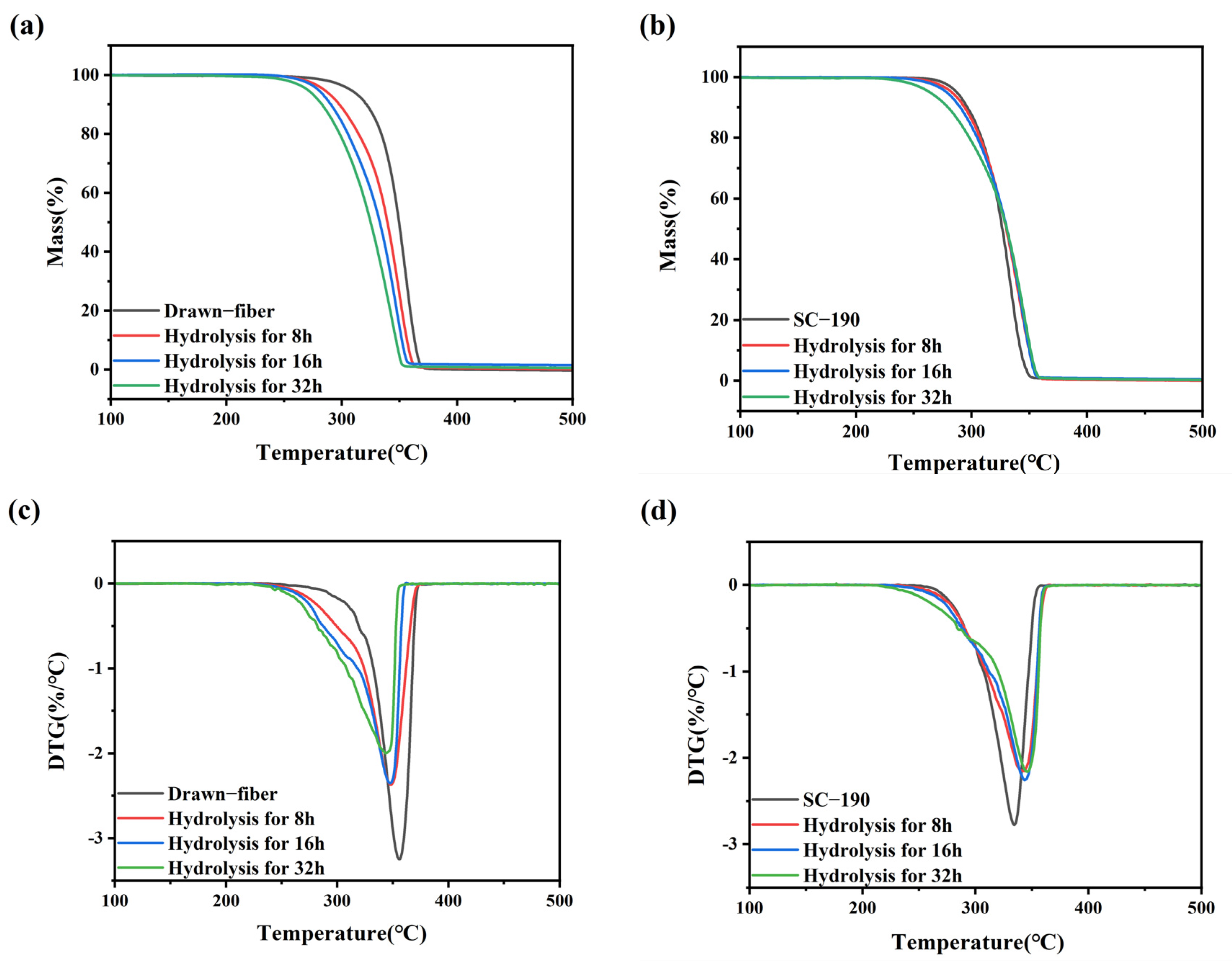
TGA (TG 209 F1, NETZSCH, Selb, Germany) was used for thermal stability tests. 5–8 mg of each sample was heated from 50 °C to 600 °C with a 10 °C/min heating rate under a nitrogen atmosphere.
3.4.4. Mechanical Performance Test
A fiber tensile tester (XQ-1C, Shanghai New Fiber Instrument Co., Ltd., Shanghai, China) was employed to test the monofilament strength of the fiber. Before the test, a 200 cN weight was used for calibration. The test conditions were as follows: pre-tension was 0.25 cN, clamping distance was 10 mm, and extension rate was 10 mm/min. Each group of fiber samples was measured 30 times.
3.4.5. Gel Permeation Chromatography (GPC)
GPC data of the samples, including the weight-average (Mw), number-average (Mn) molecular weights, and molecular weight distribution (PDI, Mw/Mn), were obtained by gel permeation chromatography (PL-GPC50, Polymer Laboratories, Church Stretton, UK). Hexafluoroisopropanol was used as the mobile phase, the flow rate was 0.3 mL/min, and polystyrene was used as the standard.
