
Pathfinder-Driven Chemical Space Exploration and Multiparameter Optimization in Tandem with Glide/IFD and QSAR-Based Active Learning Approach to Prioritize Design Ideas for FEP+ Calculations of SARS-CoV-2 PLpro Inhibitors


Abstract
:1. Introduction
2. Materials and Methods
2.1. Computational Details
2.2. Reaction-Based Enumeration Using Pathfinder
2.3. Ligand and Protein Preparation
2.3.1. Ligand Preparation
2.3.2. Protein Preparation
2.4. Glide SP Docking and Auto QSAR Active Learning Models
2.5. Induced Fit Docking (IFD) to Screen the Library of Enumerated Design Ideas
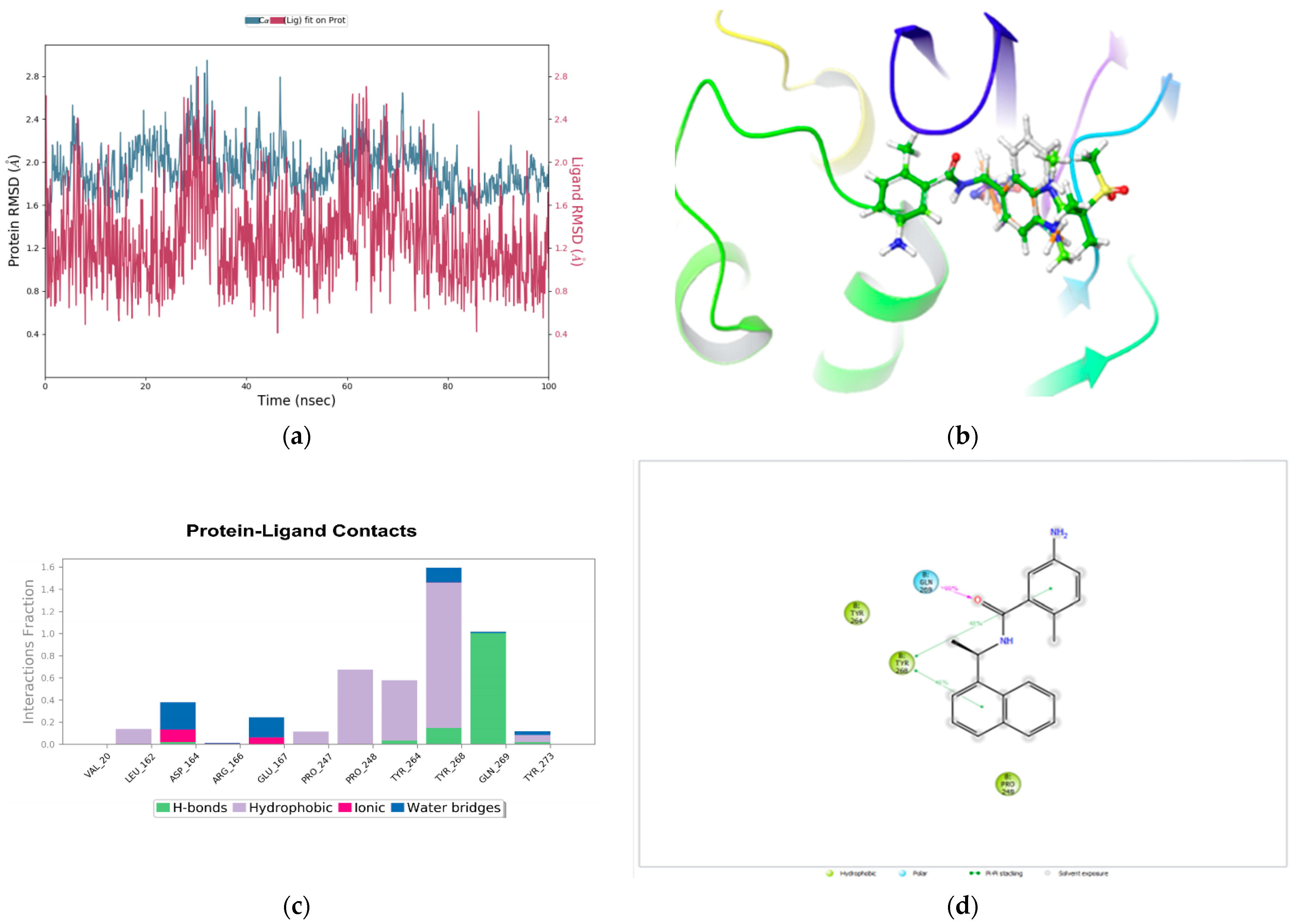
2.6. Molecular Dynamics Simulations
2.7. Maximum Common Substructural (MCS) Docking
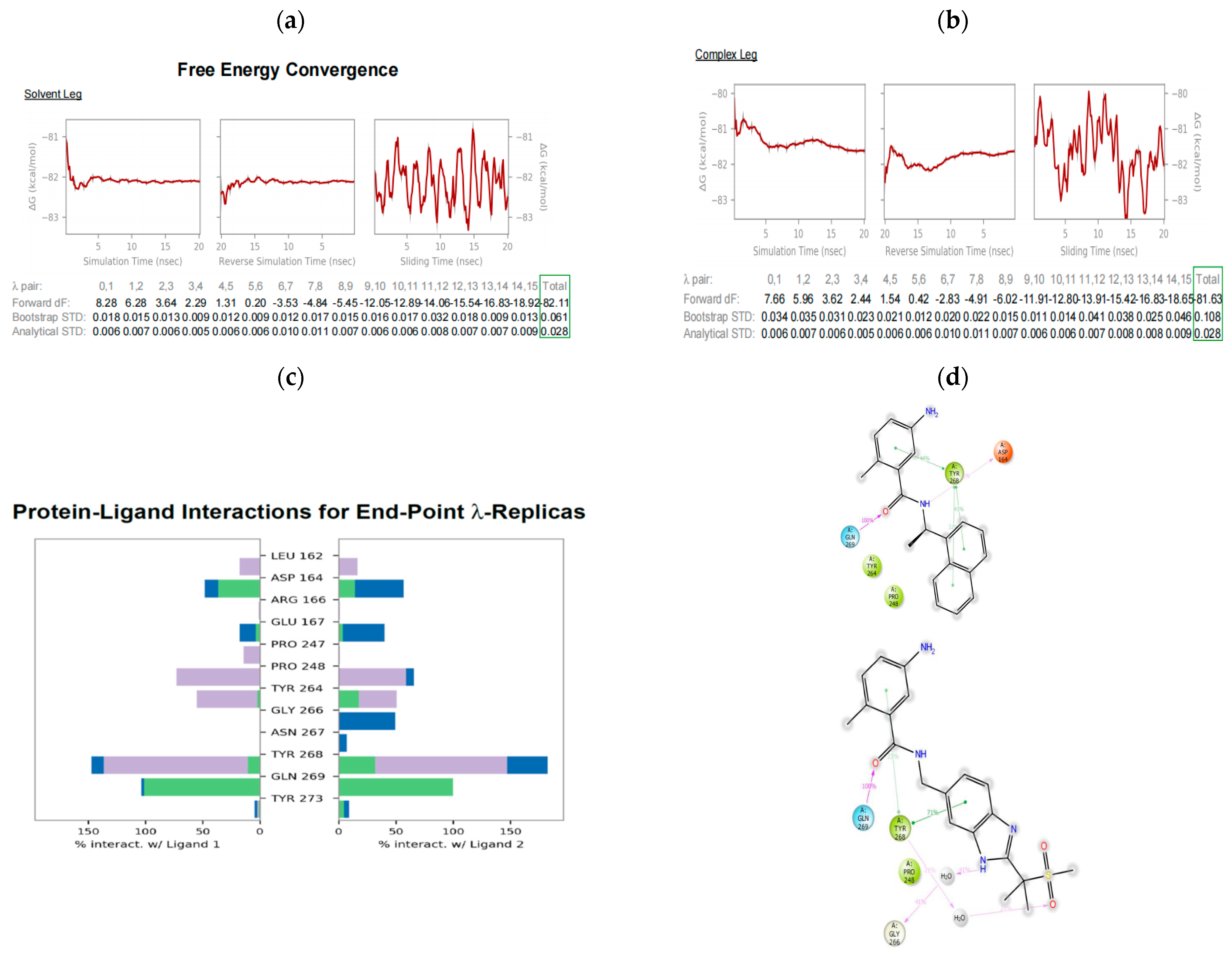
2.8. Free Energy Perturbation Plus (FEP+) to Predict the Relative Binding Affinities of the Design Ideas
3. Results and Discussions
3.1. Pathfinder Reaction-Based Enumeration
3.2. Glide SP Docking and Filtering of Enumerated Design Ideas
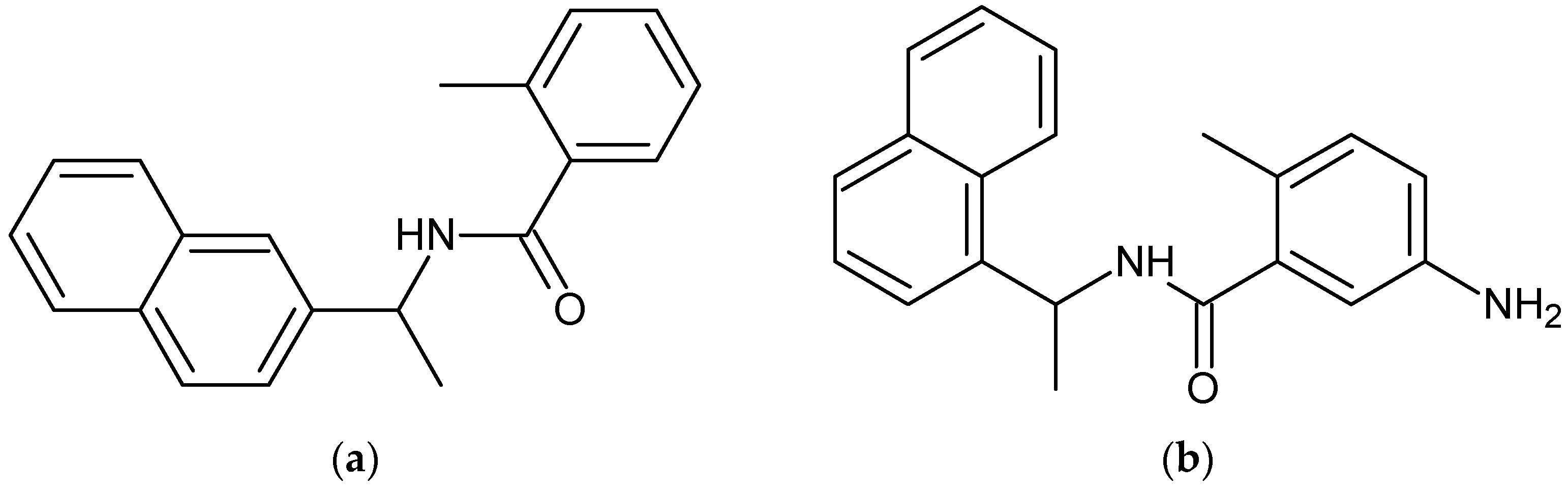
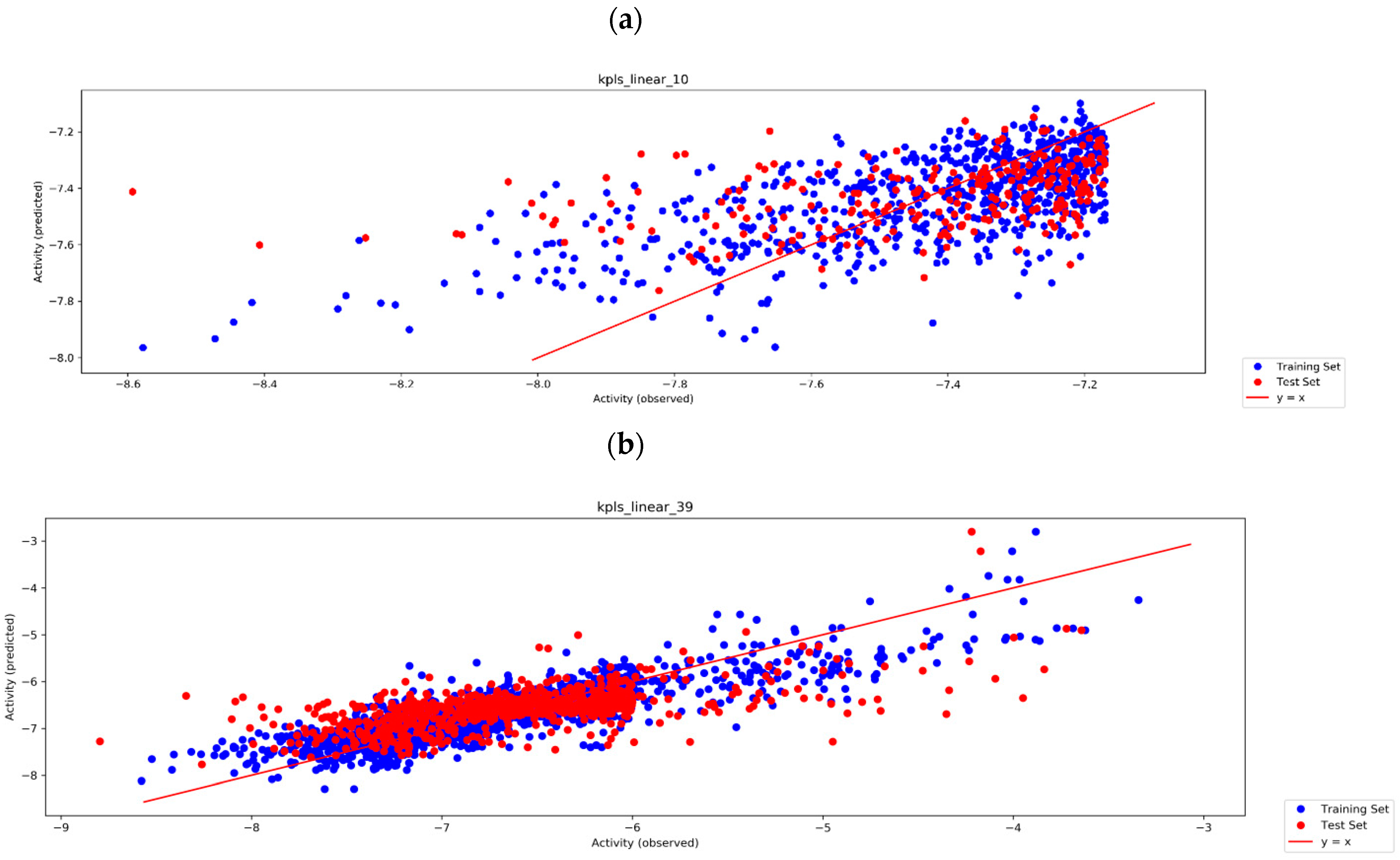
3.3. Auto QSAR-Based Active Learning Models to Prioritize Ideas for Glide/IFD XP Docking
3.4. Relative Binding Affinity Prediction Using FEP+
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Crotti, L.; Arbelo, E. COVID-19 Q2 treatments, QT interval, and arrhythmic risk: The need for an international registry on arrhythmias. Heart Rhythm. 2020, 17, 1423–1424. [Google Scholar] [CrossRef] [PubMed]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 billion Compounds. Mol. Inf. 2020, 39, 2000028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Savi, C.; Hughes, D.L.; Kvaerno, L. Quest for a COVID-19 Cure by Repurposing Small-Molecule Drugs: Mechanism of Action, Clinical Development, Synthesis at Scale, and Outlook for Supply. Org. Process. Res. Dev. 2020, 24, 940–976. [Google Scholar] [CrossRef]
