
Structure/Function Analysis of Truncated Amino-Terminal ACE2 Peptide Analogs That Bind to SARS-CoV-2 Spike Glycoprotein

 ,
,
Abstract
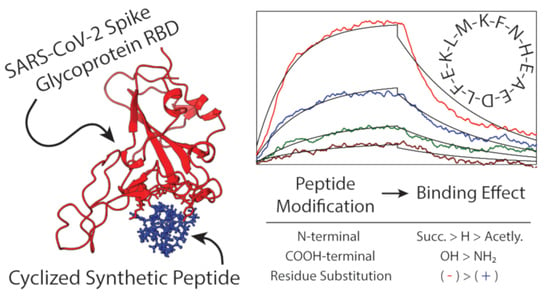
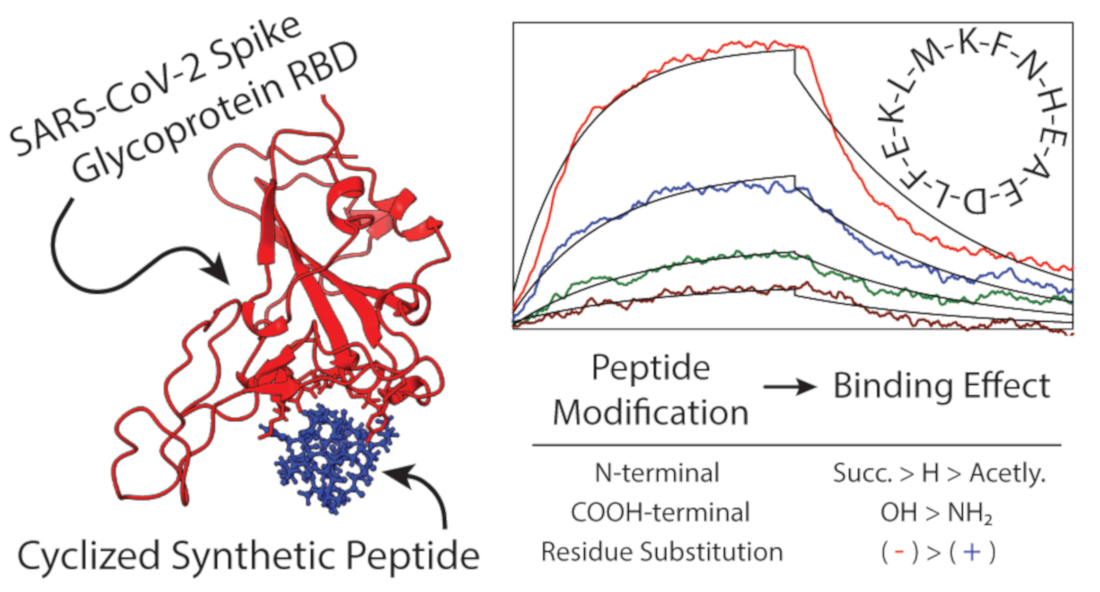
:
1. Introduction
2. Results
2.1. Development of the Synthetic Peptides
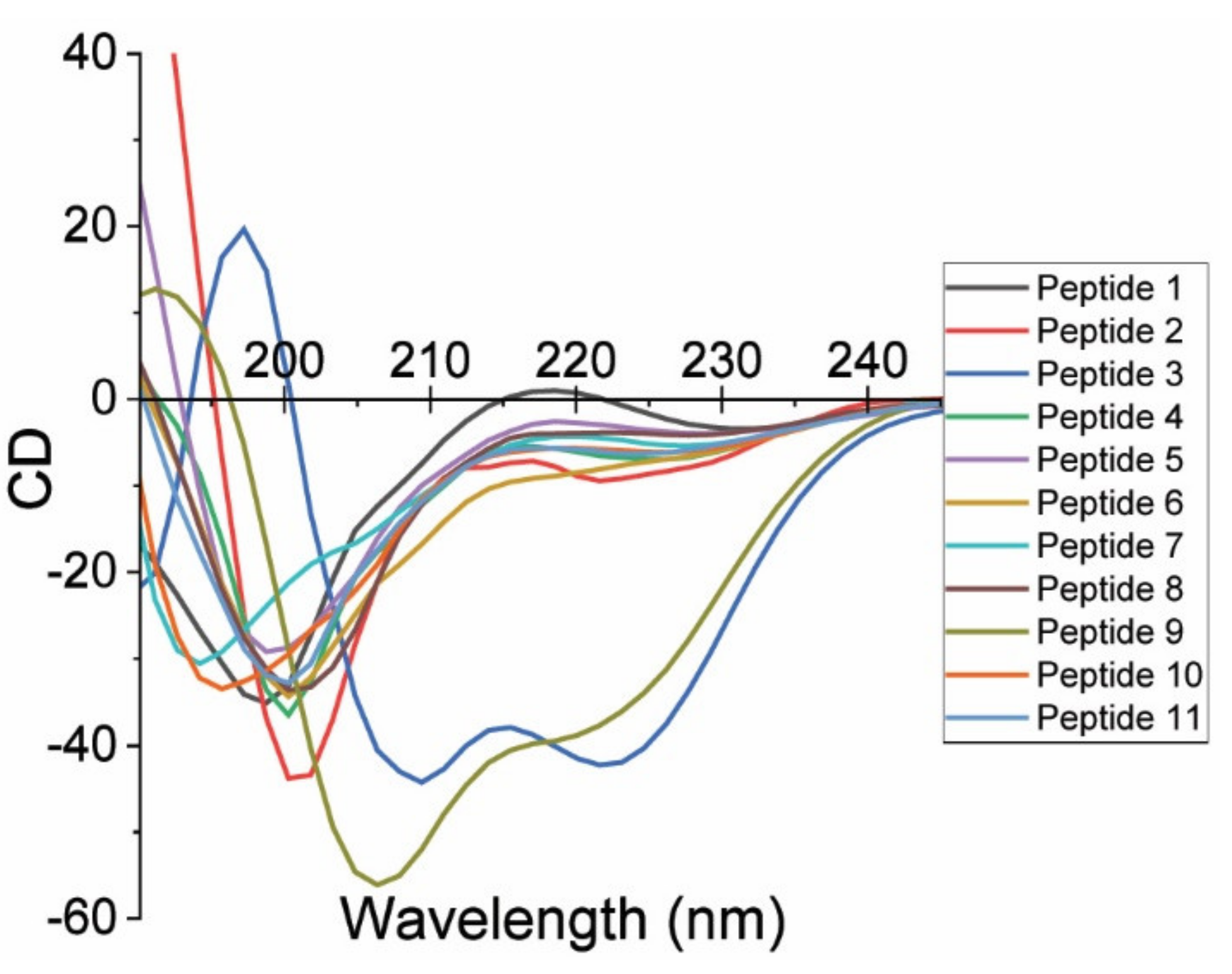
2.2. Circular Dichroism Measurements and Secondary Peptide Structure
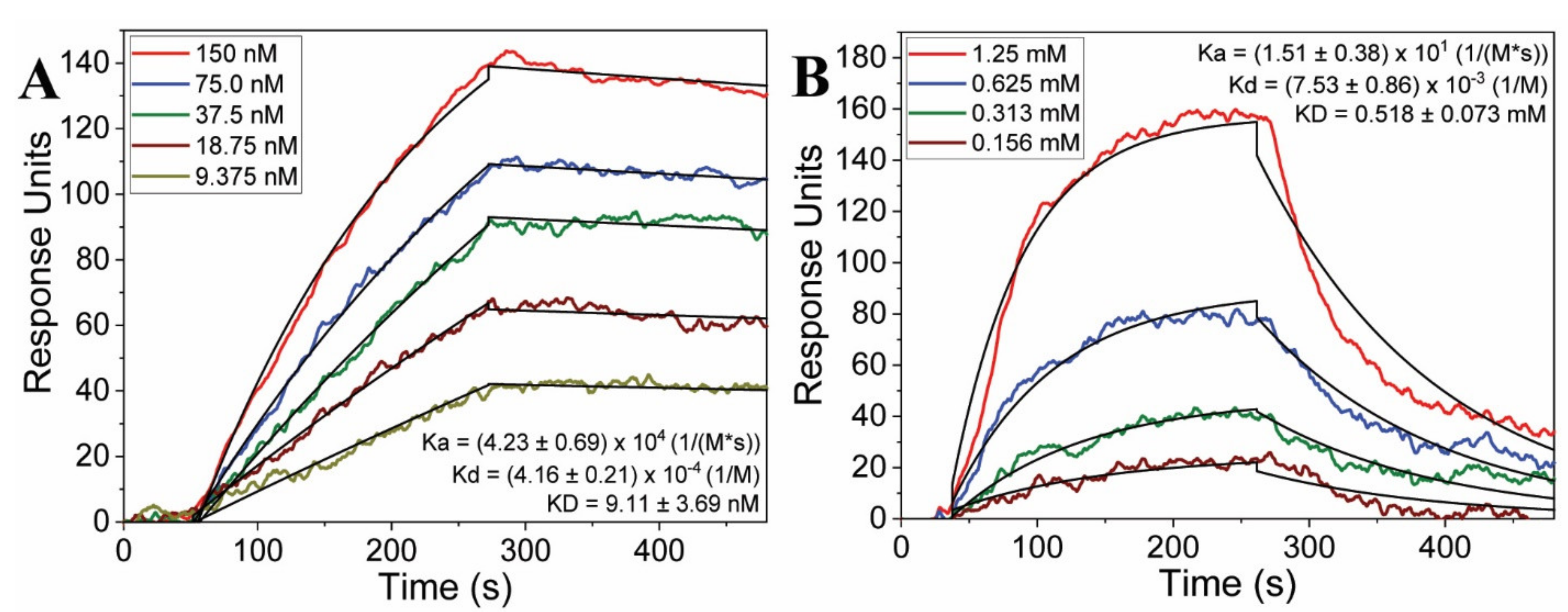
2.3. Binding Kinetics
2.4. Modeling Considerations
2.5. Residue Substitution and Electrostatic Effects
2.6. Consideration of the Influence of Charge at the RBD of SARS-CoV-2 Variants
3. Materials and Methods
3.1. Peptide Design and Synthesis
3.2. Recombinant Proteins
3.3. Peptide Circular Dichroism Measurements
3.4. Surface Plasmon Resonance (SPR) Measurements
3.5. Molecular Docking Simulations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wang, H.; Li, X.; Li, T.; Zhang, S.; Wang, L.; Wu, X.; Liu, J. The genetic sequence, origin, and diagnosis of SARS-CoV-2. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1629–1635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Z.; Holmes, E.C. A Genomic Perspective on the Origin and Emergence of SARS-CoV-2. Cell 2020, 181, 223–227. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 13 January 2022).
- Rabaan, A.A.; Al-Ahmed, S.H.; Haque, S.; Sah, R.; Tiwari, R.; Malik, Y.S.; Dhama, K.; Yatoo, M.I.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. SARS-CoV-2, SARS-CoV, and MERS-CoV: A comparative overview. Infez. Med. 2020, 28, 174–184. [Google Scholar] [PubMed]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Korteweg, C.; McNutt, M.A.; Gu, J. Pathogenetic mechanisms of severe acute respiratory syndrome. Virus Res. 2008, 133, 4–12. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.-W. Mutations Strengthened SARSCoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef]
- Amin, M.; Sorour, M.K.; Kasry, A. Comparing the Binding Interactions in the Receptor Binding Domains of SARS-CoV-2 and SARS-CoV. J. Phys. Chem. Lett. 2020, 11, 4897–4900. [Google Scholar] [CrossRef]
- Rajarshi, K.; Khan, R.; Singh, M.K.; Ranjan, T.; Ray, S.; Ray, S. Essential functional molecules associated with SARS-CoV-2 infection: Potential therapeutic targets for COVID-19. Gene 2021, 768, 145313. [Google Scholar] [CrossRef]
