
Influence of Steric Effect on the Pseudo-Multicomponent Synthesis of N-Aroylmethyl-4-Arylimidazoles

 and
and
Abstract
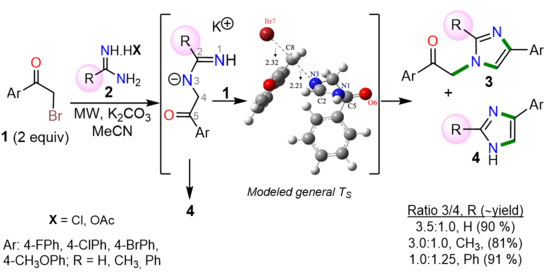
:
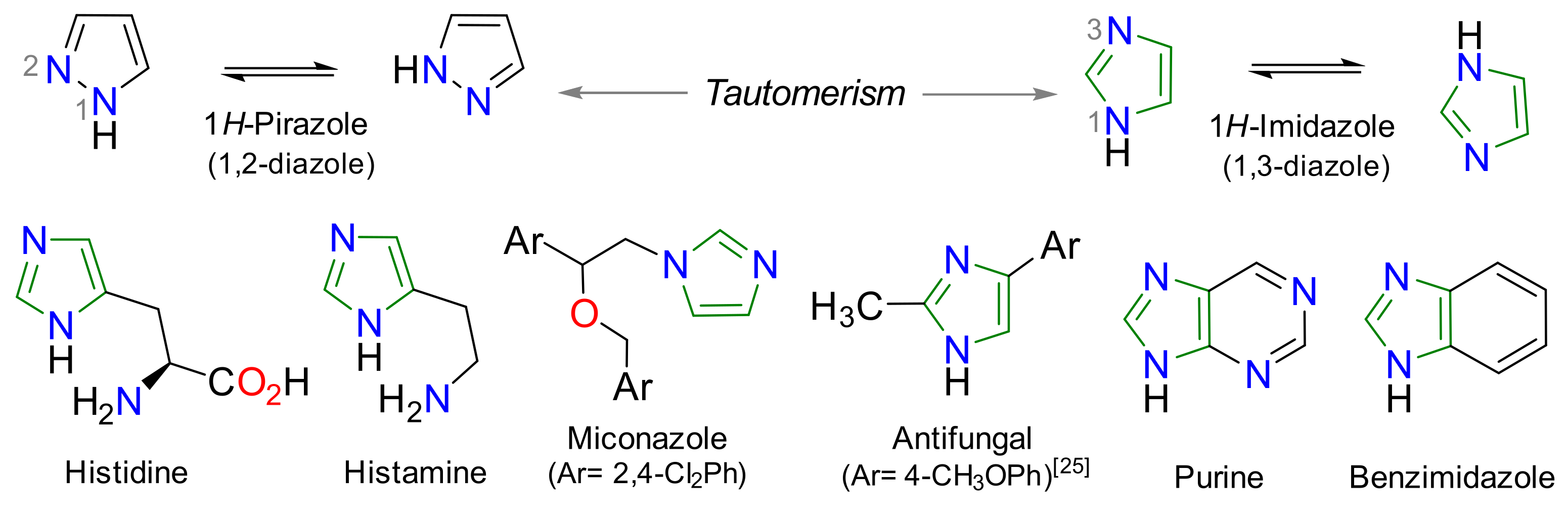
1. Introduction
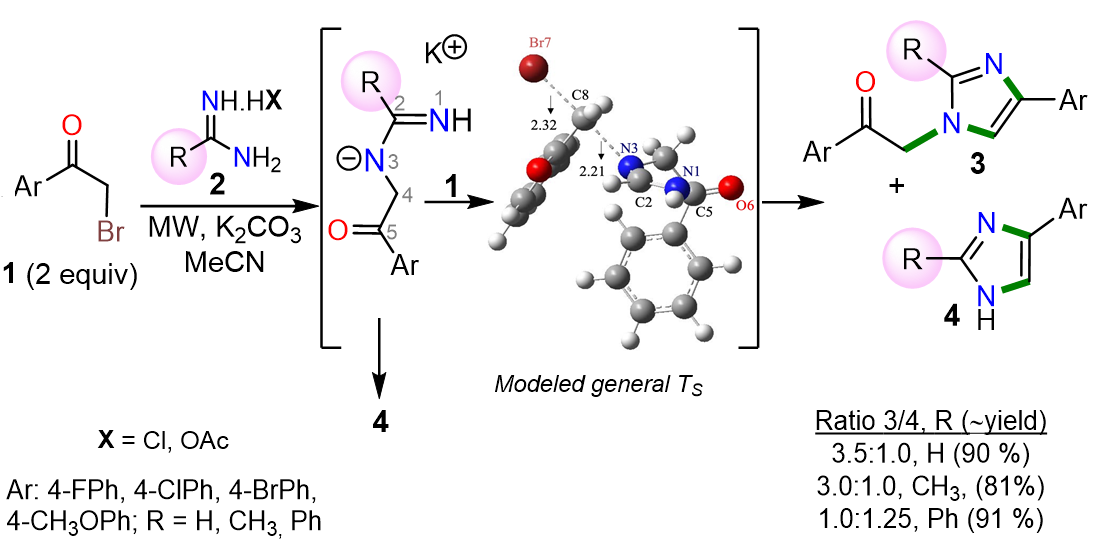
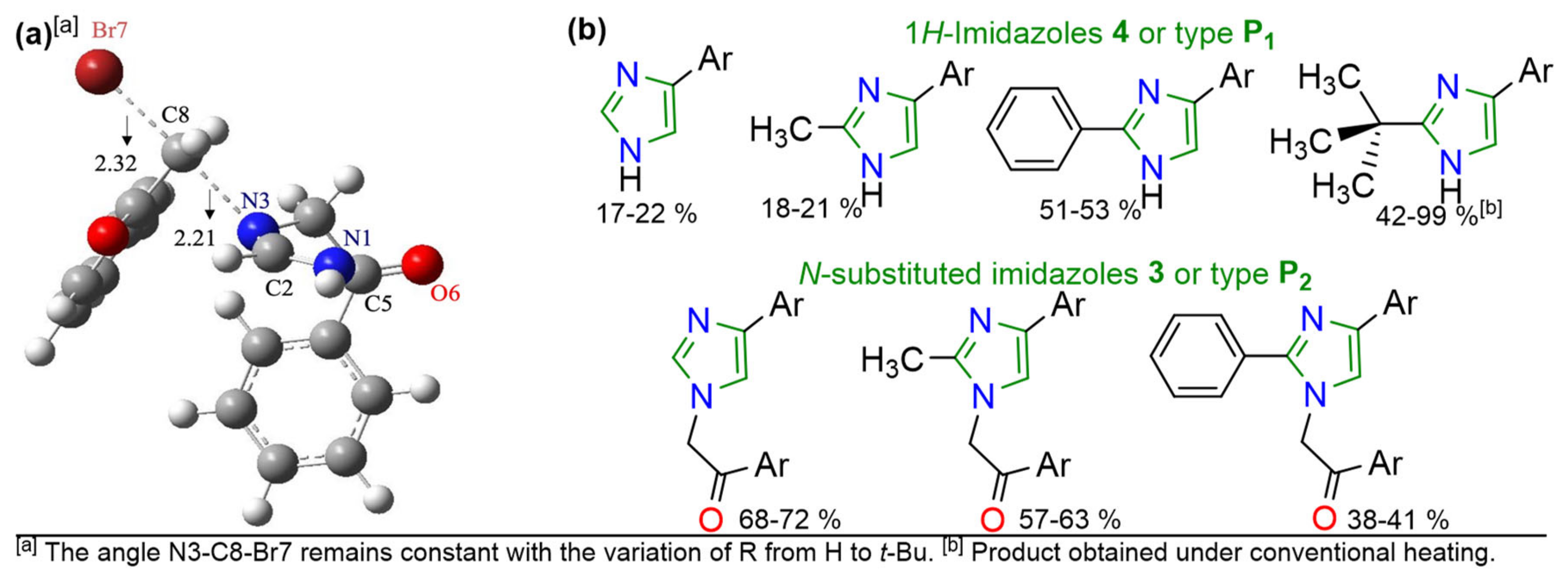
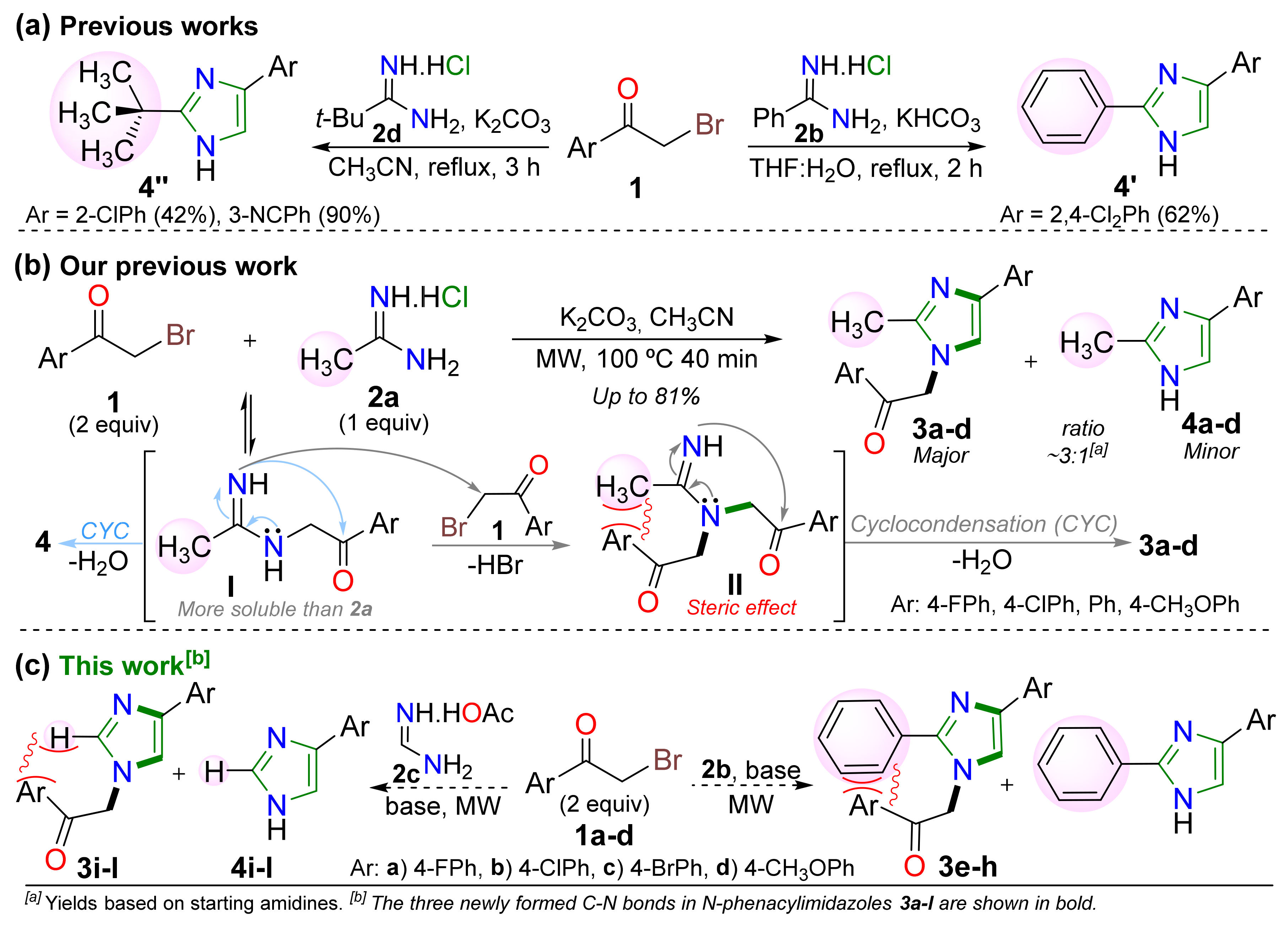
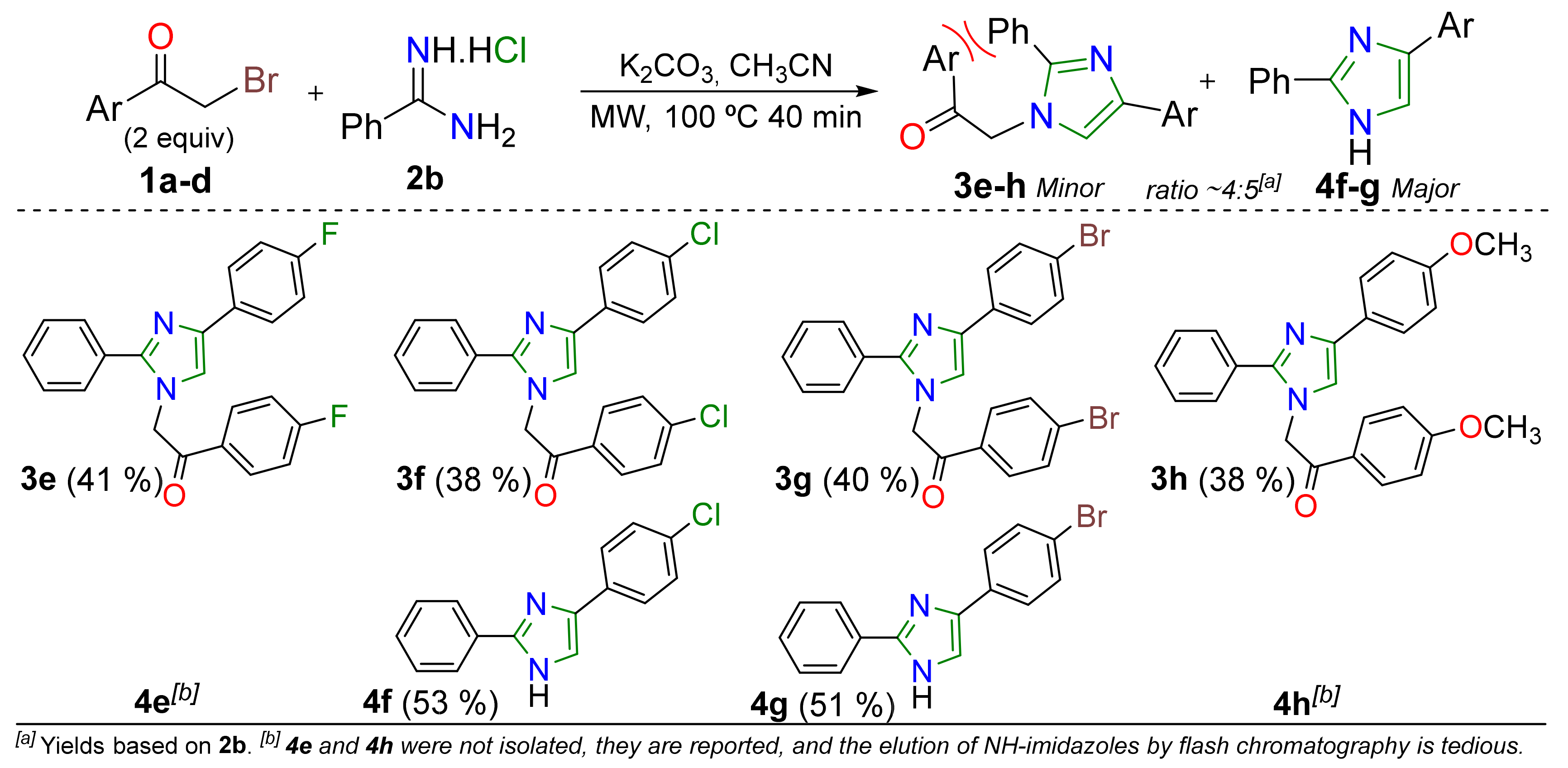
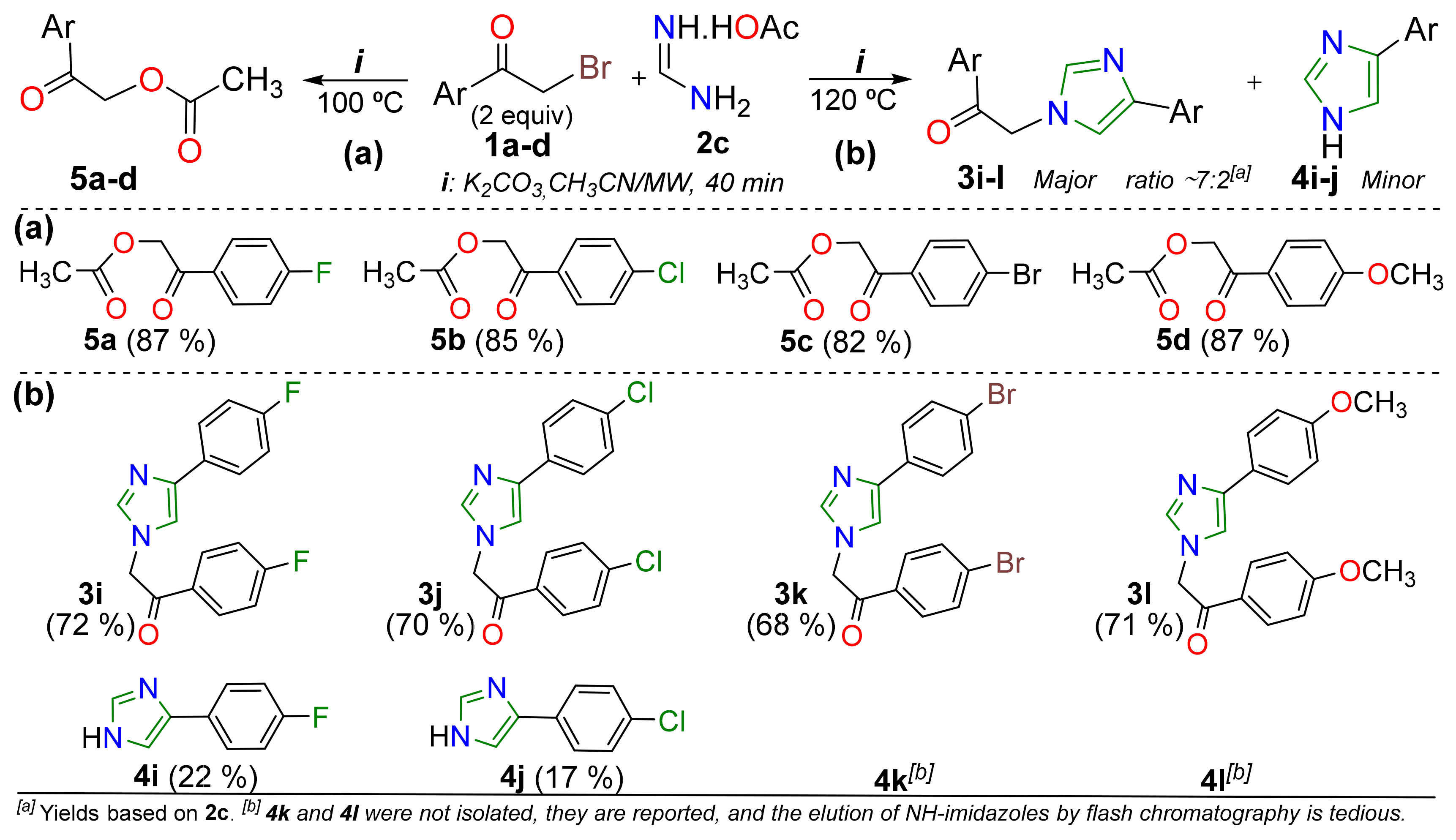
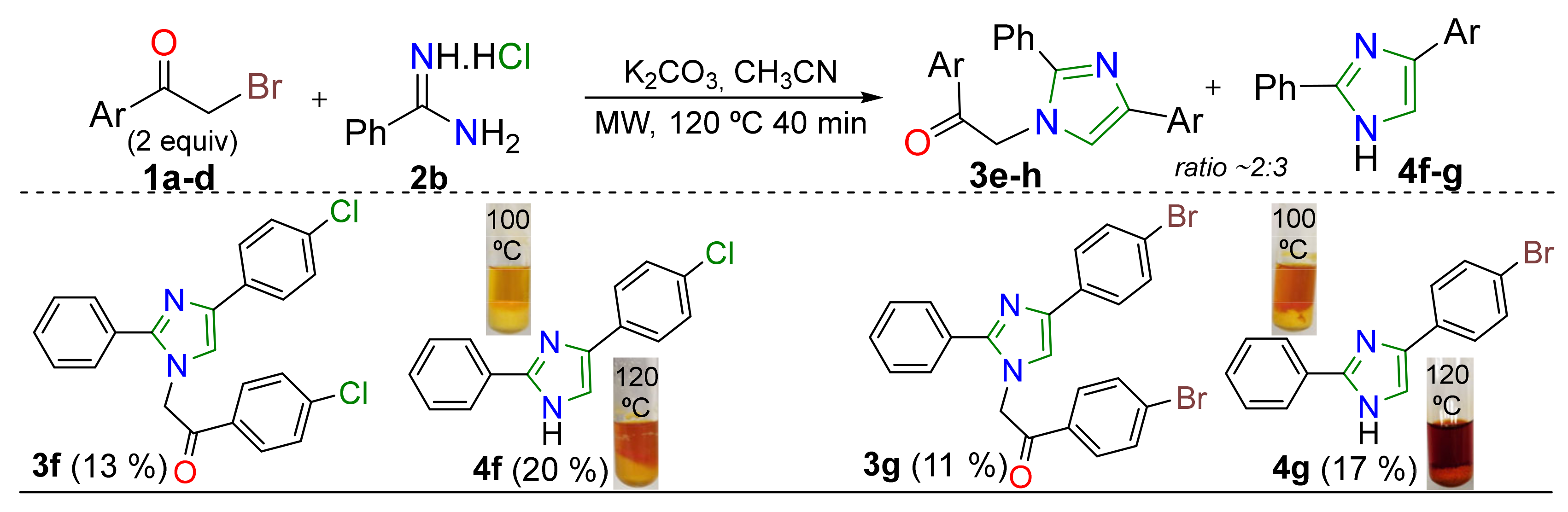
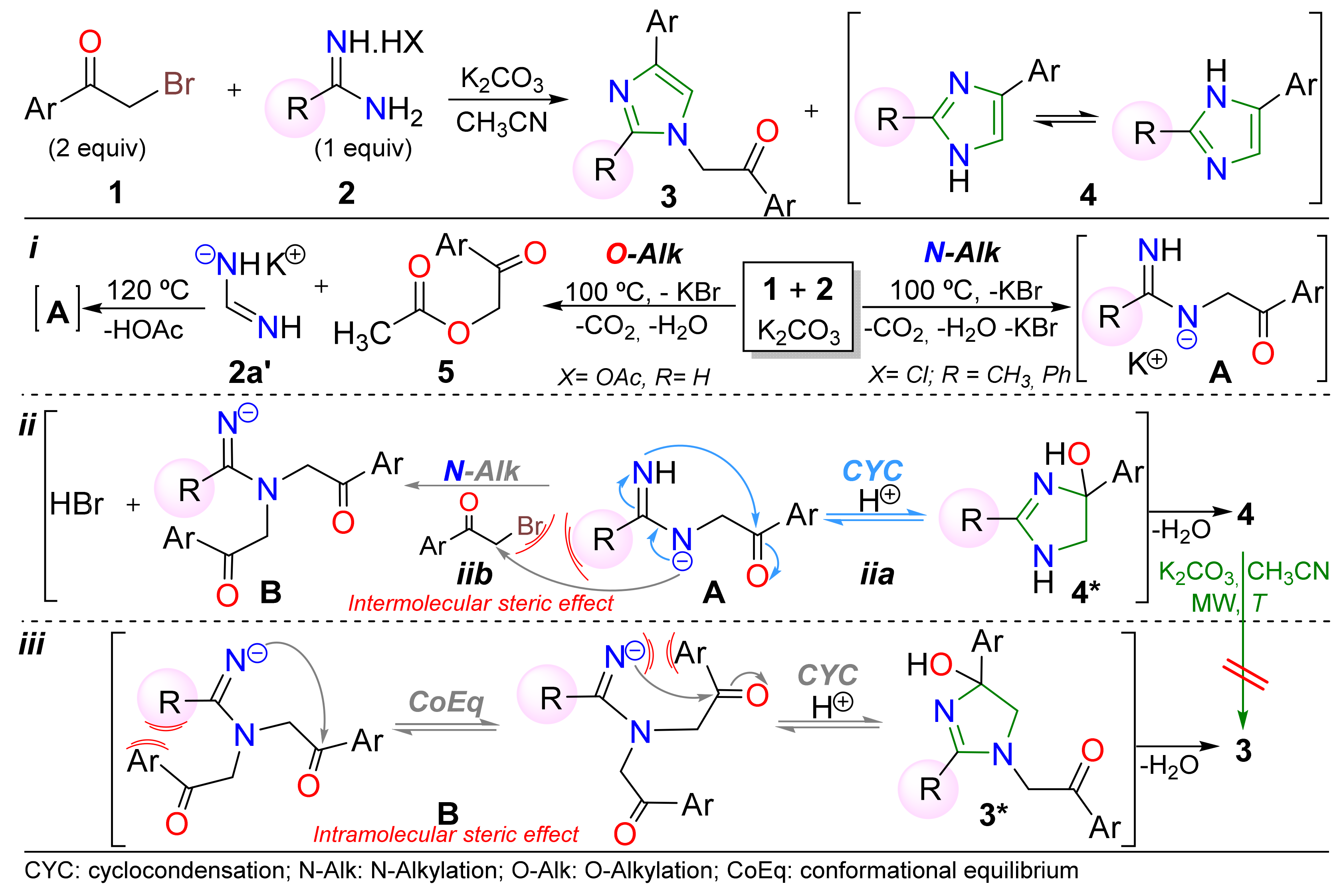
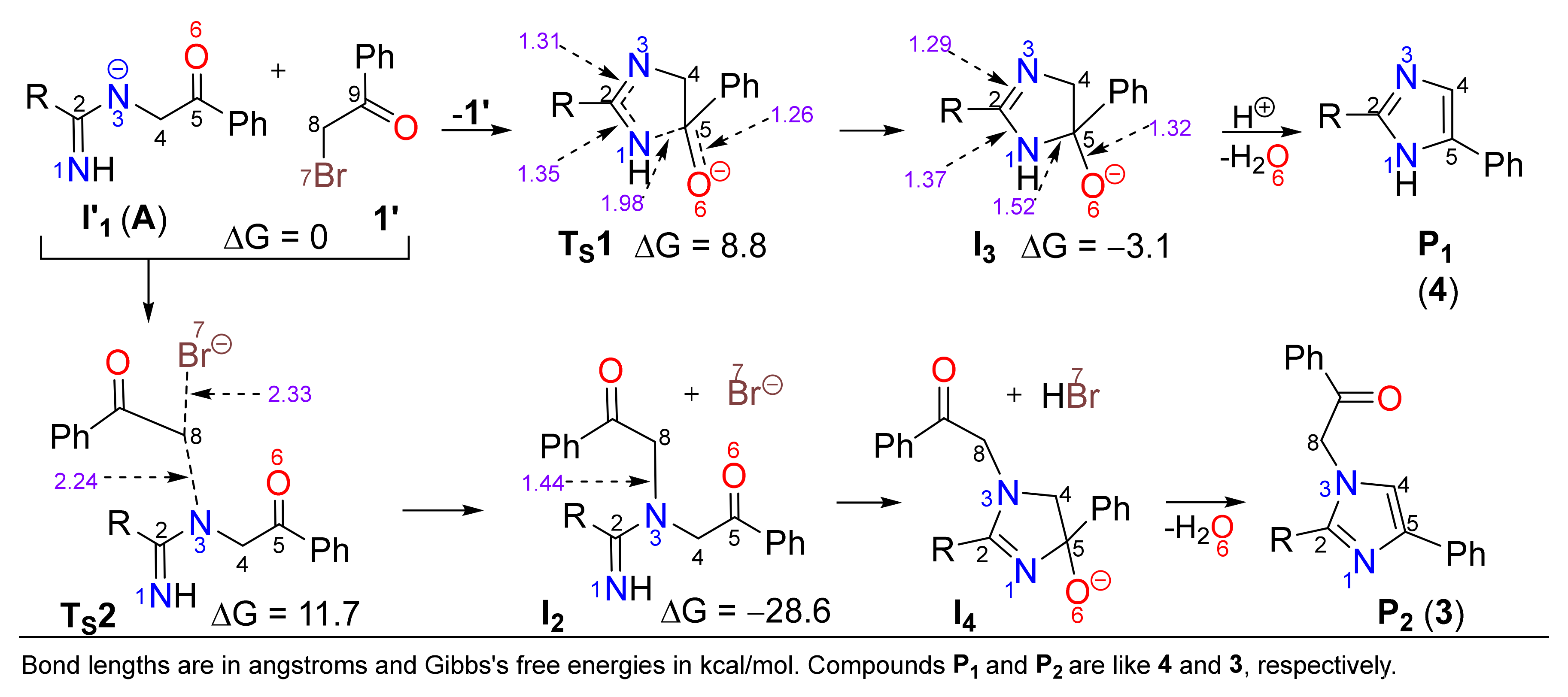
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. General Procedures
3.3. Characterization Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Castillo, J.C.; Portilla, J. Recent advances in the synthesis of new pyrazole derivatives. Targets Heterocycl. Syst. 2018, 22, 194–223. [Google Scholar] [CrossRef]
- Shabalin, D.A.; Camp, J.E. Recent advances in the synthesis of imidazoles. Org. Biomol. Chem. 2020, 18, 3950–3964. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez, A.A.; Godoy, A.; Portilla, J. Functional Pyrazolo [1,5-a]pyrimidines: Current Approaches in Synthetic Transformations and Uses as an Antitumor Scaffold. Molecules 2021, 26, 2708. [Google Scholar] [CrossRef]