- Saha, A.; Sharma, A.R.; Bhattacharya, M.; Sharma, G.; Lee, S.S.; Chakraborty, C. Tocilizumab: A Therapeutic Option for the Treatment of Cytokine Storm Syndrome in COVID-19. Arch. Med. Res. 2020, 51, 595–597. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species severe acute respiratory syndrome related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 536, 536–544. [Google Scholar]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109. [Google Scholar] [CrossRef]
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Leist, S.R.; Schäfer, A.; Won, J.; Brown, A.J.; Montgomery, S.A.; Hogg, A.; Babusis, D.; Clarke, M.O.; et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020, 11, 222. [Google Scholar] [CrossRef] [Green Version]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 33, 11876–11881. [Google Scholar] [CrossRef]
- Liu, T.; Luo, S.; Libby, P.; Shi, G.P. Cathepsin L-selective inhibitors: A potentially promising treatment for COVID-19 patients. Pharmacol. Ther. 2020, 213, 107587. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Luan, J.; Zhang, L. Molecular docking of potential SARS-CoV-2 papain-like protease Inhibitors. Biochem. Biophys. Res. Commun. 2021, 538, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Sacco, M.D.; Xia, Z.; Lambrinidis, G.; Townsend, J.A.; Hu, Y.; Meng, X.; Szeto, T.; Ba, M.; Zhang, X.; et al. Discovery of SARS-CoV-2 Papain-like Protease Inhibitors through a Combination of High-Throughput Screening and a FlipGFP-Based Reporter Assay. ACS Cent. Sci. 2021, 7, 1245–1260. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J. Med. Chem. 2022, 65, 2716–2746. [Google Scholar] [CrossRef]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal structure of SARS-CoV-2 papain-like Protease. Acta Pharm. Sin. B 2021, 11, 237–245. [Google Scholar] [CrossRef]
- Báez-Santos, Y.M.; Barraza, S.J.; Wilson, M.W.; Agius, M.P.; Mielech, A.M.; Davis, N.M.; Baker, S.C.; Larsen, S.D.; Mesecar, A.D. X-ray Structural and Biological Evaluation of a Series of Potent and Highly Selective Inhibitors of Human Coronavirus Papain-like Proteases. J. Med. Chem. 2014, 57, 2393–2412. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Takayama, J.; Aubin, Y.; Ratia, K.; Chaudhuri, R.; Baez, Y.; Sleeman, K.; Coughlin, M.; Nichols, D.B.; Mulhearn, D.C.; et al. Structure-Based Design, Synthesis, and Biological Evaluation of a Series of Novel and Reversible Inhibitors for the severe acute respiratory syndrome-Coronavirus Papain-Like Protease. J. Med. Chem. 2009, 52, 5228–5240. [Google Scholar] [CrossRef] [Green Version]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuria, R.; Fu, W.; Prabhakar, B.S.; Johnson, M.E.; et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, R.; Tang, S.; Zhao, G.; Lu, H.; Case, D.A.; Johnson, M.E. Comparison of SARS and NL63 Papain-Like Protease Binding Sites and Binding Site Dynamics: Inhibitor Design Implications. J. Mol. Biol. 2011, 414, 272–288. [Google Scholar] [CrossRef]
- Arshadi, A.K.; Webb, J.; Salem, M.; Cruz, E.; Calad-Thomson, S.; Ghadirian, N.; Collins, J.; Diez-Cecilia, E.; Kelly, B.; Goodarzi, H.; et al. Artificial Intelligence for COVID-19 Drug Discovery and Vaccine Development. Front. Artif. Intell. Appl. 2020, 3, 65. [Google Scholar] [CrossRef] [PubMed]