- Moghadas, S.M.; Vilches, T.N.; Zhang, K.; Wells, C.R.; Shoukat, A.; Singer, B.H.; Meyers, L.A.; Neuzil, K.M.; Langley, J.M.; Fitzpatrick, M.C.; et al. The Impact of Vaccination on Coronavirus Disease 2019 (COVID-19) Outbreaks in the United States. Clin. Infect. Dis. 2021, 73, 2257–2264. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Kasry, A.; Amin, M. The new SARS-CoV-2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med. Drug Discov. 2021, 10, 100086. [Google Scholar] [CrossRef] [PubMed]
- Luan, B.; Wang, H.; Huynh, T. Enhanced binding of the N501Y-mutated SARS-CoV-2 spike protein to the human ACE2 receptor: Insights from molecular dynamics simulations. FEBS Lett. 2021, 595, 1454–1461. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S. Evolutionary and structural analysis elucidates mutations on SARS-CoV2 spike protein with altered human ACE2 binding affinity. Biochem. Biophys. Res. Commun. 2021, 538, 97–103. [Google Scholar] [CrossRef]
- Hoffmann, M.; Arora, P.; Groß, R.; Seidel, A.; Hörnich, B.F.; Hahn, A.S.; Krüger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell 2021, 184, 2384–2393. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Fontanet, A.; Autran, B.; Lina, B.; Kieny, M.P.; Karim, S.S.A.; Sridhar, D. SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet 2021, 397, 952–954. [Google Scholar] [CrossRef]
- Kannan, S.R.; Spratt, A.N.; Sharma, K.; Chand, H.S.; Byrareddy, S.N.; Singh, K. Omicron SARS-CoV-2 variant: Unique features and their impact on pre-existing antibodies. J. Autoimmun. 2022, 126, 102779. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein–ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Washington, N.L.; Gangavarapu, K.; Zeller, M.; Bolze, A.; Cirulli, E.T.; Barrett, K.M.S.; Larsen, B.B.; Anderson, C.; White, S.; Cassens, T.; et al. Emergence and rapid transmission of SARS-CoV-2 B.1.1.7 in the United States. Cell 2021, 184, 2587–2594. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication Ex Vivo and transmission In Vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant. Cell 2021, 184, 3426–3437. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Dejnirattisai, W.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine-induced sera. Cell 2021, 189, 2348–2361. