
Stable Isotope Analysis of Residual Pesticides via High Performance Liquid Chromatography and Elemental Analyzer–Isotope Ratio Mass Spectrometry


 ,
,
Abstract
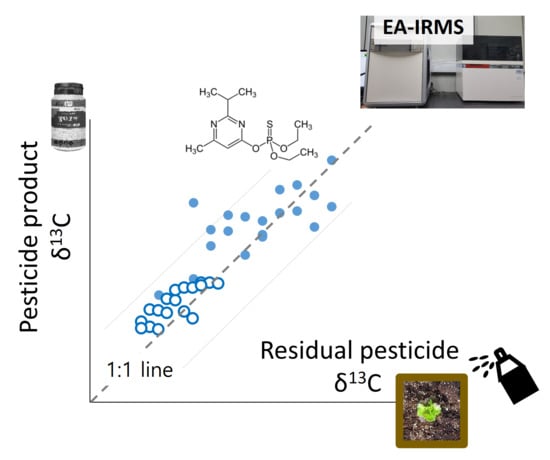
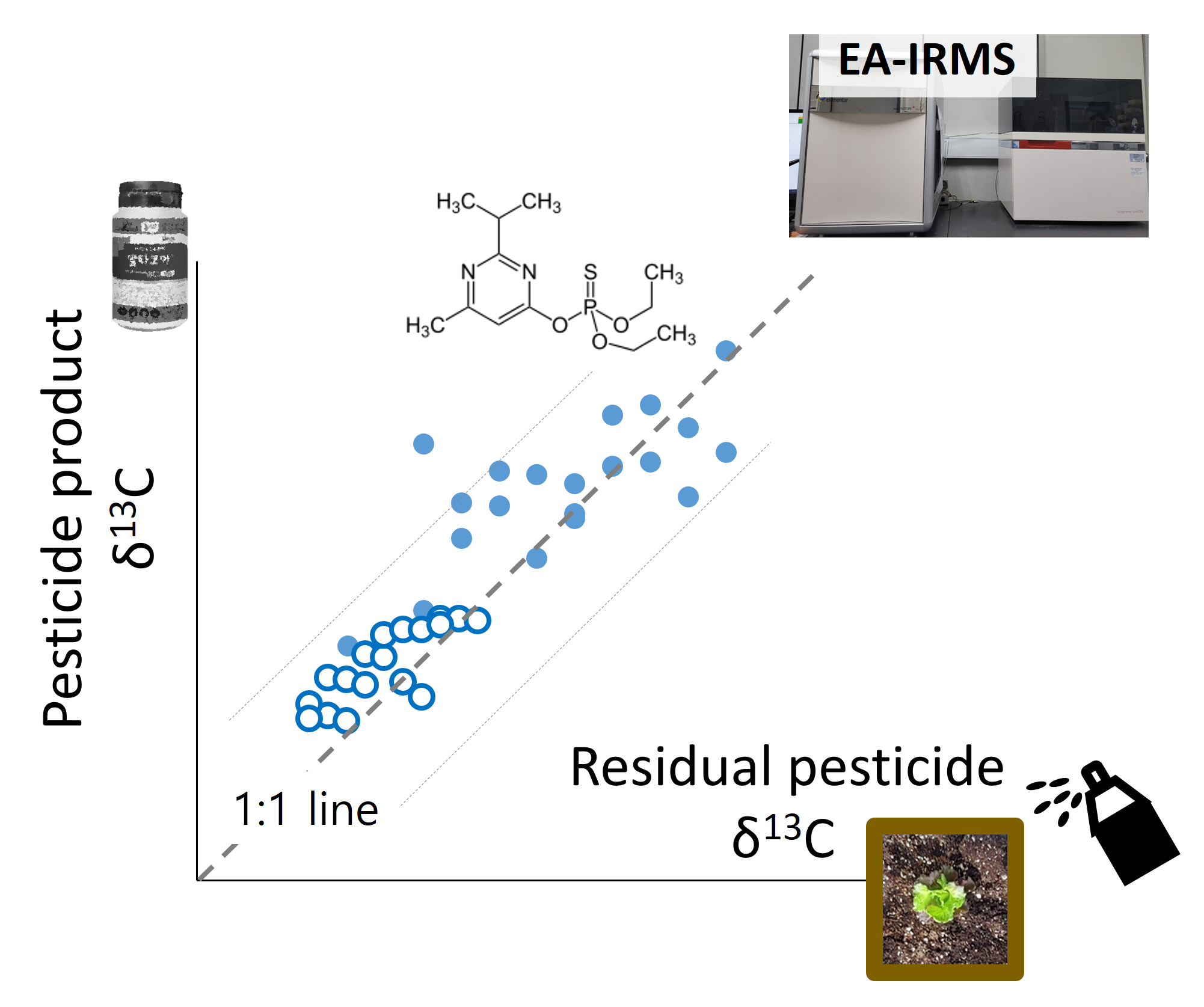
:
1. Introduction
2. Results and Discussion
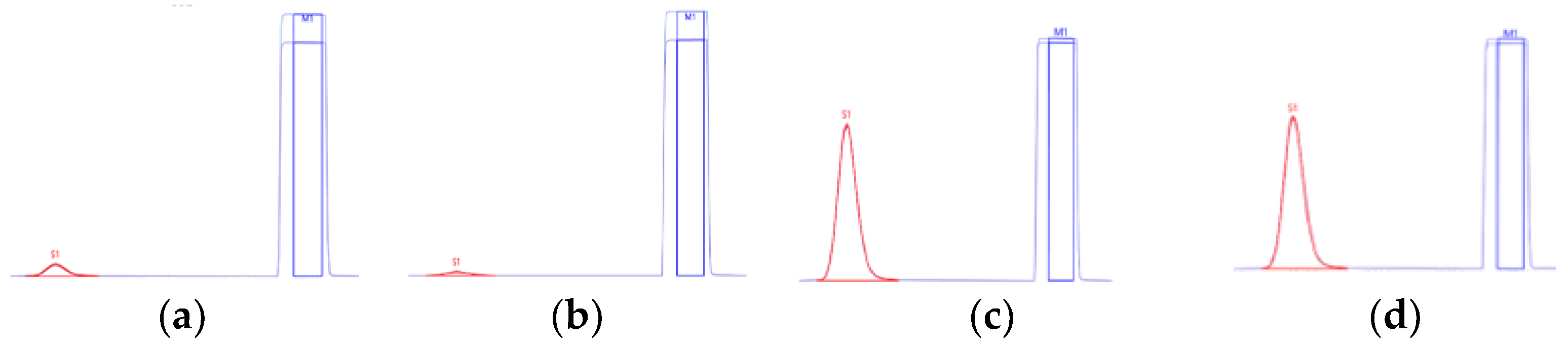
2.1. Error Evaluation during Isotope Measurement
2.2. δ13C Variations in Standard Material: Before and after HPLC Purification
2.3. Application of HPLC/EA–IRMS and δ13C Determination of Target Compounds in a Soil-Crop System
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Validation of HPLC/EA–IRMS Isotopic Measurement
3.3. Pesticide Application and Plant Growth
3.4. Sample Preparation: Isolation and Purification of Residual Pesticides In Situ Samples
3.5. Standard Calibration Curve and Estimating Pesticide Amount
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huynh, K.; Leonard, E.; Chong, J.-H.; Palmer, C.; Tharayil, N. Persistence and metabolism of the diamide insecticide cyantraniliprole in tomato plants. Sci. Rep. 2021, 11, 21570. [Google Scholar] [CrossRef]
- Nicolopoulou-Stamati, P.; Maipas, S.; Kotampasi, C.; Stamatis, P.; Hens, L. Chemical Pesticides and Human Health: The Urgent Need for a New Concept in Agriculture. Front. Public Health 2016, 4, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, E.-J.; Yun, H.-Y.; Lee, D.-H.; Shin, K.-H. Application of Compound-Specific Isotope Analysis in Environmental Forensic and Strategic Management Avenue for Pesticide Residues. Molecules 2021, 26, 4412. [Google Scholar] [CrossRef] [PubMed]
- González-Curbelo, M.Á.; Varela-Martínez, D.A.; Riaño-Herrera, D.A. Pesticide-Residue Analysis in Soils by the QuEChERS Method: A Review. Molecules 2022, 27, 4323. [Google Scholar] [CrossRef] [PubMed]
- Höhener, P.; Guers, D.; Malleret, L.; Boukaroum, O.; Martin-Laurent, F.; Masbou, J.; Payraudeau, S.; Imfeld, G. Multi-elemental compound-specific isotope analysis of pesticides for source identification and monitoring of degradation in soil: A review. Environ. Chem. Lett. 2022, 20, 3927–3942. [Google Scholar] [CrossRef]
- Cui, G.; Lartey-Young, G.; Chen, C.; Ma, L. Photodegradation of pesticides using compound-specific isotope analysis (CSIA): A review. RSC Adv. 2021, 11, 25122–25140. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, P.; Schmitt, A.-D.; Gangloff, S.; Masbou, J.; Imfeld, G. Plants affect the dissipation and leaching of anilide pesticides in soil mesocosms: Insights from compound-specific isotope analysis (CSIA). Agric. Ecosyst. Environ. 2021, 308, 107257. [Google Scholar] [CrossRef]