- Sharma, P.; Larosa, C.; Antwi, J.; Govindarajan, R.; Werbovetz, K.A. Imidazoles as potential anticancer agents: An update on recent studies. Molecules 2021, 26, 4213. [Google Scholar] [CrossRef]
- Portilla, J.; Quiroga, J.; Abonía, R.; Insuasty, B.; Nogueras, M.; Cobo, J.; Mata, E.G. Solution-phase and solid-phase synthesis of 1-pyrazol-3-ylbenzimidazoles. Synth. Stuttg. 2008, 2008, 0387–0394. [Google Scholar] [CrossRef]
- Soni, J.; Sethiya, A.; Sahiba, N.; Agarwal, D.K.; Agarwal, S. Contemporary Progress in the Synthetic Strategies of Imidazole and its Biological Activities. Curr. Org. Synth. 2019, 16, 1078–1104. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef]
- Dos Santos Nascimento, M.V.P.; Mattar Munhoz, A.C.; De Campos Facchin, A.B.M.; Fratoni, E.; Rossa, T.A.; Mandolesi Sá, M.; Campa, C.C.; Ciraolo, E.; Hirsch, E.; Dalmarco, E.M. New pre-clinical evidence of anti-inflammatory effect and safety of a substituted fluorophenyl imidazole. Biomed. Pharmacother. 2019, 111, 1399–1407. [Google Scholar] [CrossRef]
- Gao, Y.; Huang, D.C.; Liu, C.; Song, Z.L.; Liu, J.R.; Guo, S.K.; Tan, J.Y.; Qiu, R.L.; Jin, B.; Zhang, H.; et al. Streptochlorin analogues as potential antifungal agents: Design, synthesis, antifungal activity and molecular docking study. Bioorg. Med. Chem. 2021, 35, 116073. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Z.; Lu, Y.; Dalton, J.T.; Miller, D.D.; Li, W. Synthesis and antiproliferative activity of imidazole and imidazoline analogs for melanoma. Bioorg. Med. Chem. Lett. 2008, 18, 3183–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Osma, J.A.; Martínez, J.; De La Cruz-Martínez, F.; Caballero, M.P.; Fernández-Baeza, J.; Rodríguez-López, J.; Otero, A.; Lara-Sánchez, A.; Tejeda, J. Development of hydroxy-containing imidazole organocatalysts for CO2 fixation into cyclic carbonates. Catal. Sci. Technol. 2018, 8, 1981–1987. [Google Scholar] [CrossRef]
- Cosby, T.; Holt, A.; Griffin, P.J.; Wang, Y.; Sangoro, J. Proton Transport in Imidazoles: Unraveling the Role of Supramolecular Structure. J. Phys. Chem. Lett. 2015, 6, 3961–3965. [Google Scholar] [CrossRef] [PubMed]
- Naureen, S.; Chaudhry, F.; Munawar, M.A.; Ashraf, M.; Hamid, S.; Khan, M.A. Biological evaluation of new imidazole derivatives tethered with indole moiety as potent α-glucosidase inhibitors. Bioorg. Chem. 2018, 76, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Rios, M.-C.; Bravo, N.-F.; Sánchez, C.-C.; Portilla, J. Chemosensors based on N-heterocyclic dyes: Advances in sensing highly toxic ions such as CN− and Hg2+. RSC Adv. 2021, 11, 34206–34234. [Google Scholar] [CrossRef]
