
Synthesis and Biological Evaluation of Novel Pyrimidine Amine Derivatives Bearing Bicyclic Monoterpene Moieties


Abstract
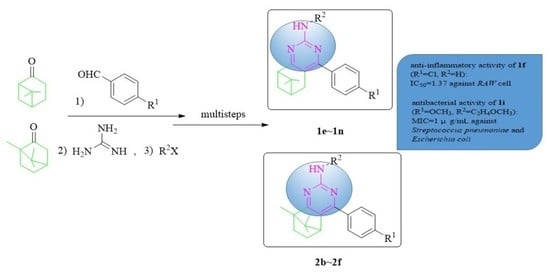
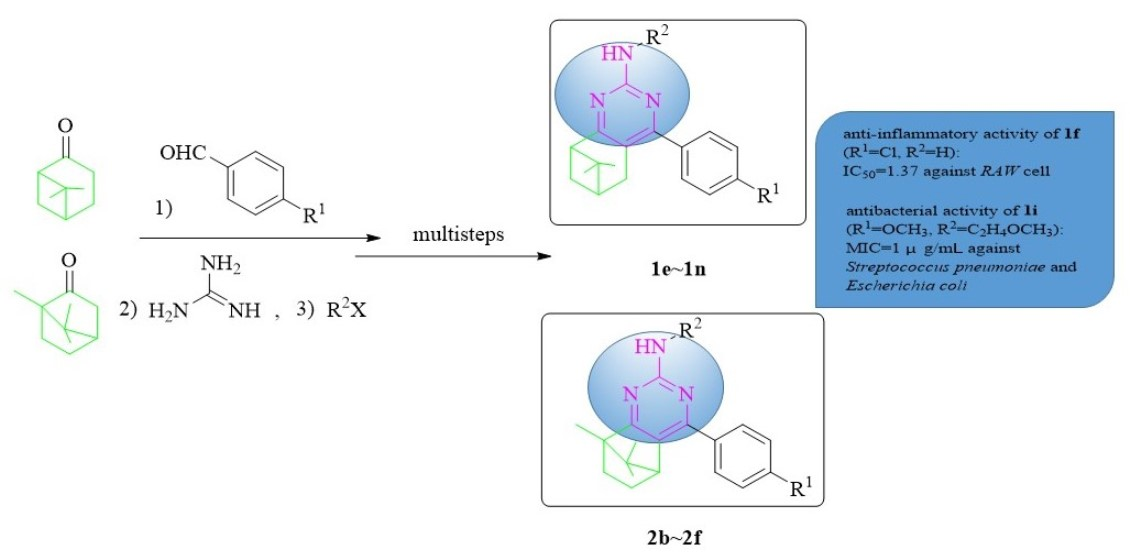
:
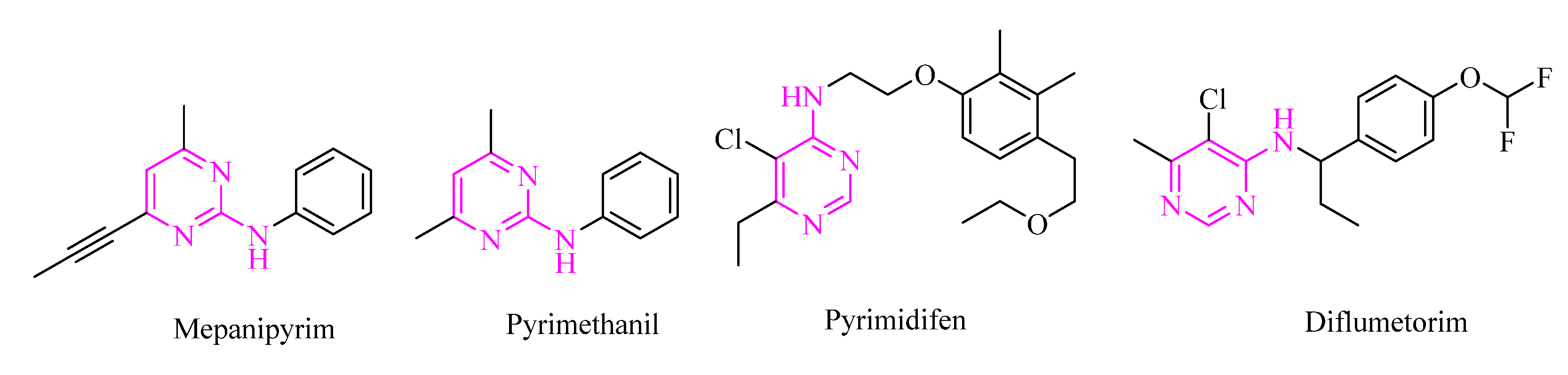
1. Introduction
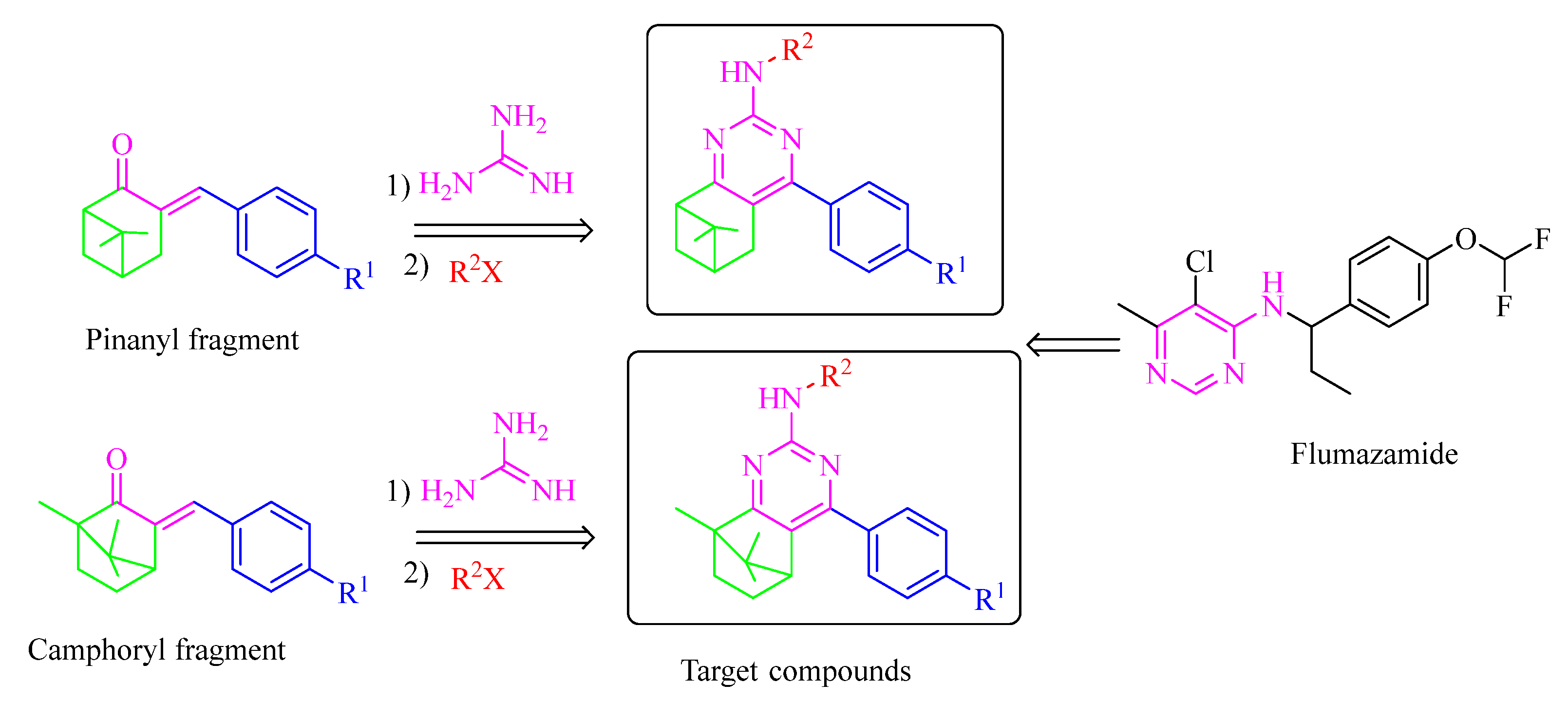
2. Results and Discussion
2.1. Chemistry
2.2. Structural Characterization of Compounds
2.3. Biological Evaluation
2.3.1. Antibacterial Activity Assay
2.3.2. Anti-Inflammatory Activity Assay
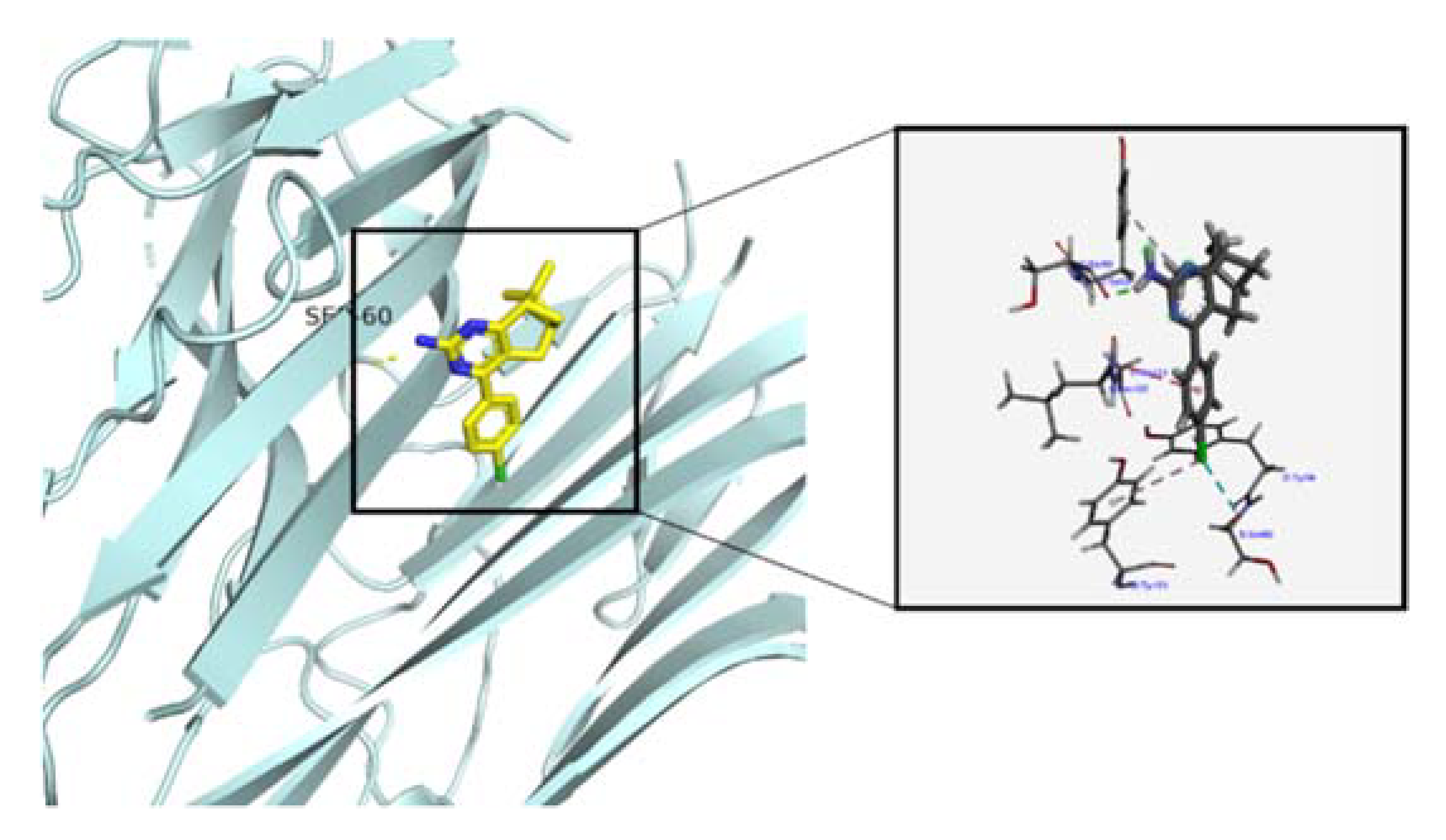
2.3.3. Molecular Docking Study
3. Conclusions
4. Experimental Section
4.1. Chemistry
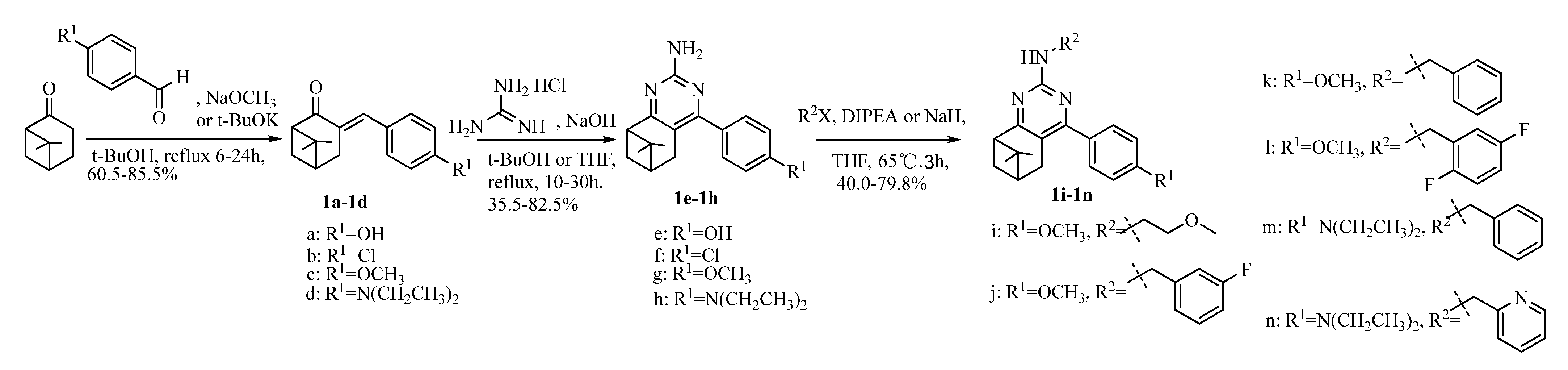
4.1.1. Synthesis of Intermediate 3-Arylbenzylidene Nopinones 1a~1d
4.1.2. Synthesis of Pinanealkylpyrimidineamines 1e~1n
4.1.3. Synthesis of Pinanylpyrimidinamines 1i~1n
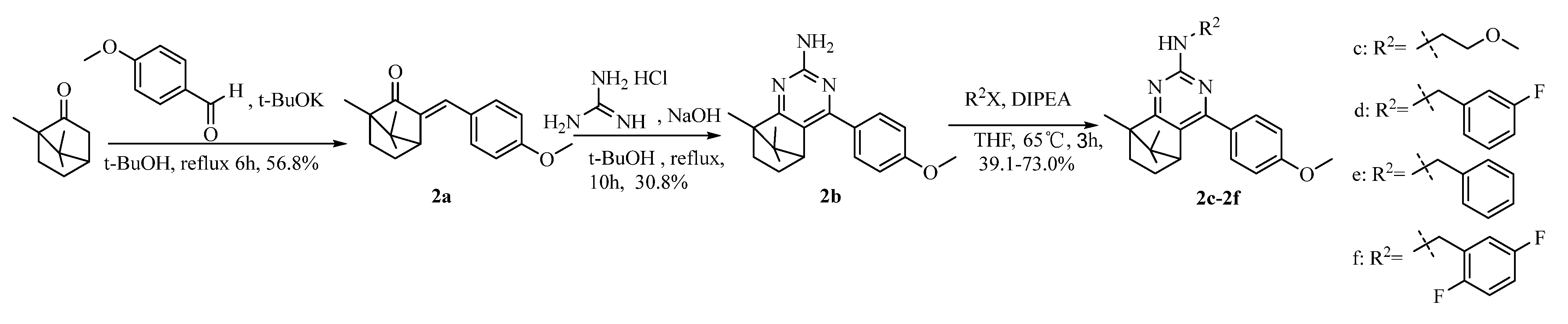
4.1.4. Synthesis of Intermediate 3-(4-Methoxybenzylidene)-1, 7, 7-Trimethylbicyclo [2.2.1] Heptan-2-One (2a)
4.1.5. Synthesis of Camphoryl Pyrimidine Amines 2b~2f
4.2. Antibacterial Activities Assay
4.3. Anti-Inflammatory Activities Assay
4.4. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giovanny, C.S.; Lucas, M.M.; Bruna, A.B.; Vitor, F.M.; Luiz, C.S.; Bruno, H.S. Heterocyclic compounds as antiviral drugs: Synthesis, structure–activity relationship and traditional applications. J. Heterocyclic. Chem. 2021, 58, 2226–2260. [Google Scholar]