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Shayestehpour, M.; Mirzaei, H. The impact of spike mutated variants of SARS-CoV2 [Alpha, Beta, Gamma, Delta, and Lambda] on the efficacy of subunit recombinant vaccines. Braz. J. Infect. Dis. 2021, 25, 101606. [Google Scholar] [CrossRef]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2—What Do TheyMean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Shaw, R.H.; Supasa, P.; Liu, C.; Stuart, A.S.; Pollard, A.J.; Liu, X.; Lambe, T.; Crook, D.; Stuart, D.I.; et al. Reduced neutralisation of SARS-CoV-2 omicron B.1.1.529 variant by post-immunisation serum. Lancet 2022, 399, 234–236. [Google Scholar] [CrossRef]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124. [Google Scholar] [CrossRef]
- Huo, J.; Le Bas, A.; Ruza, R.; Duyvesteyn, H.; Mikolajek, H.; Malinauskas, T.; Tan, T.; Rijal, P.; Dumoux, M.; Ward, P.; et al. Neutralizing nanobodies bind SARS-CoV-2 spike RBD and block interaction with ACE2. Nat. Struct. Mol. Biol. 2020, 9, 846–854. [Google Scholar] [CrossRef]
- Pomplun, S. Targeting the SARS-CoV-2-spike protein: From antibodies to miniproteins and peptides. RSC Med. Chem. 2020, 12, 197–202. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef] [PubMed]
- Qiao, B.; Cruz, M.O.d.l. Enhanced Binding of SARS-CoV-2 Spike Protein to Receptor by Distal Polybasic Cleavage Sites. ACS Nano 2020, 14, 10616–10623. [Google Scholar] [CrossRef] [PubMed]
- Rane, J.S.; Pandey, P.; Chatterjee, A.; Khan, R.; Kumar, A.; Prakash, A.; Ray, S. Targeting virus–host interaction by novel pyrimidine derivative: An in silico approach towards discovery of potential drug against COVID-19. J. Biomol. Struct. Dyn. 2020, 39, 5768–5778. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, S.; Li, D.; Wei, D.-Q.; Zhao, J.; Wang, J. Human Intestinal Defensin 5 Inhibits SARS-CoV-2 Invasion by Cloaking ACE2. Gastroenterology 2020, 159, 1145–1147. [Google Scholar] [CrossRef]
- Souza, P.F.N.; Lopes, F.E.S.; Amaral, J.L.; Freitas, C.D.T.; Oliveira, J.T.A. A molecular docking study revealed that synthetic peptides induced conformational changes in the structure of SARS-CoV-2 spike glycoprotein, disrupting the interaction with human ACE2 receptor. Int. J. Biol. Macromol. 2020, 164, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Král, P. Computational Design of ACE2-Based Peptide Inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. [Google Scholar] [CrossRef] [Green Version]
- Ngwa, W.; Kumar, R.; Thompson, D.; Lyerly, W.; Moore, R.; Reid, T.; Lowe, H.; Toyang, N. Potential of Flavonoid-Inspired Phytomedicines against COVID-19. Molecules 2020, 25, 2707. [Google Scholar] [CrossRef]
