1. Introduction
Sodium borohydride NaBH
4 in alkaline aqueous solution is a potential fuel of low-temperature fuel cell [
1]. It is regarded as an
indirect fuel (i.e., H carrier) when it is hydrolyzed to produce H
2, the as-produced H
2 then feeding a fuel cell (Equation (1)) [
2]. It is regarded as a
direct fuel (i.e., reductant) when it directly feeds a direct liquid fuel cell to be electro-oxidized (Equation (2)) [
3]:
The aqueous solution has to be alkaline, that is, stabilized [
4], because this is the only way to prevent spontaneous (exothermic, with an enthalpy of about −240 kJ mol
−1 [
5]) hydrolysis of BH
4− from occurring extensively. In hydrolysis (Equation (1)), a metal catalyst is therefore required to accelerate the production of H
2 [
6]. In electro-oxidation (Equation (2)), a metal electro-catalyst is required to promote the generation of a maximum of electrons (out of eight) [
4]. However, the electro-catalyst also acts as catalyst of hydrolysis, a reaction that is in this case regarded as heterogeneous because it is detrimental to the fuel cell faradaic efficiency [
7].
In hydrolysis (Equation (1)) as well as in electro-oxidation (Equation (2)), complete reaction implies transformation of BH
4− into B(OH)
4− via formation of short-living intermediates. For spontaneous hydrolysis, Mochalov et al., suggested in 1965 BH
3OH
−, BH
2(OH)
2−, and BH(OH)
3− as possible short-living intermediates [
8]. They showed, for instance, that the direct transformation of BH
4− into B(OH)
4− has the same kinetic constant (
k = 5.31 × 10
7 min
−1) as the transformation of BH
4− into BH
3OH
− (
k’ = 5.15 × 10
7 min
−1). The same year, Gardiner and Collat suggested the formation of BH
3, BH
3OH
−, and [H]
+[BHOH]
− as possible short-living intermediates [
9]. More recently, Guella et al. reported that, by
11B nuclear magnetic resonance (NMR) spectroscopy, they detected only BH
4− and B(OH)
4− (Equation (1)) for a Pd-catalyzed hydrolysis [
10]. The non-detection of other species was explained by the fact that the hydrolysis intermediates are excessively short-living in their experimental conditions. By quantum chemical calculations, Lu et al. [
11] confirmed Guella et al.’s explanation and modelled a multistep process involving the following hypothetical short-living intermediates (Equation (3)):
Comparable predictions were reported by Zhou et al. [
12], Andrieux et al. [
13], Churikov et al. [
14], and Choi et al. [
15] detected traces of BH
3OH
− by using
11B NMR spectroscopy. It is therefore arguable whether BH
3OH
−, as the first short-living intermediate, directly hydrolyzes into B(OH)
4−. This is a possible parallel pathway as suggested by Mochalov et al. [
8] for example.
Budroni et al. [
16] refers to BH
3OH
− as a critical short-living intermediate. As discussed above, this applies to the hydrolysis reaction (Equation (1)). Interestingly, this also applies to electro-oxidation of BH
4− (Equation (2)) on metal electrodes (e.g., Pd, Pt, and Au) [
17,
18,
19,
20,
21,
22,
23]. For instance, Molina Concha et al. [
24] studied Pt-catalyzed electro-oxidation of BH
4− by in situ Fourier Transform Infrared (FTIR) spectroscopy. They observed that: (i) BH
3OH
− formed at low potentials (<0.7 V) by hydrolysis of BH
4− and/or partial oxidation of BH
4−; (ii) BH
3OH
− quickly electro-oxidized into BH
2 intermediates such as BH
2OH and BH
2(OH)
2−; and (iii) the BH
2 intermediates electro-oxidized into BO
2− at high potentials (>0.7 V).
Similarly, Nanayakkara et al. [
25] investigated the mechanism of H
2 release of BH
3 in water and the following solvent effects by using MP2 quantum calculations. One H
2O molecule interacting with BH
3 led to an activation energy equal to 24.9 kcal mol
−1, while the energy values ranged from 29 and 32 kcal mol
−1 when one H
2O molecule interacted with BH
3 and another H
2O molecule interacted with the H
2O molecule bonded to BH
3. The resulting enthalpy was estimated at 20 kcal mol
−1 for the first configuration and ranged between 12 and 14 kcal mol
−1 for the others.