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-Based Drug Discovery. arXiv 2015, arXiv:1510.02855. [Google Scholar]
- Sugiyama, M.G.; Cui, H.; Redka, D.S.; Karimzadeh, M.; Rujas, E.; Maan, H.; Hayat, S.; Cheung, K.; Misra, R.; McPhee, J.B.; et al. Multiscale interactome analysis coupled with off-target drug predictions reveals drug repurposing candidates for human coronavirus disease. Sci. Rep. 2021, 11, 23315. [Google Scholar] [CrossRef] [PubMed]
- Arshia, A.H.; Shadravan, S.; Solhjoo, A.; Sakhteman, A.; Sam, A. De novo design of novel protease inhibitor candidates in the treatment of SARS-CoV-2 using deep learning, docking, and molecular dynamic simulations. Comput. Biol. Med. 2021, 139, 104967. [Google Scholar] [CrossRef]
- Murugesan, S.; Kottekad, S.; Crasta, I.; Sreevathsan, S.; Usharani, D.; Perumal, M.K.; Mudliar, S.N. Targeting COVID-19 (SARS-CoV-2) main protease through active phytocompounds of ayurvedic medicinal plants—Emblica officinalis (Amla), Phyllanthus niruri Linn. (Bhumi Amla) and Tinospora cordifolia (Giloy)—A molecular docking and simulation study. Comput. Biol. Med. 2021, 136, 104683. [Google Scholar] [CrossRef]
- Patel, C.N.; Jani, S.P.; Jaiswal, D.G.; Kumar, S.P.; Mangukia, N.; Parmar, R.M.; Rawal, R.M.; Pandya, H.A. Identification of antiviral phytochemicals as a potential SARS-CoV-2 main protease (Mpro) inhibitor using docking and molecular dynamics simulations. Sci. Rep. 2021, 11, 20295. [Google Scholar] [CrossRef]
- LigPrep; Schrödinger, L.L.C: New York, NY, USA, 2021.