- Shen, X.; Xu, Z.; Zhang, X.; Yang, F. Stable carbon isotope fractionation during the biodegradation of lambda-cyhalothrin. Sci. Total Environ. 2015, 532, 415–419. [Google Scholar] [CrossRef]
- Xu, Z.; Liu, W.; Yang, F. A new approach to estimate bioavailability of pyrethroids in soil by compound-specific stable isotope analysis. J. Hazard. Mater. 2018, 349, 1–9. [Google Scholar] [CrossRef]
- Elsner, M.; Imfeld, G. Compound-specific isotope analysis (CSIA) of micropollutants in the environment—Current developments and future challenges. Curr. Opin. Biotechnol. 2016, 41, 60–72. [Google Scholar] [CrossRef]
- Torrentó, C.; Bakkour, R.; Glauser, G.; Melsbach, A.; Ponsin, V.; Hofstetter, T.B.; Elsner, M.; Hunkeler, D. Solid-phase extraction method for stable isotope analysis of pesticides from large volume environmental water samples. Analyst 2019, 144, 2898–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashima, H.; Katayama, Y. Source Evaluation of Diazinon Using Stable Carbon Isotope Ratio. Environ. Forensics 2010, 11, 363–371. [Google Scholar] [CrossRef]
- Melsbach, A.; Ponsin, V.; Torrentó, C.; Lihl, C.; Hofstetter, T.B.; Hunkeler, D.; Elsner, M. 13C- and 15N-Isotope Analysis of Desphenylchloridazon by Liquid Chromatography–Isotope-Ratio Mass Spectrometry and Derivatization Gas Chromatography–Isotope-Ratio Mass Spectrometry. Anal. Chem. 2019, 91, 3412–3420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogusu, E.O.; Wolbert, J.B.; Kujawinski, D.M.; Jochmann, M.A.; Elsner, M. Dual element (15N/14N, 13C/12C) isotope analysis of glyphosate and AMPA by derivatization-gas chromatography isotope ratio mass spectrometry (GC/IRMS) combined with LC/IRMS. Anal. Bioanal. Chem. 2015, 407, 5249–5260. [Google Scholar] [CrossRef]
- Schürner, H.K.V.; Seffernick, J.L.; Grzybkowska, A.; Dybala-Defratyka, A.; Wackett, L.P.; Elsner, M. Characteristic Isotope Fractionation Patterns in s-Triazine Degradation Have Their Origin in Multiple Protonation Options in the s-Triazine Hydrolase TrzN. Environ. Sci. Technol. 2015, 49, 3490–3498. [Google Scholar] [CrossRef]
- Isaji, Y.; Ogawa, N.O.; Boreham, C.J.; Kashiyama, Y.; Ohkouchi, N. Evaluation of δ13C and δ15N Uncertainties Associated with the Compound-Specific Isotope Analysis of Geoporphyrins. Anal. Chem. 2020, 92, 3152–3160. [Google Scholar] [CrossRef]