The present study is to be seen against the background described above. Based on a systematic study using 11B NMR spectroscopy, we attempted to detect and identify any short-living intermediates in order to gain insight and better understanding of both hydrolysis and electro-oxidation of BH4−. Furthermore, theoretical investigations were performed for obtaining vibrational results, determining the sensitive frequencies and estimating the energies of the different hypothetical molecular structures.
2. Results and Discussion
2.1. Hydrolysis Conditions Where H2O Acts as Both Reactant and Solvent
In hydrolysis and electro-oxidation conditions, the fuel is an alkaline aqueous solution of BH4− for which the concentration of BH4− is usually kept low (typically < 1 M). We therefore set our experimental conditions to be in line with such practices: the concentration of NaOH was fixed as 0.1 M and the concentration of BH4− (from NaBH4) was chosen as 0.66 M.
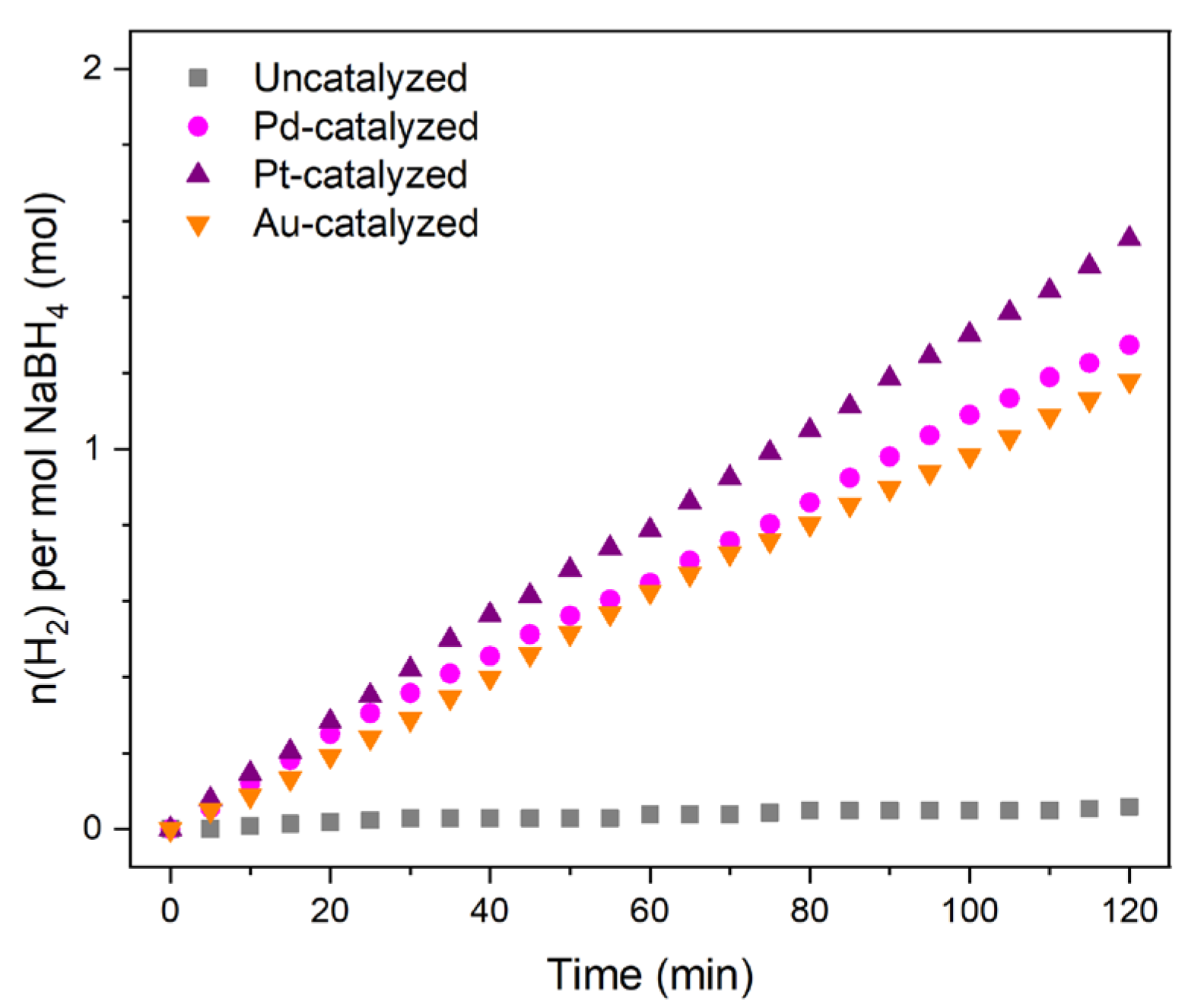
In hydrolysis and electro-oxidation conditions, the reaction is catalyzed by a metal catalyst and an electro-catalyst, respectively. We selected three bulk metals such as Pd, Pt, and Au (each as a piece of metal wire). They were selected because each has been used in hydrolysis [
26] and electro-oxidation [
22].
