
Fisetin as a Senotherapeutic Agent: Biopharmaceutical Properties and Crosstalk between Cell Senescence and Neuroprotection

 ,
, 

 and
and 
Abstract
:1. Introduction
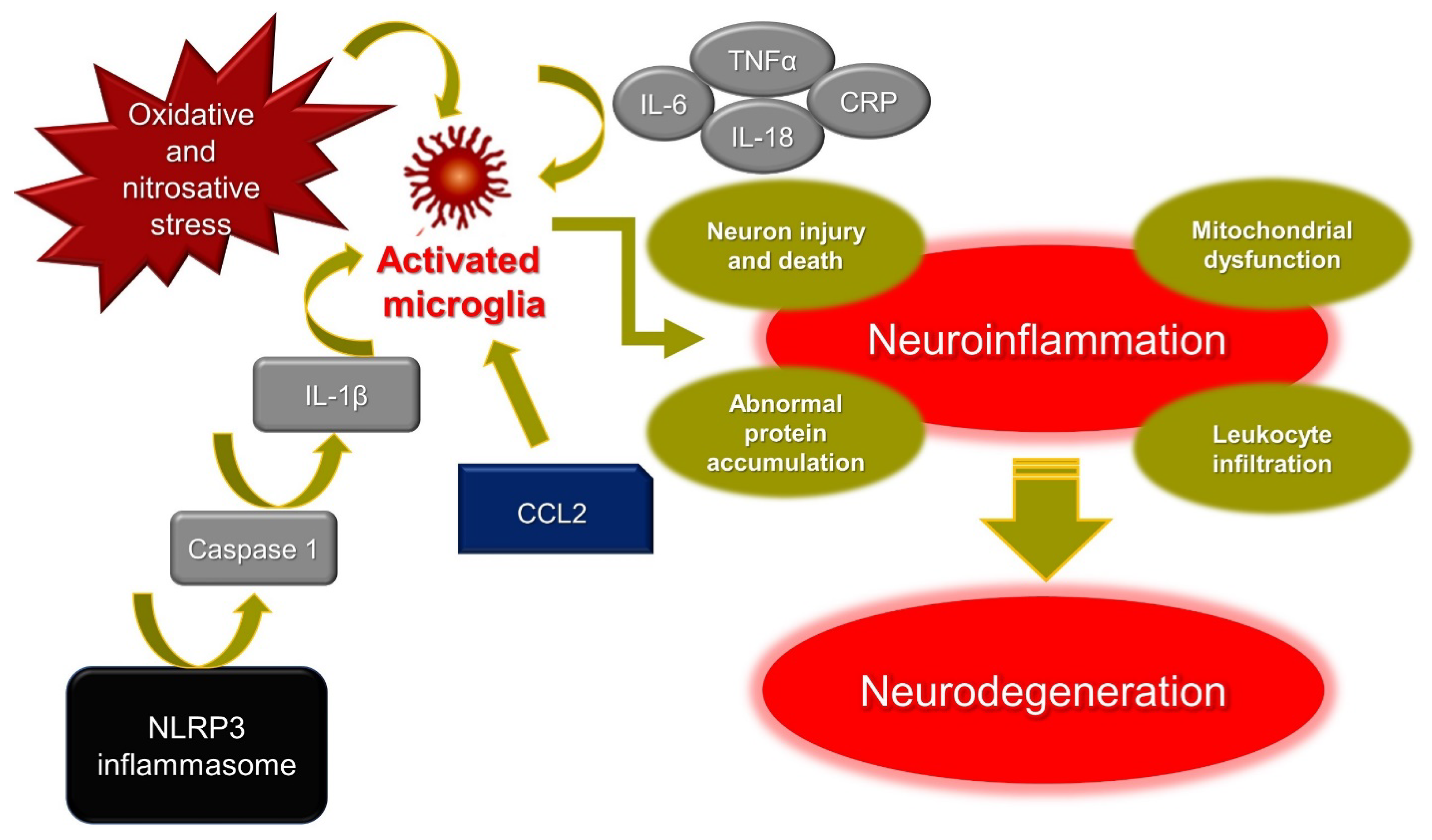
2. Crossroads of Neuroinflammation and Neurodegenerative Disorders
2.1. Neurodegenerative Disorders
2.2. The Interplay between Neuroinflammation and Neurodegeneration
3. Fisetin as a Senolytic Drug: Crosstalk between Neuroprotection and Senescence
3.1. Senescence Mechanisms
3.2. CNS Senescence
3.2.1. Neurons
3.2.2. Microglia
3.2.3. Astrocytes
3.3. Senolytic Drugs
3.4. Fisetin as Senolytic Drug in the CNS
4. Fisetin: In Silico Evaluation of Pharmacodynamics, Pharmacokinetics, and Toxicity
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Martel, J.; Ojcius, D.; Ko, Y.-F.; Ke, P.-Y.; Wu, C.-Y.; Peng, H.-H.; Young, J.D. Hormetic Effects of Phytochemicals on Health and Longevity. Trends Endocrinol. Metab. 2019, 30, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Maher, P. The Potential of Flavonoids for the Treatment of Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grynkiewicz, G.; Demchuk, O.M. New Perspectives for Fisetin. Front. Chem. 2019, 7, 697. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, D.; Sharma, A.; Sak, K.; Tuli, H.S.; Buttar, H.S.; Bishayee, A. Fisetin: A bioactive phytochemical with potential for cancer prevention and pharmacotherapy. Life Sci. 2018, 194, 75–87. [Google Scholar] [CrossRef]
- Ravula, A.R.; Teegala, S.B.; Kalakotla, S.; Pasangulapati, J.P.; Perumal, V.; Boyina, H.K. Fisetin, potential flavonoid with multifarious targets for treating neurological disorders: An updated review. Eur. J. Pharmacol. 2021, 910, 174492. [Google Scholar] [CrossRef]
- Maher, P. Preventing and Treating Neurological Disorders with the Flavonol Fisetin. Brain Plast. 2020, 6, 155–166. [Google Scholar] [CrossRef]
- Yen, F.S.; Qin, C.S.; Xuan, S.T.S.; Ying, P.J.; Le, H.Y.; Darmarajan, T.; Gunasekaran, B.; Salvamani, S. Hypoglycemic Effects of Plant Flavonoids: A Review. Evid. -Based Complement. Altern. Med. 2021, 2021, 2057333. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Roda, A.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L.; Villegas, S. Amyloid-beta peptide and tau protein crosstalk in Alzheimer′s disease. Neural Regen. Res. 2022, 17, 1666. [Google Scholar] [CrossRef]
- Jankovic, J.; Tan, E.K. Parkinson’s disease: Etiopathogenesis and treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef]
- Carradori, S.; D’Ascenzio, M.; Chimenti, P.; Secci, D.; Bolasco, A. Selective MAO-B inhibitors: A lesson from natural products. Mol. Divers. 2014, 18, 219–243. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Disabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef] [Green Version]
- Zengeler, K.E.; Lukens, J.R. Innate immunity at the crossroads of healthy brain maturation and neurodevelopmental disorders. Nat. Rev. Immunol. 2021, 21, 454–468. [Google Scholar] [CrossRef]