- Phillips, A.A.; Wu, F.; Sessions, A.L. Sulfur isotope analysis of cysteine and methionine via preparatory liquid chromatography and elemental analyzer isotope ratio mass spectrometry. Rapid Commun. Mass Spectrom. 2021, 35, e9007. [Google Scholar] [CrossRef] [PubMed]
- Broek, T.A.B.; Walker, B.D.; Andreasen, D.H.; McCarthy, M.D. High-precision measurement of phenylalanine δ15N values for environmental samples: A new approach coupling high-pressure liquid chromatography purification and elemental analyzer isotope ratio mass spectrometry. Rapid Commun. Mass Spectrom. 2013, 27, 2327–2337. [Google Scholar] [CrossRef]
- Ogawa, N.O.; Nagata, T.; Kitazato, H.; Ohkouchi, N. Ultra-sensitive elemental analyzer/isotope ratio mass spectrometer for stable nitrogen and carbon isotope analyse. In Earth, Life, and Isotopes; Ohkouchi, N., Tayasu, I., Koba, K., Eds.; Kyoto University Press: Kyoto, Japan, 2010; pp. 339–353. [Google Scholar]
- Wang, D.; Lv, W.; Yuan, Y.; Zhang, T.; Teng, H.; Losey, J.E.; Chang, X. Mechanism of the different metabolome responses between Plutella xylostella and Pieris rapae treated with the diamide insecticides. Ecotoxicol. Environ. Saf. 2020, 203, 111033. [Google Scholar] [CrossRef]
- Lee, J.W.; Kim, J.Y.; Kim, H.G.; Hur, K.J.; Kwon, C.H.; Hur, J.H. Residual Characteristics of Bistrifluron and Chlorantraniliprole in Strawberry (Fragaria ananassa Duch.) for Establishing Pre-Harvest Residue Limit. Korean J. Environ. Agriculture. Korean Soc. Environ. Agric. 2017, 36, 57–62. [Google Scholar] [CrossRef]
- Zhang, J.-M.; Chai, W.-G.; Wu, Y.-L. Residues of chlorantraniliprole in rice field ecosystem. Chemosphere 2012, 87, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Lee, S.W.; Lim, D.; Kim, S.W.; Kim, I.S. Evaluation of Cyantraniliprole Residues Translocated by Lettuce, Spinach and Radish. Korean J. Environ. Agric. 2021, 40, 335–344. [Google Scholar] [CrossRef]
- Park, J.-E.; Kwon, H.; Back, S.-J.; Ro, J.-H.; Kim, D.B.; Lee, H.-S.; Jin, Y. Development of multi-residue analytical method using QuEChERS and LC-MS/MS for 36 newly registered pesticides in Korea for major agricultural export commodities. Korean J. Pestic. Sci. 2018, 22, 378–392. [Google Scholar] [CrossRef]
- Yoon, J.; Lim, D.; Lee, S.; Kim, I. Uptake of soil-residual diazinon by rotational lettuce under greenhouse conditions. Foods 2022, 11, 3510. [Google Scholar] [CrossRef]