In the present study and unlike in common practices [
26], our objective was not to develop an active (or very active) hydrolysis catalyst. Our objective was to work with a lowly active catalyst so that the kinetics of H
2 production remains slow when analyzing the solutions by
11B NMR spectroscopy. We thus focused on metals in bulk state, which is a state that offers the desired catalytic activity. We ensured this by performing a series of hydrolysis experiments. Typically, 2 mL of the aforementioned alkaline solution of BH
4− (corresponding to 50 mg of NaBH
4) were put into contact with 16 mg of Pd, 14.5 mg of Pt, or 14.3 mg of Au at 30 °C. Regardless of the nature of the metal, it took 2 h to produce <1.6 mol H
2 per mol BH
4− (
Figure 1), that is, <53 mL H
2 (out of 132 mL for a conversion of 100%). This means a H
2 generation rate of <0.45 mL(H
2) min
−1 that is in agreement with our need. We also ensured that, in the absence of any metal, the alkaline solution of BH
4− was quite stable. At 30 °C, <0.1 mol H
2 per mol BH
4− was produced in 2 h (namely, <3 mL(H
2)).
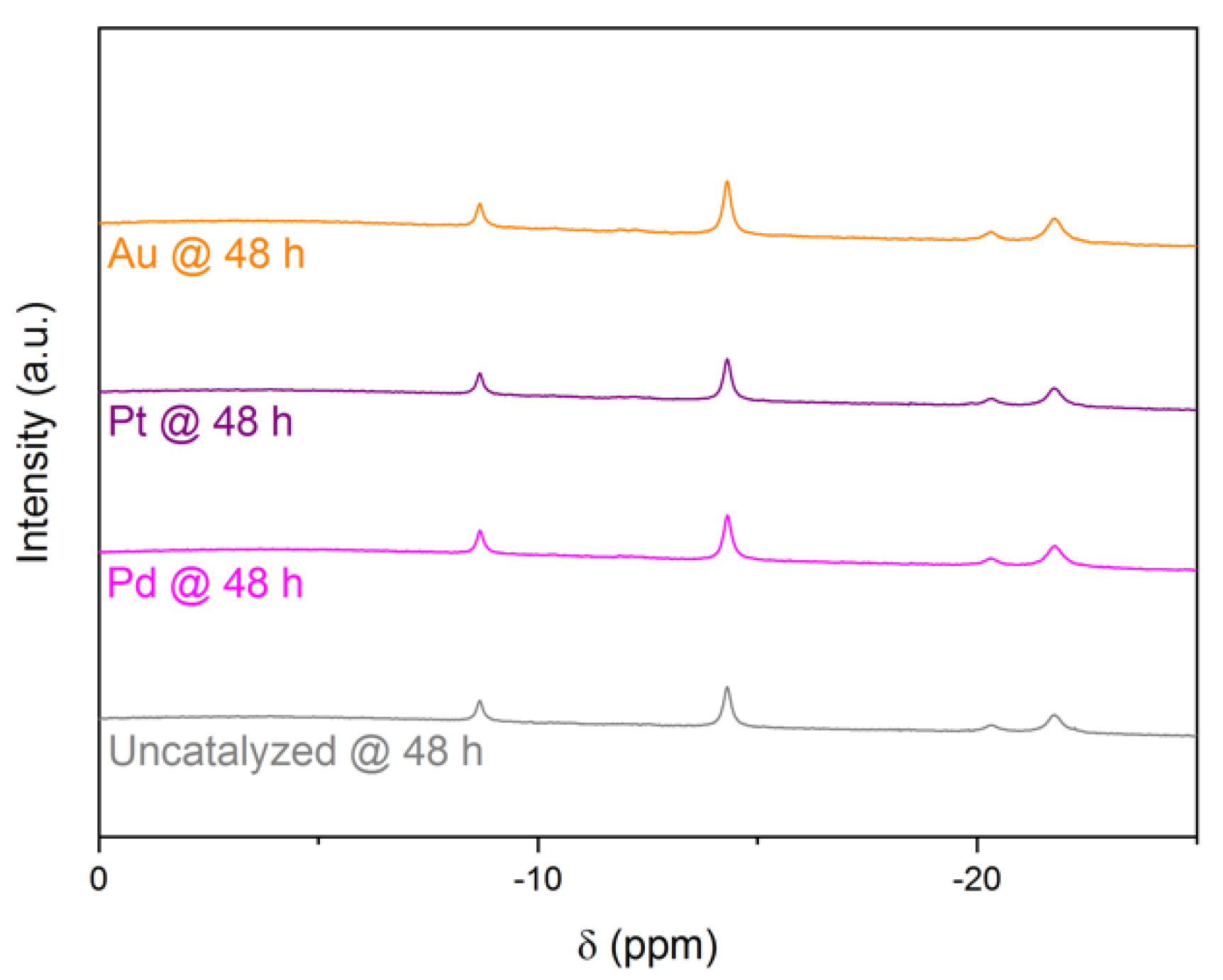
The hydrolysis tests were repeated to analyze the solution by
11B NMR spectroscopy every hour. Similar to a previous study [
13], we detected only three signals (examples of spectra in
Figure 2;
Table 1). The first main signal was a quintet at δ −41.5 ppm due to BH
4−. The second main signal was a singlet at δ +1.9 ppm evidencing the formation of B(OH)
4− (Equation (1)). There was an additional minor and almost negligible signal, a quartet at δ −12.8 ppm. It was ascribed to the short-living intermediate BH
3OH
− [
10,
15,
27].
No additional 11B NMR signals that would be attributed to other short-living intermediates were seen. This might be explained by concentrations that are below the detection limit (ca. 1 × 10−3 mol L−1) of the spectrometer. This might be also explained by low symmetry of the intermediates’ structures, which would lead to broad signals of very low intensity and thus indistinguishable from the base line. It is worth mentioning that we used Gaussian 09 software to perform geometry optimization and NMR calculations for a series of possible intermediates including BH2(OH)2− and BH(OH)3−. We found that the signals of BH2(OH)2− and BH(OH)3− should be a triplet and a doublet appearing between δ −7 and δ 0 ppm, respectively.