- Salfeena, C.T.F.; Jalaja, R.; Davis, R.; Suresh, E.; Somappa, S.B. Synthesis of 1,2,4-Trisubstituted-(1 H)-imidazoles through Cu(OTf)2-/I2-Catalyzed C-C Bond Cleavage of Chalcones and Benzylamines. ACS Omega 2018, 3, 8074–8082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Qin, M.; Yang, X.; Zhang, X.; Liu, Y.; Guo, X.; Chen, B. Acid-catalyzed synthesis of imidazole derivatives via N-phenylbenzimidamides and sulfoxonium ylides cyclization. Tetrahedron 2019, 75, 2817–2823. [Google Scholar] [CrossRef]
- Alanthadka, A.; Elango, S.D.; Thangavel, P.; Subbiah, N.; Vellaisamy, S.; Chockalingam, U.M. Construction of substituted imidazoles from aryl methyl ketones and benzylamines via N-heterocyclic carbene-catalysis. Catal. Commun. 2019, 125, 26–31. [Google Scholar] [CrossRef]
- Gelens, E.; De Kanter, F.J.J.; Schmitz, R.F.; Sliedregt, L.A.J.M.; Van Steen, B.J.; Kruse, C.G.; Leurs, R.; Groen, M.B.; Orru, R.V.A. Efficient library synthesis of imidazoles using a multicomponent reaction and microwave irradiation. Mol. Divers. 2006, 10, 17–22. [Google Scholar] [CrossRef]
- Ortiz, M.-C.; Portilla, J. Access to five-membered N-heteroaromatic compounds: Current approach based on microwave-assisted synthesis. Targets Heterocycl. Syst. 2021, 25, 436–462. [Google Scholar] [CrossRef]