- Mei, J.; Ge, Q.; Han, L.; Zhang, H.; Long, Z.; Cui, Y.; Hua, R.; Yu, Y.; Fang, H. Deposition, Distribution, Metabolism, and Reduced Application Dose of Thiamethoxam in a Pepper-Planted Ecosystem. J. Agric. Food Chem. 2019, 67, 11848–11859. [Google Scholar] [CrossRef]
- Melsbach, A.; Torrentó, C.; Ponsin, V.; Bolotin, J.; Lachat, L.; Prasuhn, V.; Hofstetter, T.B.; Hunkeler, D.; Elsner, M. Dual-Element Isotope Analysis of Desphenylchloridazon to Investigate Its Environmental Fate in a Systematic Field Study: A Long-Term Lysimeter Experiment. Environ. Sci. Technol. 2020, 54, 3929–3939. [Google Scholar] [CrossRef]
- Lee, J.; Jung, M.W.; Lee, J.; Lee, J.; Shin, Y.; Kim, J.-H. Dissipation of the Insecticide Cyantraniliprole and Its Metabolite IN-J9Z38 in Proso Millet during Cultivation. Sci. Rep. 2019, 9, 11648. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.C.; Zwank, L.; Elsner, M.; Berg, M.; Meckenstock, R.U.; Haderlein, S.B. Compound-specific stable isotope analysis of organic contaminants in natural environments: A critical review of the state of the art, prospects, and future challenges. Anal. Bioanal. Chem. 2004, 378, 283–300. [Google Scholar]
- Droz, B.; Drouin, G.; Maurer, L.; Villette, C.; Payraudeau, S.; Imfeld, G. Phase Transfer and Biodegradation of Pesticides in Water–Sediment Systems Explored by Compound-Specific Isotope Analysis and Conceptual Modeling. Environ. Sci. Technol. 2021, 55, 4720–4728. [Google Scholar] [CrossRef]
- Limon, A.W.; Moingt, M.; Widory, D. The carbon stable isotope compositions of glyphosate and aminomethylphosphonic acid (AMPA): Improved analytical sensitivity and first application to environmental water matrices. Rapid Commun. Mass Spectrom. 2021, 35, e9017. [Google Scholar] [CrossRef]
- Kuntze, K.; Eisenmann, H.; Richnow, H.-H.; Fischer, A. Compound-Specific Stable Isotope Analysis (CSIA) for Evaluating Degradation of Organic Pollutants: An Overview of Field Case Studies. In Anaerobic Utilization of Hydrocarbons, Oils, and Lipids; Boll, M., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 323–360. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soil | Crop | ||||||
|---|---|---|---|---|---|---|---|
| δ13C | Estimated C (mg/capsule) | Amount (mg/g) | N | δ13C | Estimated C (mg/capsule) | N | |
| (a) Chlorantraniliprole | |||||||
| Initial | −29.90 ± 0.02 | 0.176 ± 0.010 | 0.5 * | 2 | Not applied | ||
| 10d | −29.24 ± 0.17 | 0.171 ± 0.004 | 0.337 | 3 | −30.35 ± 0.06 | 0.268 ± 0.032 | 3 |