Another possible explanation of the absence of additional 11B NMR signals is that the experimental conditions were not suitable for detecting intermediates with a lifetime that is shorter than that of the detected BH3OH−. Based on our observations, we can state that the lifetime scale of BH3OH− is of tens of seconds, whereas it might be much shorter (e.g., microseconds scale) for the other intermediates. Yet, the hydrolysis tests described above were performed in the presence of an excess of water: we used 2 mL (mol ratio H2O/BH4− of 84) whereas about 0.1 mL (mol ratio H2O/BH4− of 4) would be enough to totally hydrolyze BH4−. Water acted as both reactant and solvent, and the excess of water could be a favorable context to promote extremely fast hydrolysis of short-living intermediates.
2.2. Hydrolysis Conditions Where H2O Is Only a Reactant
In order to move away from the conditions using water as both reactant and solvent, we drew on two ancient reports dealing with hydrolysis of BH
4−. Modler and Kreevoy investigated the hydrolysis of BH
4− (0.002 M) in moist acetonitrile (i.e., containing 0.6 M H
2O) [
28], and Taub et al. used aqueous dimethylformamide (DMF) [
29]. We thus selected DMF as aprotic solvent of NaBH
4 and used H
2O as reactant only.
We prepared four 10 mL DMF solutions of BH4− by dissolving 0.5 g of NaBH4 (1.32 M). A piece of the aforementioned Pd, Pt, and Au was added in each of three of the DMF solutions. The fourth DMF solution was kept metal-free and is denoted uncatalyzed. We then added 0.95 mL of alkaline (0.1 M NaOH) aqueous solution to each of the four DMF solutions (resulting in a concentration of H2O in DMF of 5.291 M). In these conditions, the mol ratio H2O/BH4− was about four as for the stoichiometric hydrolysis reaction (Equation (1)). The as-prepared solutions were analyzed by 11B NMR spectroscopy. It is worth mentioning that in such conditions, the hydrolysis was expected to be slow. Accordingly, the solutions were analyzed every 24 h for 3 days.