- Epik; Schrödinger, L.L.C: New York, NY, USA, 2021.
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favourable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided. Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comp. Aided. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Osipiuk, J.; Azizi, S.A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G.; et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Comm. 2021, 12, 743. [Google Scholar] [CrossRef]
- Prime; Schrödinger, L.L.C: New York, NY, USA, 2021.
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Genet. 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of Crystal Packing Forces in Determining Protein Sidechain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Glide; Schrödinger, L.L.C: New York, NY, USA, 2021.
- Dixon, S.L.; Duan, J.; Smith, E.; Von Bargen, C.D.; Sherman, W.; Repasky, M.P. AutoQSAR: An automated machine learning tool for best-practice quantitative structure-activity relationship modelling. Future. Med. Chem. 2016, 8, 1825–1839. [Google Scholar] [CrossRef]
- Induced Fit Docking Protocol; Schrödinger, L.L.C: New York, NY, USA, 2021.
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef]
- Gumede, N.J.; Singh, P.; Sabela, M.I.; Bisetty, K.; Escuder-Gilabert, L.; Medina-Hernández, M.J.; Sagrado, S. Experimental-like affinity constants and enantioselectivity estimates from flexible docking. J. Chem. Inf. Model. 2012, 52, 2754–2759. [Google Scholar] [CrossRef]
- Gumede, N.J.; Nxumalo, W.; Bisetty, K.; Escuder Gilabert, L.; Medina-Hernandez, M.J.; Sagrado, S. Prospective computational design, and in vitro bio-analytical tests of new chemical entities as potential selective CYP17A1 lyase inhibitors. Bioorg. Chem. 2020, 94, 103462. [Google Scholar] [CrossRef]
- Shaw, D.E. A fast, scalable method for the parallel evaluation of distance-limited pair wise particle interactions. J. Comput. Chem. 2005, 26, 1318. [Google Scholar] [CrossRef] [Green Version]
- Lippert, R.A.; Bowers, K.J.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Shaw, D.E. A common, avoidable source of error in molecular dynamics integrators. J. Chem. Phys. 2007, 126, 046101. [Google Scholar] [CrossRef] [Green Version]
- Cappel, D.; Jerome, S.; Hessler, G.; Matter, H. Impact of Different Automated Binding Pose Generation Approaches on Relative Binding Free Energy Simulations. J. Chem. Inf. Model. 2020, 60, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pearce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M.K.; Greenwood, J.; et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137, 2695. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Deng, Y.; Knight, J.L.; Wu, Y.; Kim, B.; Sherman, W.; Shelley, J.; Lin, T.; Abel, R. Modeling Local Structural Rearrangements Using FEP/REST: Application to Relative Binding Affinity Predictions of CDK2 Inhibitors. J. Chem. Theory Comput. 2013, 9, 1282. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Deng, Y.; Russell, E.; Wu, Y.; Abel, R.; Wang, L. Accurate Calculation of Relative Binding Free Energies between Ligands with Different Net Charges. J. Chem. Theory Comput. 2018, 14, 6346. [Google Scholar] [CrossRef] [PubMed]
- Abel, R.; Wang, L.; Mobley, D.L.; Friesner, R.A. OPLS3: A Critical Review of Validation, Blind Testing, and Real-World Use of Alchemical Protein-Ligand Binding Free Energy Calculations. Curr. Top. Med. Chem. 2017, 17, 2577. [Google Scholar] [CrossRef] [PubMed]
- Fratevy, F.; Sirimulla, S. An Improved Free Energy Perturbation FEP+ Sampling Protocol for Flexible Ligand-Binding Domains. Sci. Rep. 2019, 9, 16829. [Google Scholar] [CrossRef] [Green Version]