- Liu, X.H.; Wang, Q.; Sun, Z.H.; David, E.W.; James, J.B.; Alden, S.E.; Tan, C.X.; Weng, J.Q. Synthesis and insecticidal activity of novel pyrimidine derivatives containing urea pharmacophore against Aedes aegypti. Pest Manag. Sci. 2017, 73, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Tenma, N.; Dind, B.P.; Kogaku, K.; Yukiko, Y.; Tomoya, U.; Takaya, M.; Ryosuke, K.; Shihori, H.; Yuki, E.; Shoya, Y.; et al. Total synthesis of cynaropicrin. Org. Biomol. Chem. 2021, 19, 6038–6044. [Google Scholar]
- Santosh, S.U.; Navanath, J.V.; Ajinkya, A.P.; Dattatraya, K.J.; Sunil, S.V.; Laxman, S.W.; Govind, B.K.; Deshmukh, M.B.; Prashant, V.A. Synthesis, anti-inflammatory, ulcerogenic and cyclooxygenase activities of indenopyrimidine derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 814–818. [Google Scholar]
- Scarim, C.B.; Pavan, F.R. Recent advancement in drug development of nitro(no2)-heterocyclic compounds as lead scaffolds for the treatment of mycobacterium tuberculosis. Drug Develop. Res. 2022, 83, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Türe, A.; Ergül, M.; Ergül, M.; Altun, A.; Küçükgüzel, I. Design, synthesis, and anticancer activity of novel 4-thiazolidinone-phenylaminopyrimidine hybrids. Mol. Divers. 2021, 25, 1025–1050. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.J.; Yang, Q. Development of a practical synthesis of a pyrimidine derivative with fungicidal activity. Org. Process. Res. Dev. 2019, 23, 2157–2165. [Google Scholar] [CrossRef]
- Sharga, B.M.; Krivovjaz, A.O.; Slivka, M.V.; Lambruch, L.M.; Cheypesh, A.V.; Lendel, V.G.; Nikolaychuk, V.I.; Markovich, V.P. Synthesis and antimicrobial activity of phenylselenyl tribromide and its fused thienopyrimidine derivatives. Farmacia 2016, 64, 512–520. [Google Scholar]