The
11B NMR spectra focusing on the δ range varying from +20 to −50 ppm (
Figure S1) showed only the quintet at δ −39.7 ppm due to BH
4−. By zooming over the δ range varying from +20 to −30 ppm (
Figure S2), it was possible to distinguish an additional signal of very small intensity at δ −14.1 ppm, namely the quartet due to BH
3OH
−. The quartet could be seen after 24 h for the Pd-, Pt-, and Au-catalyzed solutions, and after 48 h for the uncatalyzed solution. These results highlighted that, in the stoichiometric conditions, the hydrolysis took place to a negligible extent. Another observation is that, even in the absence of a metal, hydrolysis spontaneously took place. The non-detection of B(OH)
4− may have up to three explanations: the amount of H
2O was too low and the H
2O molecules were very diluted in DMF, which hindered interaction-reaction with BH
4− and BH
3OH
−; borates including B(OH)
4− were practically insoluble in DMF [
30] and may have precipitated; and/or, the concentration of B(OH)
4− was below the detection limit.
2.3. Hydrolysis Conditions Where H2O Is a Reactant in Excess
We therefore repeated the experiments while increasing the water content: the mol ratio H
2O/BH
4− passed from 4 to 32. Once more, we prepared four 10 mL DMF solutions of BH
4− (1.32 M) and added 7.6 mL of alkaline (0.1 M NaOH) aqueous solution. In comparison to the experiments presented in
Section 2.1, the present series used water to a lesser extent (i.e., mol ratio H
2O/BH
4− of 32 versus 84) and the 32 equivalents of H
2O were dispersed in 10 mL of DMF, mitigating the hydrolysis of BH
4−.
As before, the
11B NMR spectra (
Figure S3) mainly showed the quintet at δ −40.5 ppm due to BH
4−, and B(OH)
4− was not observed because of the reasons listed at the end of the previous section. In contrast to the results discussed above, the
11B NMR spectra showed additional signals at δ < 0 (
Figure 3). This is discussed hereafter.
The first of the additional signals was a quartet at δ −14.4 ppm. As for our experiments discussed above, it was ascribed to BH3OH−.
The second of the additional signals was also a quartet, centered at δ −8.9 ppm. It indicated the formation of another BH3-containing intermediate.
The third of the additional signals appeared as a multiplet located between δ −18.5 ppm and δ −23.5 ppm. With the help of
1H-decoupled
11B NMR spectroscopy, we shed light on its nature. It was the result of two distinct signals peaking at δ −20.3 ppm and δ −21.8 ppm (
Figure 4). By deconvolution of the signal, we found that the two signals were more likely to be two overlapping quartets, thereby indicating the formation of two other BH
3 intermediates (
Figure S4 and Table S1).
To summarize the above: the hydrolysis of DMF-solubilized BH4− in the presence of 32 equivalents of H2O involved more intermediates than the only short-living intermediate BH3OH−. There were three additional intermediates and they all showed a quartet in 11B NMR spectroscopy, indicating that they all were made up of the BH3 group.
We therefore focused our efforts on attributing the aforementioned quartets to possible BH
3 intermediates. We thought about any species likely to form in our conditions while exploring the open literature [
31,
32,
33,
34]. The following ones were listed (
Figure 5):
The complex H2O·BH3 because H2O is a Lewis base able to complex the Lewis acid BH3;
The complex DMF·BH3 because DMF is Lewis bases able to complex BH3;
The anion BH3OH−;
The anion B2H7− (i.e., [H3B−H−BH3]− or [H4B−BH3]−); and
The pentacoordinate BH3(H2).
According to Tague and Andrews [
34], the last species BH
3(H
2) possibly acts as intermediate before the formation of BH
3OH
− by reaction of BH
4− and H
2O.