- Moraca, F.; Negri, A.; de Oliveira, C.; Abel, R. Application of Free Energy Perturbation (FEP+) to Understanding Ligand Selectivity: A Case Study to Assess Selectivity Between Pairs of Phosphodiesterases (PDE’s). J. Chem. Inf. Model. 2019, 59, 2729–2740. [Google Scholar] [CrossRef]
- Abel, R.; Wang, L.; Harder, E.D.; Berne, B.J.; Friesner, R.A. Advancing Drug Discovery through Enhanced Free Energy Calculations. Acc. Chem. Res. 2017, 50, 1625–1632. [Google Scholar] [CrossRef]
- Konze, K.D.; Bos, P.H.; Dahlgren, M.K.; Leswing, K.; Tubert-Brohman, I.; Bortolato, A.; Robbason, B.; Abel, R.; Bhat, S. Reaction-Based Enumeration, Active Learning, and Free Energy Calculations to Rapidly Explore Synthetically Tractable Chemical Space and Optimize Potency of Cyclin-Dependent Kinase 2 Inhibitors. J. Chem. Inf. Model. 2019, 59, 3782–3793. [Google Scholar] [CrossRef]
- Paulsen, J.L.; Yu, H.S.; Sindhikara, D.; Wang, L.; Appleby, T.; Villaseñor, A.G.; Schmitz, U.; Shivakumar, D. Evaluation of Free Energy Calculations for the Prioritization of Macrocycle Synthesis. J. Chem. Inf. Model. 2020, 60, 3489–3498. [Google Scholar] [CrossRef]
- Kuhn, B.; Tichy, M.; Wang, L.; Robinson, S.; Martin, R.E.; Kuglstatter, A.; Benz, J.; Giroud, M.; Schirmeister, T.; Abel, R.; et al. Prospective Evaluation of Free Energy Calculations for the Prioritization of Cathepsin L Inhibitors. J. Med. Chem. 2017, 60, 2485–2497. [Google Scholar] [CrossRef] [PubMed]
- Cleves, A.E.; Johnson, S.R.; Jain, A.N. Synergy and Complementarity between Focused Machine Learning and Physics-Based Simulation in Affinity Prediction. J. Chem. Inf. Model. 2021, 61, 5948–5966. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Allen, B.K.; Beuming, T.; Pearlman, D.A.; Radak, B.K.; Sherman, W. Rigorous Free Energy Simulations in Virtual Screening. J. Chem. Inf. Model. 2020, 60, 4153–4169. [Google Scholar] [CrossRef] [PubMed]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef] [PubMed]
- Ghanakota, P.; Bos, P.H.; Konze, K.D.; Staker, J.; Marques, G.; Marshall, K.; Leswing, K.; Abel, R.; Bhat, S. Combining Cloud-Based Free-Energy Calculations, Synthetically Aware Enumerations, and Goal-Directed Generative Machine Learning for Rapid Large-Scale Chemical Exploration and Optimization. J. Chem. Inf. Model. 2020, 60, 4311–4325. [Google Scholar] [CrossRef]
- Fischer, A.; Smieško, M.; Sellner, M.; Lill, M.A. Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results. J. Med. Chem. 2021, 64, 2489–2500. [Google Scholar] [CrossRef]
- Clark, A.J.; Tiwary, P.; Borrelli, K.; Feng, S.; Miller, E.B.; Abel, R.; Friesner, R.A.; Berne, B.J. Prediction of Protein−Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynamics Simulations. J. Chem. Theory Comput. 2016, 12, 2990–2998. [Google Scholar] [CrossRef]
- Lekgau, K.; Raphoko, L.A.; Lebepe, C.M.; Mongokoana, D.F.; Leboho, T.C.; Matsebatlela, T.M.; Gumede, N.J.; Nxumalo, W. Design, and synthesis of 6-amino-quinoxaline-alkynyl as potential aromatase (CYP19A1) inhibitors. J. Mol. Struct. 2022, 1255, 132473. [Google Scholar] [CrossRef]
- Singh, K.; Coopoosamy, R.M.; Gumede, N.J.; Sabiu, S. Computational insights, and in vitro validation of antibacterial potential of shikimate pathway-derived phenolic acids as NorA efflux pump inhibitors. Molecules 2022, 27, 2601. [Google Scholar] [CrossRef]
- Miller, E.B.; Murphy, R.B.; Sindhikara, D.; Borrelli, K.W.; Grisewood, M.J.; Ranalli, F.; Dixon, S.L.; Jerome, S.; Boyles, N.A.; Day, T.; et al. Reliable and Accurate Solution to the Induced Fit Docking Problem for Protein−Ligand Binding. J. Chem. Theory Comput. 2021, 17, 2630–2639. [Google Scholar] [CrossRef]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Boyles, F.; Deane, C.M.; Morris, G.M. Learning from Docked Ligands: Ligand-Based Features Rescue Structure-Based Scoring Functions When Trained on Docked Poses. J. Chem. Inf. Model. 2020, 62, 5329–5341. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.E.M.; Baumann, H.; Blum, A.; Böse, D.; Buchstaller, H.P.; Burgdorf, L.; Cappel, D.; Chekler, E.; Czodrowski, P.; Dorsch, D.; et al. Large-Scale Assessment of Binding Free Energy Calculations in Active Drug Discovery Projects. J. Chem. Inf. Model. 2020, 60, 5457–5474. [Google Scholar] [CrossRef]