- Huang, X.; Pearce, R.; Zhang, Y. De novo design of protein peptides to block association of the SARS-CoV-2 spike protein with human ACE2. Aging 2020, 12, 11263–11276. [Google Scholar] [CrossRef]
- Freitas, F.; Ferreira, P.; Favaro, D.; Oliveira, R. Shedding Light on the Inhibitory Mechanisms of SARS-CoV-1/CoV-2 Spike Proteins by ACE2-Designed Peptides. J. Chem. Inf. Model. 2021, 61, 1226–1243. [Google Scholar] [CrossRef]
- Rathod, S.; Prajapati, P.; Punjabi, L.; Prajapati, K.; Chauhan, N.; Mansuri, M. Peptide modelling and screening against human ACE2 and spike glycoprotein RBD of SARS-CoV-2. In Silico Pharmacol. 2020, 8, 3. [Google Scholar] [CrossRef]
- Wu, X.; Yu, K.; Wang, Y.; Xu, W.; Ma, H.; Hou, Y.; Li, Y.; Cai, B.; Zhu, L.; Zhang, M.; et al. Efficacy and Safety of Triazavirin Therapy for Coronavirus Disease 2019: A Pilot Randomized Controlled Trial. Engineering 2020, 6, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Merck & Co., Inc. Merck and Ridgeback’s Investigational Oral Antiviral Molnupiravir Reduced the Risk of Hospitalization or Death by Approximately 50 Percent Compared to Placebo for Patients with Mild or Moderate COVID-19 in Positive Interim Analysis of Phase 3 Study. Available online: https://www.merck.com/news/merck-and-ridgebacks-investigational-oral-antiviral-molnupiravir-reduced-the-risk-of-hospitalization-or-death-by-approximately-50-percent-compared-to-placebo-for-patients-with-mild-or-moderat/ (accessed on 5 October 2021).
- Angelucci, A.; Cavicchioli, M.; Cintorrino, I.A.; Lauricella, G.; Rossi, C.; Strati, S.; Aliverti, A. Smart Textiles and Sensorized Garments for Physiological Monitoring: A Review of Available Solutions and Techniques. Sensors 2021, 21, 814. [Google Scholar] [CrossRef] [PubMed]
- Ivanoska-Dacikj, A.; Stachewicz, U. Smart textiles and wearable technologies—Opportunities offered in the fight against pandemics in relation to current COVID-19 state. Rev. Adv. Mater. Sci. 2020, 59, 487–505. [Google Scholar] [CrossRef]
- Idumah, C.I. Influence of nanotechnology in polymeric textiles, applications, and fight against COVID-19. J. Text. Inst. 2020, 112, 2056–2076. [Google Scholar] [CrossRef]
- Saber, D.; El-Aziz, K.A. Advanced materials used in wearable health care devices and medical textiles in the battle against coronavirus (COVID-19): A review. J. Ind. Text. 2021, 15280837211041771. [Google Scholar] [CrossRef]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef]