We then used Gaussian 09 software to perform geometry optimization and NMR calculations for each of these possible intermediates. We found the chemical shifts listed in
Table 2. As observed in this table, a relatively good agreement between CASTEP and Gaussian 09 results was obtained considering the two investigated functionals (B3LYP for Gaussian 09 and PBE for CASTEP), except for BH
3(H
2). In the case of this species, the impact of the dispersion could be invoked but additional calculations using DFT-D in CASTEP showed a very small influence of dispersion on the calculations. It is worth mentioning that in a previous study [
31], the chemical shift of B
2H
7− in THF as solvent was reported to be δ −26 ppm. Similarly, using CASTEP calculations, we found comparable values (
Table 2). We also calculated the chemical shifts for the intermediates based on BH
4−x(OH)
x− (with x = 1, 2, 3, 4), such as: BH
4− with δ −51.5 ppm; BH
3OH
− with δ −11.4 ppm; BH
2(OH)
2− with δ +0.1 ppm; BH(OH)
3− with δ +1.1 ppm; and B(OH)
4− with δ +3.1 ppm.
Going back to the results presented in
Figure 3 and using the data in
Table 2, we ascribed the quartets at follows. The signals at δ −8.9 ppm and δ −14.4 ppm (
Figure 3) were unambiguously attributed to DMF·BH
3 and BH
3OH
−. Because the calculated chemical shift of BH
3(H
2) is much different from that of remaining signals at around δ −21 ppm, we discarded its formation. We also discarded the formation of H
2O·BH
3 due to the absence of signals at around 0 ppm in our experimental conditions. Accordingly, the partly overlapping quartets are at δ −20.3 ppm and δ −21.8 ppm and are attributed to B
2H
7− and B
2H
7− in interaction with H
2O. Indeed, the chemical shift for the quartet due to [B
2H
7·H
2O]
− was calculated as −28.1 ppm using Gaussian 09 and −32.6 ppm using CASTEP; these shifts were close to those calculated for B
2H
7− (
Table 2).
Based on the experimental results reported above and supported by the calculations performed, we suggest that the BH
4− anions dissolved in DMF are able to react with H
2O taken in excess to form BH
3-based intermediates such as DMF·BH
3, BH
3OH
−, and B
2H
7−. These intermediates are much likely to be in equilibrium. Based on the discussions reported in [
30], we thus suggest that in DMF, BH
3OH
− forms first and DMF·BH
3 and B
2H
7− forms from BH
3OH
− (by substitution of Lewis bases). This is illustrated in
Figure 6.
3. Materials and Methods
Sodium borohydride NaBH4 (99%), sodium hydroxide NaOH (≥98%), N,N-dimethylformamide C3H7NO (DMF; 99.8%, anhydrous), Pt wire (99.9%, Ø 1.0 mm), Pd wire (99.9%, Ø 1.0 mm), and Au wire (99.95%, Ø 1.0 mm) all from Sigma-Aldrich were used as received. We stored and handled them in our argon-filled glove box (MBraun M200B, with O2/H2O < 0.1 ppm). We used Milli-Q deionized water (18.2 MΩ cm) and it was degassed by bubbling argon for 30 min before its use.
In a first step, the hydrolysis conditions were such that water acted as both reactant and solvent. The H2 evolution experiments were performed as follows. Under argon, 50 mg of NaBH4 were transferred in a Schlenk tube (used as hydrolysis reactor). For the catalyzed experiments, a piece of metal wire (16.1 mg of Pd, 14.5 mg of Pt, or 14.3 mg of Au) was also transferred in the tube. The tube was sealed and the glove box was taken out, installed to our hydrolysis set-up (reactor connected to an inverted burette via a cold trap kept at 0 °C), and immersed in an oil bath at 30 °C. The hydrolysis reaction was started by injecting 2 mL of an aqueous alkaline (0.1 M NaOH) solution. In these conditions, the mol ratio H2O/BH4− was 84. The displacement of the blue-colored liquid in the inverted burette due to the generated H2 was video monitored. The H2 evolution experiments were repeated to analyze the solution by 11B NMR spectroscopy (Bruker Avance 400 NMR spectrometer equipped with a BBOF probe; BF3·OEt2 as reference; acetonitrile-d3 such as ≥99.8 atom % D and from Sigma-Aldrich).
In a second step, the hydrolysis conditions were modified such that water only acted as reactant. To do so, 10 mL of DMF was used as solvent of 50 mg of NaBH4. To this solution prepared under argon, a piece of metal was added to catalyze the reaction. The hydrolysis reaction was started by injecting 0.95 mL of alkaline (0.1 M NaOH) aqueous solution. The concentration of H2O in DMF was 5.291 M and the mol ratio H2O/BH4− was about 4. The solutions were analyzed by 11B NMR spectroscopy every 24 h for 3 days.