- McKim, D.B.; Weber, M.D.; Niraula, A.; Sawicki, C.M.; Liu, X.; Jarrett, B.L.; Ramirez-Chan, K.; Wang, Y.; Roeth, R.M.; Sucaldito, A.D.; et al. Microglial recruitment of IL-1β-producing monocytes to brain endothelium causes stress-induced anxiety. Mol. Psychiatry 2018, 23, 1421–1431. [Google Scholar] [CrossRef] [Green Version]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; Van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Ng, A.; Tam, W.W.; Zhang, M.W.; Ho, C.S.; Husain, S.F.; McIntyre, R.S.; Ho, R.C. IL-1β, IL-6, TNF- α and CRP in Elderly Patients with Depression or Alzheimer’s disease: Systematic Review and Meta-Analysis. Sci. Rep. 2018, 8, 12050. [Google Scholar] [CrossRef]
- Yan, Y.-Q.; Fang, Y.; Zheng, R.; Pu, J.-L.; Zhang, B.-R. NLRP3 Inflammasomes in Parkinson’s disease and their Regulation by Parkin. Neuroscience 2020, 446, 323–334. [Google Scholar] [CrossRef]
- Curzytek, K.; Leśkiewicz, M. Targeting the CCL2-CCR2 axis in depressive disorders. Pharmacol. Rep. 2021, 73, 1052–1062. [Google Scholar] [CrossRef]
- Murcia, J.D.G.; Weinert, A.; Freitas, C.M.T.; Arens, D.K.; Ferrel, M.N.; Grose, J.H.; Ridge, P.G.; Wilson, E.; Kauwe, J.S.K.; Weber, K.S. Atypical chemokine receptor ACKR2-V41A has decreased CCL2 binding, scavenging, and activation, supporting sustained inflammation and increased Alzheimer’s disease risk. Sci. Rep. 2020, 10, 8019. [Google Scholar] [CrossRef]
- Nunes, C.; Laranjinha, J. Nitric oxide and dopamine metabolism converge via mitochondrial dysfunction in the mechanisms of neurodegeneration in Parkinson’s disease. Arch. Biochem. Biophys. 2021, 704, 108877. [Google Scholar] [CrossRef]
- Dubey, H.; Gulati, K.; Ray, A. Alzheimer’s Disease: A Contextual Link with Nitric Oxide Synthase. Curr. Mol. Med. 2020, 20, 505–515. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Gruendler, R.; Hippe, B.; Jengic, V.S.; Peterlin, B.; Haslberger, A.G. Nutraceutical Approaches of Autophagy and Neuroinflammation in Alzheimer’s Disease: A Systematic Review. Molecules 2020, 25, 6018. [Google Scholar] [CrossRef]
- Spagnuolo, C.; Moccia, S.; Russo, G.L. Anti-inflammatory effects of flavonoids in neurodegenerative disorders. Eur. J. Med. Chem. 2018, 153, 105–115. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Braidy, N.; Habtemariam, S.; Sureda, A.; Manayi, A.; Nabavi, S.M. Neuroprotective Effects of Fisetin in Alzheimer’s and Parkinson’s Diseases: From Chemistry to Medicine. Curr. Top. Med. Chem. 2016, 16, 1910–1915. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. eBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, J.L. Translating advances from the basic biology of aging into clinical application. Exp. Gerontol. 2013, 48, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.A. Extending Life: Scientific Prospects and Political Obstacles. Milbank Q. 2002, 80, 155–174. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, E.O.W.; Zhu, Y.; Tchkonia, T.; Kirkland, J.L. Discovery, development, and future application of senolytics: Theories and predictions. FEBS J. 2020, 287, 2418–2427. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.-P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of Life-Span by Introduction of Telomerase into Normal Human Cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Alimonti, A.; Nardella, C.; Chen, Z.; Clohessy, J.; Carracedo, A.; Trotman, L.C.; Cheng, K.; Varmeh, S.; Kozma, S.C.; Thomas, G.; et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J. Clin. Investig. 2010, 120, 681–693. [Google Scholar] [CrossRef]