- Glasgow, A.; Glasgow, J.; Limonta, D.; Solomon, P.; Lui, I.; Zhang, Y.; Nix, M.; Rettko, N.; Zha, S.; Yamin, R.; et al. Engineered ACE2 receptor traps potently neutralize SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 28046–28055. [Google Scholar] [CrossRef]
- Larue, R.C.; Xing, E.; Kenney, A.D.; Zhang, Y.; Tuazon, J.A.; Li, J.; Yount, J.S.; Li, P.-K.; Sharma, A. Rationally Designed ACE2-Derived Peptides Inhibit SARS-CoV-2. Bioconjugate Chem. 2021, 32, 215–223. [Google Scholar] [CrossRef]
- Curreli, F.; Victor, S.; Ahmed, S.; Drelich, A.; Tong, X.; Tseng, C.; Hillyer, C.; Debnath, A. Stapled Peptides Based on Human Angiotensin-Converting Enzyme 2 (ACE2) Potently Inhibit SARS-CoV-2 Infection In Vitro. mBio 2020, 11, e02451-20. [Google Scholar] [CrossRef]
- Morgan, D.C.; Morris, C.; Mahindra, A.; Blair, C.M.; Tejeda, G.; Herbert, I.; Turnbull, M.L.; Lieber, G.; Willett, B.J.; Logan, N.; et al. Stapled ACE2 peptidomimetics designed to target the SARS-CoV-2 spike protein do not prevent virus internalization. Pept. Sci. 2021, 113, e24217. [Google Scholar] [CrossRef] [PubMed]
- Chitsike, L.; Krstenansky, J.; Duerksen-Hughes, P.J. ACE2: S1 RBD Interaction-Targeted Peptides and Small Molecules as Potential COVID-19 Therapeutics. Adv. Pharmacol. Pharm. Sci. 2021, 2021, 1828792. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.H.; Ma, K.; Leeuwen, H.C.v.; Wasson, M.C.; Wang, X.; Idrees, K.B.; Gong, W.; Cao, R.; Mahle, J.J.; Islamoglu, T.; et al. Immobilized Regenerable Active Chlorine within a Zirconium-Based MOF Textile Composite to Eliminate Biological and Chemical Threats. J. Am. Chem. Soc. 2021, 143, 16777–16785. [Google Scholar] [CrossRef]
- Ali, A.; Vijayan, R. Dynamics of the ACE2–SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Sci. Rep. 2020, 10, 14214. [Google Scholar] [CrossRef]
- Rajpoot, S.; Ohishi, T.; Kumar, A.; Pan, Q.; Banerjee, S.; Zhang, K.; Baig, M. A Novel Therapeutic Peptide Blocks SARS-CoV-2 Spike Protein Binding with Host Cell ACE2 Receptor. Drugs R&D 2021, 29, 273–283. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Barh, D.; Tiwari, S.; Silva Andrade, B.; Giovanetti, M.; Almeida Costa, E.; Kumavath, R.; Ghosh, P.; Góes-Neto, A.; Carlos Junior Alcantara, L.; Azevedo, V. Potential chimeric peptides to block the SARS-CoV-2 spike receptor-binding domain. F1000Research 2020, 9, 576. [Google Scholar] [CrossRef]