In a third step, the hydrolysis conditions were once again modified. They were such that the water amount in DMF was increased and the mol ratio H2O/BH4− passed from 4 to 32. Otherwise, the solutions were prepared similarly and they were analyzed by 11B NMR spectroscopy every 24 h for 3 days.
We finalized the attribution of the
11B NMR signals using theory and calculations. We used Gaussian 09 software to perform geometry optimization, vibrational analysis, and the NMR calculations. The molecular structures were determined by density functional theory calculations. A gas phase geometry optimization of the Gibbs free energy was calculated using B3LYP hybrid density functional with 6-311(++)G(2d,p) basis set at 298.15 K. NMR spectra (NMR references: TMS and BF
3-OEt
2) were predicted by using the same level of theory (B3LYP/6-311++G(2d,p)). Additional computational methods were used to probe the structural properties of the different intermediates (
Figure 5 and
Table 2). As the reactions are difficult to stop, to isolate the structures, molecular simulations appeared to be the most powerful strategy to determine the corresponding spectroscopic properties. In complement of Gaussian 09 calculations to determine the NMR chemical shifts, calculations consisting into geometry optimization and NMR properties determination were performed using CASTEP implemented in Materials Studio 2020 [
35]. This is a DFT-based code using the projector-augmented waves (PAW) and gauge-included projector-augmented waves (GIPAW) algorithms for NMR chemical shifts, respectively. Here, the PBE functional was used in the generalized gradient approximation (GGA) for the exchange correlation energy. The core−valence interactions were described by norm-conserving pseudopotentials within the NMR CASTEP package and without implementation of any additional corrections. A kinetic energy cut-off was considered and the size of the box was fixed at 10 Å (additional calculations have been performed by considering a box size fixed at 20 Å and leading to similar results), which produced converged results for geometry optimization and NMR shielding determination. The convergence of the self-consistent field (SCF) calculations were reached when the total energy variation of the system was lower than 10
−5 eV/atom, the maximum force variation was lower than 0.03 eV/Å, and the maximal displacement was lower than 0.001 Å. In order to compare the GIPAW calculated
11B shielding values with the corresponding experimental values, the following expression was used:
δiso, calc =
σref −
σiso, where
σref corresponds to the value obtained for
11B (BH
3-OEt
2) and
σiso is the computational value for the investigated species.
Additional calculations were performed with CASTEP to investigate the effect of the dispersion (by considering DFT-D corrections (suing OBS method implemented in Materials Studio)) and the use of ultrasoft pseudo-potentials. A small influence on the NMR properties was observed if the dispersion was taken into account, while the use of ultrasoft pseudo-potentials led to stronger variations.
4. Conclusions
When hydrolysis of BH4− took place in water that acted as both solvent and reactant, only one short-living intermediate was detected. It was the well-known BH3OH−. In such conditions, the amount of water was excessive, offering a favorable environment to the complete hydrolysis of each BH4− into B(OH)4−. When hydrolysis of BH4− took place in DMF in the presence of a stoichiometric amount of water, only BH3OH− was detected again. In these conditions, the amount of water was too low and, if any, the other intermediates were not detected because of too low concentrations (below the detection limit). When hydrolysis of BH4− took place in DMF as solvent and in the presence of an excess of water, four BH3-based intermediates were detected, as evidenced by 11B NMR quartets peaking at δ −8.9, δ −14.4, δ −20.3, and δ −21.8 ppm. Using geometry optimization and calculations, these signals could be ascribed to DMF·BH3, BH3OH−, and B2H7− (in two conformations or in interaction with DMF or H2O) that are likely to be equilibrium. This illustrates the capacity of the Lewis acid BH3 to interact with Lewis bases such as DMF, OH−, and BH4−. We also suggest that in DMF, BH3OH− forms first and DMF·BH3 and B2H7− forms from BH3OH−. These findings are important from a fundamental point of view for a better understanding of hydrolysis of BH4− at the molecular level. These findings are also important for a better understanding of production of boron-based impurities in synthesis of boranes; boranes can be produced from BH4− in an organic solvent like DMF that may contain traces of moisture.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}