- Murai, T.; Kikukawa, Y.; Hirao, H. Novel Imidazole Compound and Usage Thereof. Japan Patent EP1605078 (A1), 14 December 2005. [Google Scholar]
- Osakada, N.; Osakada, M.; Sawada, T.; Kaneko, S.; Mizutani, A.; Uesaka, N.; Nakasato, Y.; Katayama, K.; Sugawara, M.; Kitamura, Y. Imidazole Derivative. Japan Patent EP2090570 (A1), 19 August 2009. [Google Scholar]
- Vargas-Oviedo, D.; Charris-Molina, A.; Portilla, J. Efficient Access to o-Phenylendiamines and Their Use in the Synthesis of a 1,2-Dialkyl-5-trifluoromethylbenzimidazoles Library Under Microwave Conditions. ChemistrySelect 2017, 2, 3896–3901. [Google Scholar] [CrossRef]
- Elejalde, N.R.; Macías, M.; Castillo, J.C.; Sortino, M.; Svetaz, L.; Zacchino, S.; Portilla, J. Synthesis and in vitro Antifungal Evaluation of Novel N-Substituted 4-Aryl-2-methylimidazoles. ChemistrySelect 2018, 3, 5220–5227. [Google Scholar] [CrossRef]
- Macías, M.A.; Elejalde, N.R.; Butassi, E.; Zacchino, S.; Portilla, J. Studies via X-ray analysis on intermolecular interactions and energy frameworks based on the effects of substituents of three 4-aryl-2-methyl-1H-imidazoles of different electronic nature and their in vitro antifungal evaluation. Acta Crystallogr. Sect. C Struct. Chem. 2018, 74, 1447–1458. [Google Scholar] [CrossRef] [PubMed]