- Chou, P.Y.; Fasman, G.D. Prediction of protein conformation. Biochemistry 1974, 13, 222–245. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.V.; French, A.D.; Jacks, T.; Rajasekaran, K. pH-Directed Self-Assembling Helical Peptide Conformation. In Small Wonders: Peptides for Disease Control; Rajasekaran, K., Cary, J.W., Jaynes, J.M., Montesinos, E., Eds.; ACS Symposium Series: Washington, DC, USA, 2012; Volume 1095, pp. 203–213. [Google Scholar]
- Woody, R.W. Chapter 2—Circular Dichroism of Peptides. In Conformation in Biology and Drug Design; Hruby, V.J., Ed.; Academic Press: Cambridge, MA, USA, 1985; Volume 7, pp. 15–144. [Google Scholar]
- Sim, S.; Kim, Y.; Kim, T.; Lim, S.; Lee, M. Directional Assembly of α-Helical Peptides Induced by Cyclization. J. Am. Chem. Soc. 2012, 134, 20270–20272. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odolczyk, N.; Marzec, E.; Winiewska-Szajewska, M.; Poznański, J.; Zielenkiewicz, P. Native Structure-Based Peptides as Potential Protein-Protein Interaction Inhibitors of SARS-CoV-2 Spike Protein and Human ACE2 Receptor. Molecules 2021, 26, 2157. [Google Scholar] [CrossRef] [PubMed]
- Matsoukas, J.; Apostolopoulos, V.; Lazoura, E.; Deraos, G.; Matsoukas, M.-T.; Katsara, M.; Tselios, T.; Deraos, S. Round and Round We Go: Cyclic Peptides in Disease. Curr. Med. Chem. 2006, 13, 2221–2232. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.Z.; Rhodes, C.A.; McCroskey, L.C.; Wen, J.; Appiah-Kubi, G.; Wang, D.J.; Guttridge, D.C.; Pei, D. Enhancing the Cell Permeability and Metabolic Stability of Peptidyl Drugs by Reversible Bicyclization. Angew. Chem. Int. Ed. 2017, 56, 1525–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, K.; Walport, L.J.; Walshe, J.L.; Solomon, P.D.; Low, J.K.K.; Tran, D.H.; Mouradian, K.S.; Silva, A.P.G.; Wilkinson-White, L.; Norman, A.; et al. Cyclic peptides can engage a single binding pocket through highly divergent modes. Proc. Natl. Acad. Sci. USA 2020, 117, 26728–26738. [Google Scholar] [CrossRef]
- Choi, J.-S.; Joo, S.H. Recent Trends in Cyclic Peptides as Therapeutic Agents and Biochemical Tools. Biomol. Ther. 2021, 28, 18–24. [Google Scholar] [CrossRef]
- Du Vigneaud, V.; Ressler, C.; Swan, J.M.; Roberts, C.W.; Katsoyannis, P.G. The synthesis of oxytocin. J. Am. Chem. Soc. 1954, 76, 3115–3121. [Google Scholar] [CrossRef]