- Weig, A.W.; Barlock, S.L.; O’Connor, P.M.; Marciano, O.M.; Smith, R.; Ernst, R.K.; Melander, R.J.; Melander, C. A scaffold hopping strategy to generate new aryl-2-amino pyrimidine MRSA biofilm inhibitors. RSC Med. Chem. 2021, 12, 293–296. [Google Scholar] [CrossRef] [PubMed]
- El-Dean, A.M.K.; Abd-Ella, A.A.; Hassanien, R.; El-Sayed, M.E.A.; Zaki, R.M.; Abdel-Raheem, S.A.A. Chemical design and toxicity evaluation of new pyrimidothienotetrahydroisoquinolines as potential in-secticidal agent. Toxicol. Rep. 2019, 6, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.Z.; Liu, A.P.; Huang, M.Z.; Liu, M.H.; Pei, H.; Huang, L.; Yi, H.B.; Liu, W.D.; Hu, A.X. Design, synthesis, DFT study and antifungal activity of the derivatives of pyrazolecarboxamide containing thiazole or oxazole ring. Eur. J. Med. Chem. 2018, 149, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Hassaneen, H.M.; Saleh, F.M.; Abdallah, T.A.; Mohamed, Y.S.; Awad, E.M. Synthesis, reactions, and antimicrobial activity of some novel pyrazolo[3,4-d]pyrimidine, pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine, and pyrazolo[4,3-e][1,2,4]triazolo[3,4-c]pyrimidine derivatives. J. Heterocyclic. Chem. 2020, 57, 892–912. [Google Scholar] [CrossRef]
- Madhvi Utreja, D.; Sharma, S. Barbiturates: A review of synthesis and antimicrobial research progress. Curr. Org Synth. 2022, 19, 31–55. [Google Scholar] [CrossRef]
- Lewis, P.O.; PharmD, B. Risk factor evaluation for methicillin-resistant staphylococcus aureus and pseudomonas aeruginosain community-acquired pneumonia. Ann. Pharmacother. 2021, 55, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Tressler, A.B.; Markwei, M.; Fortin, C.; Yao, M.; Procop, G.E.; Soper, D.E.; Goje, O. Risks for recurrent vulvovaginal candidiasis caused by non-albicans Candida versus Candida Albicans. J. Womens Health 2021, 30, 1588–1596. [Google Scholar] [CrossRef]