- Elejalde, N.R.; Butassi, E.; Zacchino, S.; Macías, M.A.; Portilla, J. Inter molecular inter action energies and molecular conformations in N -substituted 4-aryl-2-methyl imidazoles with promising in vitro anti fungal activity. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2019, 75, 1–12. [Google Scholar] [CrossRef]
- Vargas-Oviedo, D.; Butassi, E.; Zacchino, S.; Portilla, J. Eco-friendly synthesis and antifungal evaluation of N-substituted benzimidazoles. Mon. Fur. Chem. 2020, 151, 575–588. [Google Scholar] [CrossRef]
- Gopi, E.; Kumar, T.; Menna-Barreto, R.F.S.; Valença, W.O.; Da Silva Júnior, E.N.; Namboothiri, I.N.N. Imidazoles from nitroallylic acetates and α-bromonitroalkenes with amidines: Synthesis and trypanocidal activity studies. Org. Biomol. Chem. 2015, 13, 9862–9871. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.F.; Chen, Z.C. Hypervalent iodine in synthesis. 48. A one-pot convenient procedure for the synthesis of 2-mercaptothiazoles by cyclocondensation of ketones with [hydroxy(tosyloxy)iodo]-benzene and ammonium dithiocarbamate. Synth. Commun. 2001, 31, 415–420. [Google Scholar] [CrossRef]