- Baker, D.J.; Perez-Terzic, C.; Jin, F.; Pitel, K.S.; Niederländer, N.J.; Jeganathan, K.; Yamada, S.; Reyes, S.; Rowe, L.; Hiddinga, H.J.; et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat. Cell Biol. 2008, 10, 825–836. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Romanov, V.S.; Abramova, M.V.; Svetlikova, S.B.; Bykova, T.V.; Zubova, S.G.; Aksenov, N.D.; Fornace, A.J.; Pospelova, T.V.; Pospelov, V.A. p21Waf1is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle 2010, 9, 3945–3955. [Google Scholar] [CrossRef] [Green Version]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef]
- von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eischen, C.M.; Lozano, G. The Mdm network and its regulation of p53 activities: A rheostat of cancer risk. Hum. Mutat. 2014, 35, 728–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Wei, W.; Dutriaux, A.; Jobling, W.A.; Sedivy, J.M. Real-time imaging of transcriptional activation in live cells reveals rapid up-regulation of the cyclin-dependent kinase inhibitor gene CDKN1A in replicative cellular senescence. Aging Cell 2003, 2, 295–304. [Google Scholar] [CrossRef]
- Wong, E.S.M.; Le Guezennec, X.; Demidov, O.; Marshall, N.T.; Wang, S.T.; Krishnamurthy, J.; Sharpless, N.; Dunn, N.R.; Bulavin, D.V. p38MAPK Controls Expression of Multiple Cell Cycle Inhibitors and Islet Proliferation with Advancing Age. Dev. Cell 2009, 17, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Ohtani, N.; Zebedee, Z.; Huot, T.J.G.; Stinson, J.A.; Sugimoto, M.; Ohashi, Y.; Sharrocks, A.D.; Peters, G.; Hara, E. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature 2001, 409, 1067–1070. [Google Scholar] [CrossRef]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Tavana, O.; Benjamin, C.L.; Puebla-Osorio, N.; Sang, M.; Ullrich, S.E.; Ananthaswamy, H.; Zhu, C. Absence of p53-dependent apoptosis leads to UV radiation hypersensitivity, enhanced immunosuppression and cellular senescence. Cell Cycle 2010, 9, 3348–3356. [Google Scholar] [CrossRef] [Green Version]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forero, D.A.; González-Giraldo, Y.; López-Quintero, C.; Castro-Vega, L.J.; Barreto, G.E.; Perry, G. Meta-analysis of Telomere Length in Alzheimer’s Disease. J. Gerontol. Ser. A 2016, 71, 1069–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forero, D.A.; Gonzalez, Y.; López-Quintero, C.; Castro-Vega, L.J.; Barreto, G.E.; Perry, G. Telomere length in Parkinson’s disease: A meta-analysis. Exp. Gerontol. 2016, 75, 53–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; DeMaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Doorn, K.J.; Lucassen, P.J.; Boddeke, H.W.; Prins, M.; Berendse, H.W.; Drukarch, B.; van Dam, A.-M. Emerging roles of microglial activation and non-motor symptoms in Parkinson’s disease. Prog. Neurobiol. 2012, 98, 222–238. [Google Scholar] [CrossRef]