- Riina, M.; Ulla, A.; Jenni, T.; Hannu, U.; Tytti, S. Antibacterial and oxidative stress-protective effects of five monoterpenes from softwood. Molecules 2022, 27, 1–15. [Google Scholar]
- Mahajan, A.; Singh, H.; Singh, A.; Agrawal, D.K.; Arora, A.; Chundawat, T.S. Trifluoromethylated quinolone-hydantoin hybrids: Synthesis and antibacterial evaluation. Science 2022, 4, 30. [Google Scholar] [CrossRef]
- Ana, C.M.; Pedro, L.R.; Irlan, A.F.; Luciana, S.; Marcus, T.S.; Sabrina, G.A.; Ricardo, D.C. Antifungal activity, mode of action, docking prediction and anti-biofilm effects of (+)-β-pinene enantiomers against candida spp. Curr. Top. Med. Chem. 2018, 18, 2481–2490. [Google Scholar]
- Rosa, T.; Mariarosaria, L.; Marco, B.; Simone, R.; Nicodemo, G.P. Salvia officinalis L. from Italy: A comparative chemical and biological study of its essential oil in the mediterranean context. Molecules 2021, 25, 5826. [Google Scholar]
- Chen, W.Y.; Vermaak, I.; Viljoen, A. Camphor-A fumigant during the black death and a coveted fragrant wood in ancient Egypt and Babylon-A Review. Molecules 2013, 18, 5434–5454. [Google Scholar] [CrossRef] [Green Version]