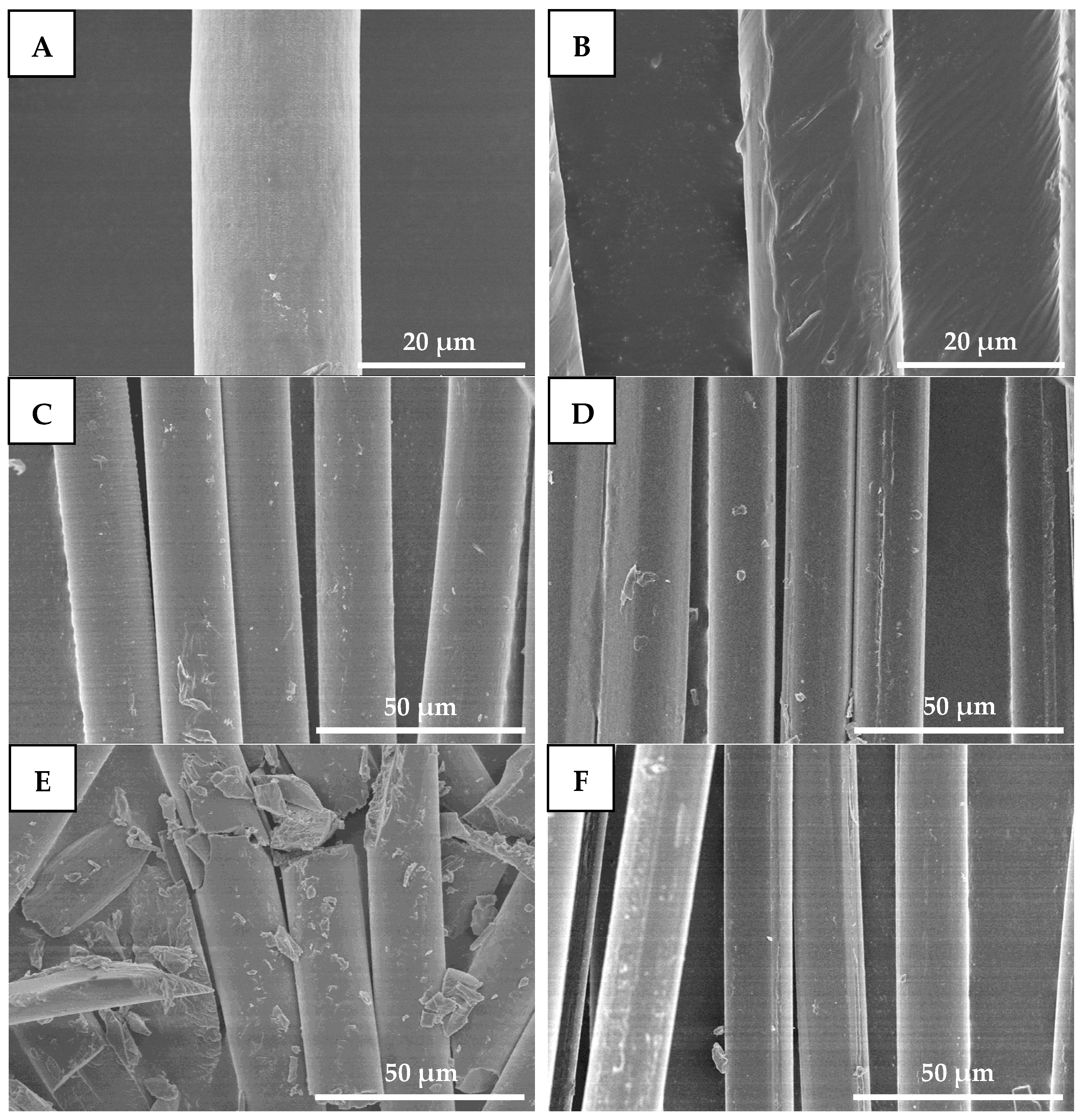
3.4.6. Morphological Observation
The morphology of the fiber was observed by a scanning electron microscope (SEM) (Quanta 250, FEI Company, Brno, Czech Republic) at 5 kV. And coated with a thin layer of gold before examination.