- Peccati, F.; Jiménez-Osés, G. Enthalpy−Entropy Compensation in Biomolecular Recognition: A Computational Perspective. ACS Omega 2021, 6, 11122–11130. [Google Scholar] [CrossRef] [PubMed]
- Biswal, J.; Jayaprakash, P.; Rayala, S.K.; Venkatraman, G.; Rangaswamy, R.; Jeyaraman, J. WaterMap and Molecular Dynamic Simulation-Guided Discovery of Potential PAK1 Inhibitors Using Repurposing Approaches. ACS Omega 2021, 6, 26829–26845. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Name | Model Code | STDEV | RMSE | R2 | Q2 | #Factors | #Training Set | #Test Set |
|---|---|---|---|---|---|---|---|---|
| Auto QSAR _model_1 | KPLS_linear_10 | 0.1805 | 0.2230 | 0.4310 | 0.1571 | 1 | 478 | 160 |
| Auto QSAR _model_2 | KPLS_radial_44 | 0.2261 | 0.2354 | 0.6262 | 0.5929 | 3 | 816 | 273 |
| Auto QSAR _model_3 | KPLS_radial_26 | 0.2284 | 0.2398 | 0.6090 | 0.5630 | 3 | 865 | 289 |
| Auto QSAR _model_4 | KPLS_radial_32 | 0.2474 | 0.2615 | 0.6227 | 0.5793 | 3 | 906 | 303 |
| Auto QSAR _model_5 | KPLS_molprint2D_8 | 0.2743 | 0.2983 | 0.6980 | 0.6450 | 3 | 1073 | 358 |
| Auto QSAR _model_6 | KPLS_molprint2D_48 | 0.2853 | 0.3040 | 0.7084 | 0.6676 | 3 | 1142 | 381 |
| Auto QSAR _model_7 | KPLS_molprint2D_47 | 0.3014 | 0.3091 | 0.7086 | 0.6910 | 3 | 1252 | 418 |
| Auto QSAR _model_8 | KPLS_radial_42 | 0.2833 | 0.3053 | 0.7579 | 0.7191 | 4 | 1362 | 454 |
| Auto QSAR _model_9 | KPLS_molprint2D_15 | 0.3323 | 0.3424 | 0.6718 | 0.6476 | 3 | 1662 | 554 |
| Auto QSAR_model_10 | KPLS_linear_39 | 0.3444 | 0.5292 | 0.7410 | 0.3868 | 3 | 2391 | 797 |
| Rank Order No. | Enumeration Coupling Building Blocks/Pathway | Compound ID | TPSA | Num | Num | Mol | Mol | Pred Y | Pred Y | Docking | Glide | IFD | Exp IC50 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HDonors | HAcceptors | LogP | Wt | SD | Score | E-Model | Score | (uM) | |||||
| 1 | N-Heterocycles | 58 | 109.98 | 3 | 6 | 1.32 | 394.456 | ND * | ND * | −12.39 | −103.831 | −664.209 | |
| 2 | R1–R2_Carboxylates | 39 | 110.52 | 3 | 5 | 3.60 | 425.529 | −6.86 | 0.098 | −10.93 | −95.015 | −664.167 | |
| 3 | Pathway 10 | 1 | 135.08 | 3 | 8 | 2.64 | 546.481 | −6.62 | 0.072 | −10.87 | −113.770 | −673.770 | |
| 4 | N-Heterocycles | 128 | 83.8 | 3 | 3 | 3.01 | 320.396 | ND * | ND * | −9.91 | −78.744 | −660.799 | |
| 5 | N-Heterocycles | 96 | 141.17 | 4 | 7 | 2.20 | 396.451 | ND * | ND * | −9.64 | −102.840 | −660.007 | |
| 6 | Carboxylates | 5 | 82.05 | 2 | 5 | 1.26 | 310.357 | −6.60 | 0.075 | −9.44 | −71.375 | −660.109 | |
| 7 | R1 (vinyl and aryl halides) R2 (Carboxylates) | 2 | 82.05 | 2 | 5 | 1.26 | 310.357 | −6.60 | 0.075 | −9.44 | −71.375 | −660.109 | |
| 8 | R1–R2_Carboxylates | 36 | 123.13 | 3 | 7 | 3.02 | 449.845 | −6.64 | 0.213 | −9.12 | −80.291 | −659.220 | |
| 9 | Carboxylates | 1 | 75.43 | 2 | 4 | 2.63 | 309.369 | −6.68 | 0.032 | −8.81 | −63.709 | −658.183 | |
| 10 | 25 a | ND * | ND * | ND * | ND * | ND * | ND * | ND * | −8.69 | −76.159 | −658.503 | 2.64 | |
| 11 | R1–R2_Carboxylates | 49 | 120.56 | 3 | 7 | 2.61 | 388.431 | −6.62 | 0.150 | −8.55 | −69.600 | −653.725 | |
| 12 | Pathway 10 | 24 | 99.42 | 2 | 4 | 5.13 | 448.906 | −6.61 | 0.122 | −8.53 | −80.814 | −658.802 | |
| 13 | Primary and secondary amines | 30 | 87.9 | 2 | 4 | 3.84 | 377.492 | −7.60 | 0.090 | −8.40 | −76.477 | −657.629 | |
| 14 | vinyl and aryl halides_Trifluoroborates | 24 | 53.71 | 1 | 4 | 4.53 | 403.469 | −6.79 | 0.065 | −8.39 | −78.794 | −658.165 | |