- Sokolova, A.S.; Yarovaya, O.I.; Shernyukov, A.V.; Gatilov, Y.V.; Razumova, Y.V.; Zarubaevc, V.V.; Tretiak, D.S.; Pokrovsky, A.G.; Kiselev, O.L.; Salakhutdinov, N.F. Discovery of a new class of antiviral compounds: Camphorimine derivatives. Eur. J. Med. Chem. 2015, 105, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Q.; Liu, Y.; Li, M.; Mao, J.W.; Zhang, L.R.; Huang, R.B.; Jin, X.B.; Ye, L.B. Anti-tumor effect of α-pinene on human hepatoma cell lines through inducing G2/M cell cycle arrest. J. Pharmacol. Sci. 2015, 127, 332–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, S.S.; Yu, H.; Xie, Y.F.; Guo, Y.H.; Fan, J.J.; Yao, W.R. The anti-inflammatory potential of Cinnamomum camphora (L.) J.Presl essential oil in vitro and in vivo. J. Ethnopharmacol. 2021, 267, 113516. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.Y. A preliminary study on the relation between camphor wood volatile matters and corrosion of museum metallic objects. IOP Conf. Ser. Earth Environ. Sci. 2020, 467, 012147. [Google Scholar] [CrossRef] [Green Version]
- Bryleva, Y.A.; Ustimenko, Y.P.; Plyusnin, V.F.; Mikheilis, A.V.; Shubin, A.A.; Glinskaya, L.A.; Komarov, V.Y.; Agafontsev, A.M.; Tkachev, A.V. Ln(III) complexes with a chiral 1H-pyrazolo[3,4-b]pyridine derivative fused with a -α-pinene moiety: Synthesis, crystal structure, and photophysical studies in solution and in the solid state. New. J. Chem. 2021, 45, 2276–2284. [Google Scholar] [CrossRef]
- Yang, J.L.; Fang, H.; Fang, X.Y.; Xu, X.; Yang, Y.Q.; Rui, J.; Wu, C.L.; Wang, S.F.; Xu, H.J. A novel tetrahydroquinazolin-2-amine-based high selective fluorescent sensor for Zn2+ from nopinone. Tetrahedron 2016, 72, 4503–4509. [Google Scholar] [CrossRef]