- Chen, X.Y.; Englert, U.; Bolm, C. Base-Mediated Syntheses of Di- and Trisubstituted Imidazoles from Amidine Hydrochlorides and Bromoacetylenes. Chem.-A Eur. J. 2015, 21, 13221–13224. [Google Scholar] [CrossRef]
- Kumar, S.; Jaller, D.; Patel, B.; Lalonde, J.M.; Duhadaway, J.B.; Malachowski, W.P.; Prendergast, G.C.; Muller, A.J. Structure Based Development of Phenylimidazole-Derived Inhibitors of Indoleamine 2,3-Dioxygenase. J. Med. Chem. 2008, 4968–4977. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Y.; Chen, Y.; Gu, L.; Luo, Y.; Yang, Z.; He, L. Syn thesis Preparation of Imidazole Derivatives via Bisfunctionalization of Alkynes Catalyzed by Ruthenium Carbonyl. Synthesis 2019, 51, 3520–3528. [Google Scholar] [CrossRef]
- Zhou, X.; Ma, H.; Cao, J.; Liu, X.; Huang, G. Novel and efficient transformation of enamides into α-acyloxy ketones via an acyl intramolecular migration process. Org. Biomol. Chem. 2016, 14, 10070–10073. [Google Scholar] [CrossRef] [PubMed]
- Mizar, P.; Wirth, T. Flexible stereoselective functionalizations of ketones through umpolung with hypervalent iodine reagents. Angew. Chem.-Int. Ed. 2014, 53, 5993–5997. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substituent (R) | ΔΔG(TS1/P1–TS2/P2) (kcal/mol) | Ratio 4/3 (Total Yield %) |
|---|---|---|---|
| 1 | H | 0.85 | 1:3.5 (90) [b] |
| 2 | CH3 | −0.78 | 1:3.0 (81) [b] |