| 20d | −28.37 ± 0.13 | 0.222 ± 0.031 | 0.370 | 3 | −30.27 ± 0.50 | 0.206 ± 0.017 | 3 |
| 30d | −28.85 ± 0.10 | 0.093 ± 0.012 | 0.334 | 3 | −30.56 ± 0.40 | 0.085 ± 0.010 | 5 |
| 45d | −28.24 ± 0.28 | 0.110 ± 0.018 | 0.254 | 3 | −31.11 ± 0.10 | 0.130 ± 0.037 | 3 |
| Overall | −28.92 | 0.154 | −30.57 | ||||
| (b) Cyantraniliprole | |||||||
| Initial | −29.42 ± 0.27 | 0.159 ± 0.023 | 1 * | 3 | Not applied | 1 | |
| 10d | −28.74 ± 0.17 | 0.212 ± 0.019 | 0.93 | 3 | −29.03 | 0.043 | 1 |
| 20d | −29.15 ± 0.05 | 0.173 ± 0.051 | 0.97 | 3 | −28.79 | 0.042 | 1 |
| 30d | −28.28 ± 0.58 | 0.142 ± 0.013 | 0.81 | 3 | −28.54 | 0.070 | 1 |
| 45d | −28.80 ± 0.56 | 0.143 ± 0.026 | 0.80 | 3 | −28.45 | 0.047 | 1 |
| Overall | −28.88 | 0.166 | −28.70 | ||||
| HPLC System | Cyantraniliprole | Chlorantraniliprole |
|---|---|---|
| Column | Zorbax Eclips XDB C18 column (4.6 × 250 mm, 5 µm) | Zorbax Eclips XDB C18 column (4.6 × 250 mm, 5 µm) |
| Mobile phase (duration time) | 45% Acetonitrile in water (15 min) | 55% Acetonitrile in water (15 min) |
| Flow rate | 1 mL/min | 1 mL/min |
| Column temperature | 30 °C | 30 °C |
| Detection | 264 nm | 254 nm |
| Injected volume | 0.05mL to 0.50 mL | 0.05mL to 0.50 mL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, H.Y.; Won, E.-J.; Choi, J.; Cho, Y.; Lim, D.-J.; Kim, I.-S.; Shin, K.-H. Stable Isotope Analysis of Residual Pesticides via High Performance Liquid Chromatography and Elemental Analyzer–Isotope Ratio Mass Spectrometry. Molecules 2022, 27, 8587. https://doi.org/10.3390/molecules27238587
Yun HY, Won E-J, Choi J, Cho Y, Lim D-J, Kim I-S, Shin K-H. Stable Isotope Analysis of Residual Pesticides via High Performance Liquid Chromatography and Elemental Analyzer–Isotope Ratio Mass Spectrometry. Molecules. 2022; 27(23):8587. https://doi.org/10.3390/molecules27238587
Chicago/Turabian StyleYun, Hee Young, Eun-Ji Won, Jisoo Choi, Yusang Cho, Da-Jung Lim, In-Seon Kim, and Kyung-Hoon Shin. 2022. "Stable Isotope Analysis of Residual Pesticides via High Performance Liquid Chromatography and Elemental Analyzer–Isotope Ratio Mass Spectrometry" Molecules 27, no. 23: 8587. https://doi.org/10.3390/molecules27238587
APA StyleYun, H. Y., Won, E. -J., Choi, J., Cho, Y., Lim, D. -J., Kim, I. -S., & Shin, K. -H. (2022). Stable Isotope Analysis of Residual Pesticides via High Performance Liquid Chromatography and Elemental Analyzer–Isotope Ratio Mass Spectrometry. Molecules, 27(23), 8587. https://doi.org/10.3390/molecules27238587