- Ponomarev, K.Y.; Morozova, E.A.; Suslov, E.V.; Korchagina, D.V.; Tolstikova, T.G.; Volcho, K.P.; Salakhutdinov, N.F. Synthesis and analgesic activity of 5,7- and 6-substituted diazaadamantanes containing monoterpene moieties. Chem. Nat. Comp. 2017, 53, 1131–1136. [Google Scholar] [CrossRef]
- Lin, G.S.; Bai, X.; Duan, W.G.; Cen, B.; Huang, M.; Lu, S.Z. High value-added application of sustainable natural forest product α pinene: Synthesis of myrtenal oxime esters as potential KARI inhibitors. ACS Sustain. Chem. Eng. 2019, 7, 7862–7868. [Google Scholar] [CrossRef]
- Rui, J.; Zhang, Q.; Wang, X.; Xu, X.; Xu, H.J.; Rao, W.D.; Wang, S.F. Synthesis and biological activity of novel pinanyl pyrazole acetamide derivatives. Chin. J. Org. Chem. 2017, 37, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.J.; Wang, Y.Y.; Zhao, Y.X.; Ma, C.H.; Yang, Y.Q.; Xu, X.; Gu, W.; Wang, S.F. Synthesis and α-amylase inhibitory potential and molecular docking study of nopinone-based thiazolylhydrazone derivatives. Chin. J. Org. Chem. 2019, 39, 1372–1382. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Gu, W.; Shan, Y.; Liu, F.; Xu, X.; Yang, Y.Q.; Zhang, Q.J.; Zhang, Y.; Kuang, H.B.; Wang, Z.L.; et al. Design, synthesis and anticancer activity of novel nopinone-based thiosemicarbazone derivatives. Bioorg. Med. Chem. 2017, 27, 2360–2363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.Y.; Zhao, Y.X.; Gu, W.; Zhu, Y.Q.; Wang, S.F. Novel camphor-based pyrimidine derivatives induced cancer cell death through a ROS-mediated mitochondrial apoptosis pathway. RSC Adv. 2019, 9, 29711–29720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raji, M.; Le, T.M.; Csámpai, A.; Nagy, V.; Zupkó, I.; Szakonyi, Z. Stereoselective synthesis and applications of pinane-based chiral 1,4-amino alcohol derivatives. Synthesis 2022, 54, 3831–3844. [Google Scholar]