| 3 | Ph | −2.84 | 1.25:1 (91) [b] |
| 4 | t-Bu | −7.12 | 42/99: −/− (42/99) [c] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elejalde-Cadena, N.R.; García-Olave, M.; Figueroa, D.; Vidossich, P.; Miscione, G.P.; Portilla, J. Influence of Steric Effect on the Pseudo-Multicomponent Synthesis of N-Aroylmethyl-4-Arylimidazoles. Molecules 2022, 27, 1165. https://doi.org/10.3390/molecules27041165
Elejalde-Cadena NR, García-Olave M, Figueroa D, Vidossich P, Miscione GP, Portilla J. Influence of Steric Effect on the Pseudo-Multicomponent Synthesis of N-Aroylmethyl-4-Arylimidazoles. Molecules. 2022; 27(4):1165. https://doi.org/10.3390/molecules27041165
Chicago/Turabian StyleElejalde-Cadena, Nerith Rocio, Mayra García-Olave, David Figueroa, Pietro Vidossich, Gian Pietro Miscione, and Jaime Portilla. 2022. "Influence of Steric Effect on the Pseudo-Multicomponent Synthesis of N-Aroylmethyl-4-Arylimidazoles" Molecules 27, no. 4: 1165. https://doi.org/10.3390/molecules27041165
APA StyleElejalde-Cadena, N. R., García-Olave, M., Figueroa, D., Vidossich, P., Miscione, G. P., & Portilla, J. (2022). Influence of Steric Effect on the Pseudo-Multicomponent Synthesis of N-Aroylmethyl-4-Arylimidazoles. Molecules, 27(4), 1165. https://doi.org/10.3390/molecules27041165