| 15 | N-Heterocycles | 67 | 103.77 | 4 | 3 | 3.34 | 372.428 | ND * | ND * | −8.34 | −90.436 | −658.944 | |
| 16 | R1 (vinyl and aryl halides) R2 (Carboxylates) | 4 | 63.4 | 1 | 3 | 2.06 | 252.273 | −6.76 | 0.054 | −8.31 | −53.957 | −657.165 | |
| 17 | Pathway 10 | 27 | 107.69 | 4 | 4 | 4.90 | 500.643 | −6.79 | 0.091 | −8.26 | −97.525 | −659.060 | |
| 18 | 2 a | ND * | ND * | ND * | ND * | ND * | ND * | ND * | −8.25 | −56.924 | −656.513 | 8.70 | |
| 19 | 24 a | ND * | ND * | ND * | ND * | ND * | ND * | ND * | −8.14 | −72.533 | −658.903 | 0.56 | |
| 20 | N-Heterocycles | 116 | 83.8 | 3 | 3 | 3.61 | 358.804 | ND * | ND * | −8.10 | −75.467 | −658.695 | |
| 21 | N-Heterocycles | 57 | 70.91 | 3 | 2 | 4.49 | 377.822 | ND * | ND * | −7.98 | −76.563 | −659.233 | |
| 22 | N-Heterocycles | 172 | 95.24 | 3 | 4 | 3.47 | 364.449 | ND * | ND * | −7.94 | −54.584 | −656.907 | |
| 23 | R1 (vinyl and aryl halides) R2 (Carboxylates) | 1 | 89.34 | 3 | 4 | 0.72 | 273.13 | −6.69 | 0.035 | −7.77 | −54.137 | −660.284 | |
| 24 | N-Heterocycles | 91 | 83.8 | 3 | 3 | 3.13 | 320.396 | ND * | ND * | −7.58 | −87.783 | −657.771 | |
| 25 | 1 a | ND * | ND * | ND * | ND * | ND * | ND * | ND * | −7.48 | −59.646 | −655.252 | 200 | |
| 26 | N-Heterocycles | 37 | 122.71 | 4 | 5 | 2.45 | 356.817 | ND * | ND * | −7.46 | −73.976 | −657.600 | |
| 27 | Primary and secondary amines | 23 | 114.01 | 3 | 5 | 1.91 | 342.33 | −7.55 | 0.077 | −7.29 | −60.112 | −659.115 | |
| 28 | Hydrazine-aryl | 15 | 119.39 | 3 | 7 | 2.51 | 378,436 | −7.55 | 0.068 | −7.28 | −66.860 | −652.823 | |
| 29 | Primary and secondary amines | 24 | 80.9 | 2 | 4 | 3.81 | 360.461 | −7.42 | 0.123 | −7.25 | −70.170 | −657.073 | |
| 30 | R1–R2_ Carboxylates | 5 | 130.21 | 4 | 5 | 2.25 | 378.388 | −6.62 | 0.090 | −7.10 | −59.593 | −661.117 | |
| 31 | Hydrazine-aryl | 54 | 110.16 | 3 | 6 | 3.71 | 396.882 | −7.55 | 0.068 | −6.67 | −57.155 | −653.216 | |
| 32 | vinyl and aryl halides_Trifluoroborates | 11 | 92.51 | 3 | 7 | 3.94 | 425.558 | −6.86 | 0.084 | −6.52 | −87.781 | −658.149 | |
| 33 | N-Heterocycles | 45 | 117.94 | 3 | 5 | 2.66 | 400.504 | ND * | ND * | −6.52 | −74.218 | −657.538 | |
| 34 | N-Heterocycles | 204 | 83.8 | 3 | 3 | 4.37 | 342.402 | ND * | ND * | −6,56 | −75.146 | −655.866 | |
| 35 | Hydrazine-aryl | 35 | 110.16 | 3 | 6 | 2.95 | 380.427 | −7.51 | 0.056 | −6.51 | −66.176 | −653.539 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gumede, N.J. Pathfinder-Driven Chemical Space Exploration and Multiparameter Optimization in Tandem with Glide/IFD and QSAR-Based Active Learning Approach to Prioritize Design Ideas for FEP+ Calculations of SARS-CoV-2 PLpro Inhibitors. Molecules 2022, 27, 8569. https://doi.org/10.3390/molecules27238569
Gumede NJ. Pathfinder-Driven Chemical Space Exploration and Multiparameter Optimization in Tandem with Glide/IFD and QSAR-Based Active Learning Approach to Prioritize Design Ideas for FEP+ Calculations of SARS-CoV-2 PLpro Inhibitors. Molecules. 2022; 27(23):8569. https://doi.org/10.3390/molecules27238569
Chicago/Turabian StyleGumede, Njabulo Joyfull. 2022. "Pathfinder-Driven Chemical Space Exploration and Multiparameter Optimization in Tandem with Glide/IFD and QSAR-Based Active Learning Approach to Prioritize Design Ideas for FEP+ Calculations of SARS-CoV-2 PLpro Inhibitors" Molecules 27, no. 23: 8569. https://doi.org/10.3390/molecules27238569
APA StyleGumede, N. J. (2022). Pathfinder-Driven Chemical Space Exploration and Multiparameter Optimization in Tandem with Glide/IFD and QSAR-Based Active Learning Approach to Prioritize Design Ideas for FEP+ Calculations of SARS-CoV-2 PLpro Inhibitors. Molecules, 27(23), 8569. https://doi.org/10.3390/molecules27238569