- Deyno, S.; Mtewa, A.G.; Hope, D.; Bazira, J.; Makonnen, E.; Alete, P.E. Antibacterial activities of Echinops kebericho Mesfin Tuber extracts and isolation of the most active compound, dehydrocostus Lactone. Front. Pharmacol. 2021, 11, 1–13. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | R1 | R2 | Bacterial | Fungi | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| K. pneumoniae | S. pneumoniae | P. aeruginosa | S. aureus | E. coli | MRSA | B. cereus | C. albicans | |||
| 1e | OH | H | 32 | 32 | 16 | 32 | 8 | 64 | 8 | >1024 |
| 1f | Cl | H | 32 | 8 | 16 | 32 | 8 | 8 | 8 | >1024 |
| 1g | OCH3 | H | 64 | 32 | 64 | 1 | 8 | 8 | 32 | >1024 |
| 1h | N(C2H5)2 | H | 256 | >1024 | 128 | 16 | 128 | 8 | >1024 | >1024 |
| 1i | OCH3 |  | 128 | 1 | 32 | >1024 | 1 | 64 | 64 | 32 |
| 1j | OCH3 |  | 256 | 64 | 128 | 512 | 64 | 64 | >1024 | 32 |
| 1k | OCH3 |  | 512 | >1024 | 128 | 512 | 128 | 64 | >1024 | 64 |
| 1l | OCH3 |  | >1024 | >1024 | 128 | 16 | 512 | 8 | >1024 | 16 |
| 1m | N(C2H5)2 |  | >1024 | 32 | 128 | 512 | 64 | 64 | >1024 | >1024 |
| 1n | N(C2H5)2 |  | >1024 | >1024 | >1024 | >1024 | >1024 | >1024 | >1024 | >1024 |
| 2b | OCH3 | H | >1024 | 32 | 16 | >1024 | 8 | 64 | 32 | >1024 |
| 2c | OCH3 |  | 64 | >1024 | 256 | 16 | 32 | 128 | >1024 | 64 |
| 2d | OCH3 |  | >1024 | >1024 | 128 | 256 | 64 | 64 | >1024 | 64 |
| 2e | OCH3 |  | >1024 | >1024 | 128 | 512 | 128 | 32 | >1024 | 128 |
| 2f | OCH3 |  | 32 | 16 | 16 | 32 | 8 | 8 | 16 | 32 |
| amikacin b | - | - | 32 | 2 | 32 | 1 | 2 | 1 | 4 | -a |