4. Conclusions
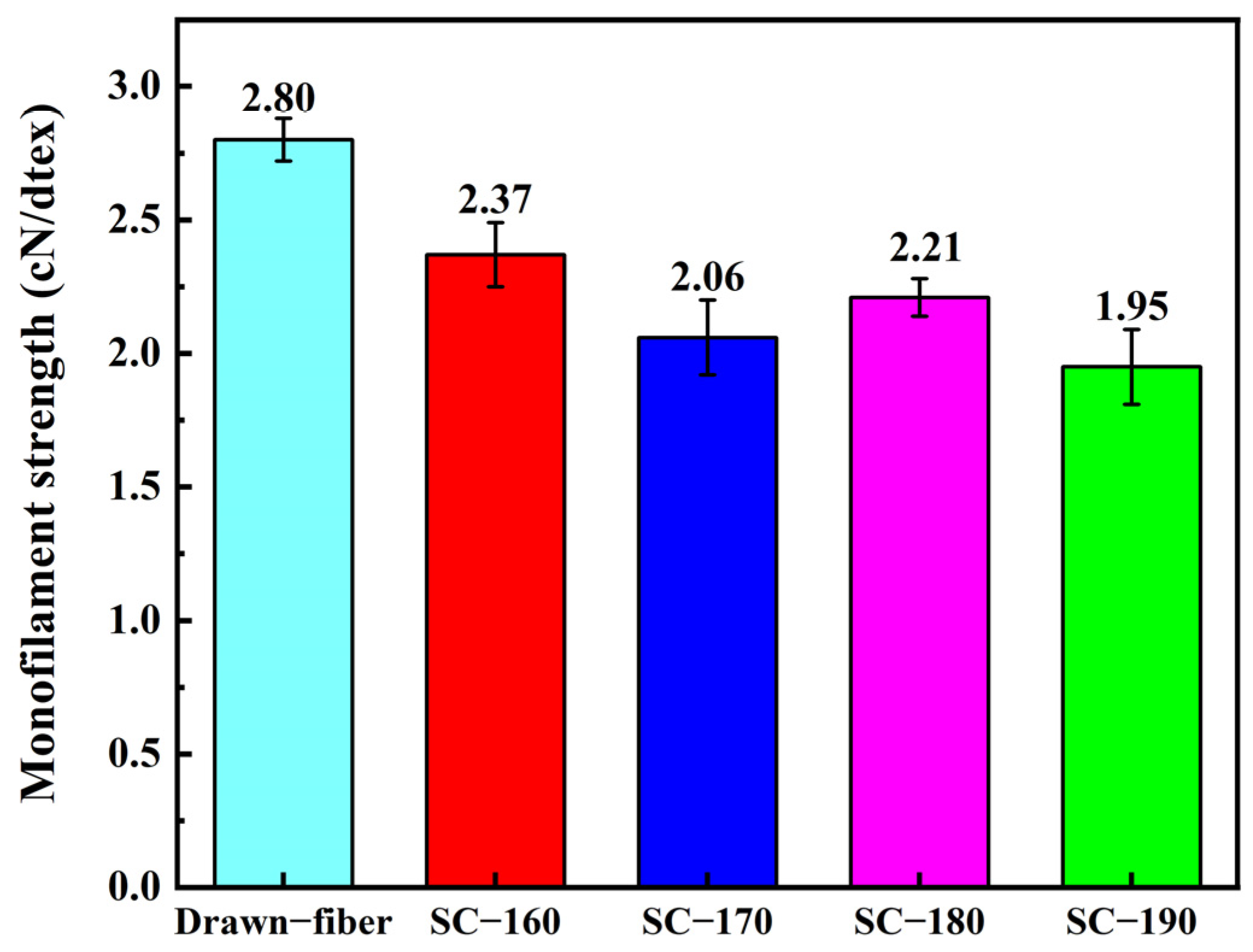
In this work, PLLA/PDLA fiber with different stereo-complex rates were prepared using a polyester fiber production line. The effects of tension heat-setting temperature on the structure and properties of PLLA/PDLA fiber were systematically investigated. As the tension heat-setting temperature increased, the content of HC crystallites gradually decreased and even disappeared, while the content of SC crystallites increased. When the tension heat-setting temperature reached 190 °C, the fiber achieved almost complete SC structure, which had a melting point of 222.5 °C. However, the monofilament strength of this fiber was relatively low, only 1.95 cN/dtex. The hydrolysis resistance of PLLA/PDLA fiber samples was studied using an accelerated hydrolysis experiment at 95 °C in a water bath. The drawn−fiber was found to exhibit a significant increase in crystallinity and the proportion of SC crystallites with the extension of the hydrolysis time. After 24 h of accelerated hydrolysis, the monofilament strength retention rate of SC−190 fiber was 78.5%, indicating better hydrolysis resistance, while the monofilament strength of other PLLA/PDLA fiber was lost to the point that it could not be tested. This study highlights the use of a polyester fiber production line to produce complete stereo-complexation PLA fiber by blended melt-spinning of PLLA and PDLA in equal proportion. Herein, the tension heat-setting at an appropriate temperature is believed to be the key to the complete stereo-complexation in the PLA fiber with favorable heat resistance and hydrolysis resistance. However, it is necessary to balance the relationship between heat resistance, hydrolysis resistance, and mechanical properties to find suitable application areas (such as filling fiber fields).


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}