- Bachstetter, A.D.; Xing, B.; de Almeida, L.; Dimayuga, E.R.; Watterson, D.M.; Van Eldik, L.J. Microglial p38α MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Aβ). J. Neuroinflammation 2011, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Sierra, A.; Gottfried-Blackmore, A.C.; McEwen, B.S.; Bulloch, K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 2007, 55, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Flanary, B.E.; Sammons, N.W.; Nguyen, C.; Walker, D.; Streit, W.J. Evidence That Aging and Amyloid Promote Microglial Cell Senescence. Rejuvenation Res. 2007, 10, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.-M.; Zhao, Y.-M.; Luo, X.; Feng, Y.; Ren, Y.; Shang, H.; He, Z.-Y.; Luo, X.M.; Chen, S.-D.; Wang, X.-Y. Repeated Lipopolysaccharide Stimulation Induces Cellular Senescence in BV2 Cells. Neuroimmunomodulation 2012, 19, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Tichauer, J.E.; Flores, B.; Soler, B.; Bernhardi, L.E.-V.; Ramírez, G.; Von Bernhardi, R. Age-dependent changes on TGFβ1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav. Immun. 2014, 37, 187–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.-M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [Green Version]
- Laberge, R.-M.; Adler, D.; DeMaria, M.; Mechtouf, N.; Teachenor, R.; Cardin, G.B.; Desprez, P.-Y.; Campisi, J.; Rodier, F. Mitochondrial DNA damage induces apoptosis in senescent cells. Cell Death Dis. 2013, 4, e727. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Jeon, O.H.; Kim, C.; Laberge, R.-M.; DeMaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef]
- Zhu, Y.; Doornebal, E.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.; Robbins, P.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.-M.; Marquess, D.; Dananberg, J.; Van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; DeMaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Luna-Vargas, M.P.; Chipuk, J.E. Physiological and Pharmacological Control of BAK, BAX, and Beyond. Trends Cell Biol. 2016, 26, 906–917. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Singh, A.K.; Garg, G.; Rizvi, S.I. Fisetin as a caloric restriction mimetic protects rat brain against aging induced oxidative stress, apoptosis and neurodegeneration. Life Sci. 2018, 193, 171–179. [Google Scholar] [CrossRef]
- Cho, I.; Song, H.; Cho, J.H. Flavonoids mitigate neurodegeneration in agedCaenorhabditis elegansby mitochondrial uncoupling. Food Sci. Nutr. 2020, 8, 6633–6642. [Google Scholar] [CrossRef]
- Chen, C.; Yao, L.; Cui, J.; Liu, B. Fisetin Protects against Intracerebral Hemorrhage-Induced Neuroinflammation in Aged Mice. Cerebrovasc. Dis. 2018, 45, 154–161. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Evans, S.A.; Fielder, E.; Victorelli, S.; Kruger, P.; Salmonowicz, H.; Weigand, B.M.; Patel, A.D.; Pirtskhalava, T.; Inman, C.L.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, 20, e13296. [Google Scholar] [CrossRef]
- Das, J.; Singh, R.; Ladol, S.; Nayak, S.K.; Sharma, D. Fisetin prevents the aging-associated decline in relative spectral power of α, β and linked MUA in the cortex and behavioral alterations. Exp. Gerontol. 2020, 138, 111006. [Google Scholar] [CrossRef]