| ketoconazole b | - | - | - | - | - | - | - | - | - | 16 |
| Compd. | MW a | CLogP b | pKa c | tPSA d |
|---|---|---|---|---|
| 1e | 281 | 3.73 | 8.81 | 70.97 |
| 1f | 300 | 4.86 | - | 50.74 |
| 1g | 295 | 4.18 | - | 59.97 |
| 1i | 353 | 5.01 | - | 55.21 |
| 2f | 435 | 7.26 | - | 45.98 |
| amikacin | 585 | −4.12 | 13.32 | 331.94 |
| 1l | 421 | 6.74 | - | 45.98 |
| ketoconazole | 531 | 3.64 | - | 66.84 |
| Compd. | (Anti-Inflammatory Activity)/μmol·L−1 (IC50 ± SD) |
|---|---|
| 1e | 2.32 ± 0.9 |
| 1f | 1.37 ± 0.41 |
| 1g | 3.0 ± 0.71 |
| 1i | 4.86 ± 0.84 |
| 2f | 1.87 ± 0.38 |
| Aspirin | 1.91 ± 0.35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Wang, Y.; Wang, S.; Wu, H. Synthesis and Biological Evaluation of Novel Pyrimidine Amine Derivatives Bearing Bicyclic Monoterpene Moieties. Molecules 2022, 27, 8104. https://doi.org/10.3390/molecules27228104
Zhang M, Wang Y, Wang S, Wu H. Synthesis and Biological Evaluation of Novel Pyrimidine Amine Derivatives Bearing Bicyclic Monoterpene Moieties. Molecules. 2022; 27(22):8104. https://doi.org/10.3390/molecules27228104
Chicago/Turabian StyleZhang, Mingguang, Yunyun Wang, Shifa Wang, and Hongyan Wu. 2022. "Synthesis and Biological Evaluation of Novel Pyrimidine Amine Derivatives Bearing Bicyclic Monoterpene Moieties" Molecules 27, no. 22: 8104. https://doi.org/10.3390/molecules27228104
APA StyleZhang, M., Wang, Y., Wang, S., & Wu, H. (2022). Synthesis and Biological Evaluation of Novel Pyrimidine Amine Derivatives Bearing Bicyclic Monoterpene Moieties. Molecules, 27(22), 8104. https://doi.org/10.3390/molecules27228104