- Currais, A.; Farrokhi, C.; Dargusch, R.; Armando, A.; Quehenberger, O.; Schubert, D.; Maher, P. Fisetin Reduces the Impact of Aging on Behavior and Physiology in the Rapidly Aging SAMP8 Mouse. J. Gerontol. Ser. A Boil. Sci. Med Sci. 2018, 73, 299–307. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, A.; Ali, W.; Jo, M.H.; Park, J.; Ikram, M.; Kim, M.O. Fisetin Rescues the Mice Brains Against D-Galactose-Induced Oxidative Stress, Neuroinflammation and Memory Impairment. Front. Pharmacol. 2021, 12, 57. [Google Scholar] [CrossRef]
- Mohite, R.; Mehta, P.; Arulmozhi, S.; Kamble, R.; Pawar, A.; Bothiraja, C. Synthesis of fisetin co-crystals with caffeine and nicotinamide using the cooling crystallization technique: Biopharmaceutical studies. New J. Chem. 2019, 43, 13471–13479. [Google Scholar] [CrossRef]
- Herrero-Martinez, J.M.; Sanmartin, M.; Roses, M.; Bosch, E.; Ràfols, C. Determination of dissociation constants of flavonoids by capillary electrophoresis. Electrophoresis 2005, 26, 1886–1895. [Google Scholar] [CrossRef]
- Moridani, M.Y.; Galati, G.; O’Brien, P.J. Comparative quantitative structure toxicity relationships for flavonoids evaluated in isolated rat hepatocytes and HeLa tumor cells. Chem.-Biol. Interact. 2002, 139, 251–264. [Google Scholar] [CrossRef]
- Xiao, J.; Högger, P. Stability of Dietary Polyphenols under the Cell Culture Conditions: Avoiding Erroneous Conclusions. J. Agric. Food Chem. 2015, 63, 1547–1557. [Google Scholar] [CrossRef]
- Bothiraja, C.; Yojana, B.D.; Pawar, A.P.; Shaikh, K.; Thorat, U.H. Fisetin-loaded nanocochleates: Formulation, characterisation, in vitroanticancer testing, bioavailability and biodistribution study. Expert Opin. Drug Deliv. 2013, 11, 17–29. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Cruz, N.; Medina-Franco, J.L. Epigenetic Target Profiler: A Web Server to Predict Epigenetic Targets of Small Molecules. J. Chem. Inf. Model. 2021, 61, 1550–1554. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kops, C.D.B.; Šícho, M.; Mazzolari, A.; Kirchmair, J. GLORYx: Prediction of the Metabolites Resulting from Phase 1 and Phase 2 Biotransformations of Xenobiotics. Chem. Res. Toxicol. 2021, 34, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Ai, H.; Chen, W.; Yin, Z.; Hu, H.; Zhu, J.; Zhao, J.; Zhao, Q.; Liu, H. CarcinoPred-EL: Novel models for predicting the carcinogenicity of chemicals using molecular fingerprints and ensemble learning methods. Sci. Rep. 2017, 7, 2118. [Google Scholar] [CrossRef] [PubMed]
- Moolakkadath, T.; Aqil, M.; Ahad, A.; Imam, S.S.; Praveen, A.; Sultana, Y.; Mujeeb, M. Preparation and optimization of fisetin loaded glycerol based soft nanovesicles by Box-Behnken design. Int. J. Pharm. 2020, 578, 119125. [Google Scholar] [CrossRef]
- Sengupta, B.; Coleman, J.; Johnson, J.; Feng, M. Graphene oxide as selective transporter of flavonols for physiological target DNA: A two-color fluorescence approach. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 214, 192–198. [Google Scholar] [CrossRef]
- Pawar, A.; Singh, S.; Rajalakshmi, S.; Shaikh, K.; Bothiraja, C. Development of fisetin-loaded folate functionalized pluronic micelles for breast cancer targeting. Artif. Cells Nanomed. Biotechnol. 2018, 46, 347–361. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
 | ||
|---|---|---|
| Human Target | Probability Score (SwissTarget Prediction) | Target Also Confirmed by |
| Xanthine dehydrogenase | 1.0 | ZINC |
| Cyclin-dependent kinases 1, 2, 5 and 6 | 1.0 | ZINC and Epigenetic Target Profiler |
| Acetylcholinesterase | 1.0 | ChEMBL |
| Arachidonate 12 and 15-lipoxygenases | 1.0 | ZINC |
| Arginase-1 | 1.0 | |
| Multidrug resistance-associated protein 1 | 0.84 | |
| NADPH oxidase 4 | 0.73 | |
| Aldose reductase | 0.73 | |
| Tyrosine-protein kinase receptor | 0.73 | |
| Carbonic anhydrases II, VI, VII and XII | 0.73 | ChEMBL |
| Arachidonate 5-lipoxygenase | 0.73 | |
| Estradiol 17β-dehydrogenase 2 | 0.73 | ChEMBL |
| P-glycoprotein 1 | 0.73 | |
| Cytochrome P450 1B1 | 0.73 | |
| ATP-binding cassette sub-family G member 2 | 0.73 | |
| Monoamine oxidase A | 0.73 | ChEMBL |
| Adenosine A1 receptor | 0.73 | |
| Glyoxalase I | 0.73 | |
| Tyrosine-protein kinase SYK | 0.73 | |
| Glycogen synthase kinase-3β | 0.73 | |
| Matrix metalloproteinases 2 and 9 | 0.73 | |
| Parameter | Fisetin | Online Tool | |
|---|---|---|---|
| Molecular properties | Molecular weight | 286.24 | SwissADME, PROTOX-II, and OSIRIS Property Explorer |
| Consensus LogP | 1.55 | ||
| N° of H-bond acceptors | 6 | ||
| N° of H-bond donors | 4 | ||
| N° of heavy atoms | 21 | ||
| N° of aromatic heavy atoms | 16 | ||
| Fraction Csp3 | 0.00 | ||
| Molar Refractivity | 76.01 | ||
| Topological Polar Surface Area | 107.22–111.13 Å2 | ||
| Lead likeness | Yes | ||
| Pan Assay Interference Structures or Structural Alert | yes (catechol) | ||
| N° rotatable bonds | 1 | ||
| Absorption | Water solubility (log mol/L) | −3.153 | pkCSM |
| Caco2 permeability expressed as log Papp in 10−6 cm/s | 0.716 | ||
| Intestinal absorption (human) (% adsorbed) | 85.46 79.43 (HIA) | pkCSM PreADMET | |
| Skin permeability (log Kp) | −6.65 cm/s −2.74 cm/s −4.33 cm/s | SwissADME pkCSM PreADMET | |
| P-glycoprotein substrate | no yes | SwissADME and PreADMET pkCSM | |
| P-glycoprotein I inhibitor | no | pkCSM | |
| P-glycoprotein II inhibitor | no | ||
| Gastrointestinal absorption | high | BOILED-Egg | |
| Distribution | Volume of distribution at steady state (VDss) (human) (log L/Kg) | 0.127 | pkCSM |
| Fraction unbound (human) | 0.045 | ||
| CNS permeability (log PS) | −2.417 | ||
| Blood–brain barrier (BBB) permeability (log BB) | −1.114 | ||
| BBB permeation | no | BOILED-Egg | |
| In vivo BBB penetration ([brain]/[blood]) | 0.32 (moderate absorption if in the range 0.1–2.0) | PreADMET | |
| Caco2 permeability | 9.57 | ||
| Pure water solubility (mg/L) | 63.3725 | ||
| Plasma protein binding (PPB) | 88.73% (weakly bound if <90%) | ||
| Metabolism | CYP2D6 substrate | no | pkCSM and PreADMET |
| CYP3A4 substrate | no | pkCSM and PreADMET | |
| CYP1A2 inhibitor | yes | SwissADME and pkCSM | |
| CYP2C19 inhibitor | no yes | SwissADME and pkCSM PreADMET | |
| CYP2C9 inhibitor | no yes | SwissADME and pkCSM PreADMET | |
| CYP2D6 inhibitor | yes no | SwissADME pkCSM and PreADMET | |
| CYP3A4 inhibitor | yes | SwissADME, PreADMET and pkCSM | |
| Excretion | Total clearance (log mL/min/Kg) | 0.557 | pkCSM |
| Renal organic cationic transporter (OCT2) substrate | no | pkCSM | |
| Parameter/Target | Fisetin (Probability) | Online Tool |
|---|---|---|
| Predicted LD50 (mg/Kg) in rodents | 159 | ProTox-II |
| Predicted toxicity class (according to GHS) | 3 | |
| Hepatotoxicity | inactive (0.70) | |
| Carcinogenicity | active (0.71) | |
| Immunogenicity | inactive (0.51) | |
| Mutagenicity | inactive (0.53) | |
| Cytotoxicity | inactive (0.98) | |
| Aryl hydrocarbon receptor | active (0.84) | Nuclear receptor signaling and stress response pathways (ProTox-II) |
| Androgen receptor | inactive (0.99) | |
| Androgen receptor ligand-binding domain | inactive (0.72) | |
| Aromatase | inactive (0.88) | |
| Estrogen receptor α | active (0.69) | |
| Estrogen receptor ligand-binding domain | active (0.86) | |
| Peroxisome-proliferator activated receptor γ | inactive (0.98) | |
| Nrf2/ARE | inactive (0.98) | |
| Heat Shock Factor Response Element | inactive (0.98) | |
| Mitochondria Membrane Potential | active (0.82) | |
| p53 | inactive (0.97) | |
| ATPase family AAA domain-containing protein 5 | inactive (0.77) | |
| Carcinogenicity | no | CarcinoPred-EL |
| Mutagenic | yes | OSIRIS Property Explorer |
| Tumorigenic | yes | |
| Irritant | yes | |
| Reproductive effective | yes | |
| Ames test | mutagen non-mutagen (pkCSM) | PreADMET |
| Carcino_mouse | no | |
| Carcino_rat | yes | |
| Algae_at | 0.0495876 | |
| Daphnia_at | 0.200841 | |
| hERG inhibition | medium risk | |
| Medaka_at | 0.0667019 | |
| Minnow_at | 0.0294925 | |
| TA100_10RLI | no | |
| TA100_NA | yes | |
| TA1535_NA | no | |
| Max. tolerated dose (human) (log mg/Kg/day) | 0.973 | pkCSM |
| hERG I inhibitor | no | |
| hERG II inhibitor | yes | |
| Oral rat acute toxicity (LD50 mol/Kg) | 2.111 | |
| Oral rat chronic toxicity (NOAEL log mg/Kg_bw/day) | 3.014 | |
| Hepatotoxicity | no | |
| Skin sensitization | no | |
| T. pyriformis toxicity (log µg/L) | 0.341 | |
| Minnow toxicity (log mM) | 0.976 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsallabi, O.; Patruno, A.; Pesce, M.; Cataldi, A.; Carradori, S.; Gallorini, M. Fisetin as a Senotherapeutic Agent: Biopharmaceutical Properties and Crosstalk between Cell Senescence and Neuroprotection. Molecules 2022, 27, 738. https://doi.org/10.3390/molecules27030738
Elsallabi O, Patruno A, Pesce M, Cataldi A, Carradori S, Gallorini M. Fisetin as a Senotherapeutic Agent: Biopharmaceutical Properties and Crosstalk between Cell Senescence and Neuroprotection. Molecules. 2022; 27(3):738. https://doi.org/10.3390/molecules27030738
Chicago/Turabian StyleElsallabi, Osama, Antonia Patruno, Mirko Pesce, Amelia Cataldi, Simone Carradori, and Marialucia Gallorini. 2022. "Fisetin as a Senotherapeutic Agent: Biopharmaceutical Properties and Crosstalk between Cell Senescence and Neuroprotection" Molecules 27, no. 3: 738. https://doi.org/10.3390/molecules27030738
APA StyleElsallabi, O., Patruno, A., Pesce, M., Cataldi, A., Carradori, S., & Gallorini, M. (2022). Fisetin as a Senotherapeutic Agent: Biopharmaceutical Properties and Crosstalk between Cell Senescence and Neuroprotection. Molecules, 27(3), 738. https://doi.org/10.3390/molecules27030738