- Remesic, M.; Lee, Y.S.; Hruby, V.J. Cyclic Opioid Peptides. Curr. Med. Chem. 2016, 23, 1288–1303. [Google Scholar] [CrossRef] [Green Version]
- Mastaglio, S.; Ruggeri, A.; Risitano, A.M.; Angelillo, P.; Yancopoulou, D.; Mastellos, D.C.; Huber-Lang, M.; Piemontese, S.; Assanelli, A.; Garlanda, C.; et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin. Immunol. 2020, 215, 108450. [Google Scholar] [CrossRef]
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.R.; Spratt, A.N.; Cohen, A.R.; Naqvi, S.H.; Chand, H.S.; Quinn, T.P.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Evolutionary analysis of the Delta and Delta Plus variants of the SARS-CoV-2 viruses. J. Autoimmun. 2021, 124, 102715. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Claro, I.M.; Candido, D.; Franco, L.A.M.; Andrade, P.S.; Coletti, T.M.; Silva, C.A.M.; Sales, F.C.; Manuli, E.R.; Gaburo, R.S.A.N.; et al. Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in Manaus: Preliminary Findings. Available online: https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586 (accessed on 14 October 2021).
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Zhou, S.; Tuttle, K.S.; Kim, A.; Li, W.; Dimitrov, D.S.; et al. Structural analysis of receptor binding domain mutations in SARS-CoV-2 variants of concern that modulate ACE2 and antibody binding. Cell Rep. 2021, 37, 110156. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Yahi, N.; Azzaz, F.; Chahinian, H. Structural dynamics of SARS-CoV-2 variants: A health monitoring strategy for anticipating COVID-19 outbreaks. J. Infect. 2021, 83, 197–206. [Google Scholar] [CrossRef]
- Jaafar, R.; Boschi, C.; Aherfi, S.; Bancod, A.; Bideau, M.L.; Edouard, S.; Colson, P.; Chahinian, H.; Raoult, D.; Yahi, N.; et al. High Individual Heterogeneity of Neutralizing Activities against the Original Strain and Nine Different Variants of SARS-CoV-2. Viruses 2021, 13, 2177. [Google Scholar] [CrossRef]
- Prevention, Centers for Disease Control and Prevention. COVID Data Tracker. Available online: https://covid.cdc.gov/covid-data-tracker/#variant-proportions (accessed on 18 October 2021).
- Franklin, J. Omicron Is Now the Dominant COVID Strain in the U.S., Making Up 73% of New Infections. Available online: https://www.npr.org/sections/coronavirus-live-updates/2021/12/20/1066083896/omicron-is-now-the-dominant-covid-strain-in-the-u-s-making-up-73-of-cases (accessed on 2 February 2022).
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
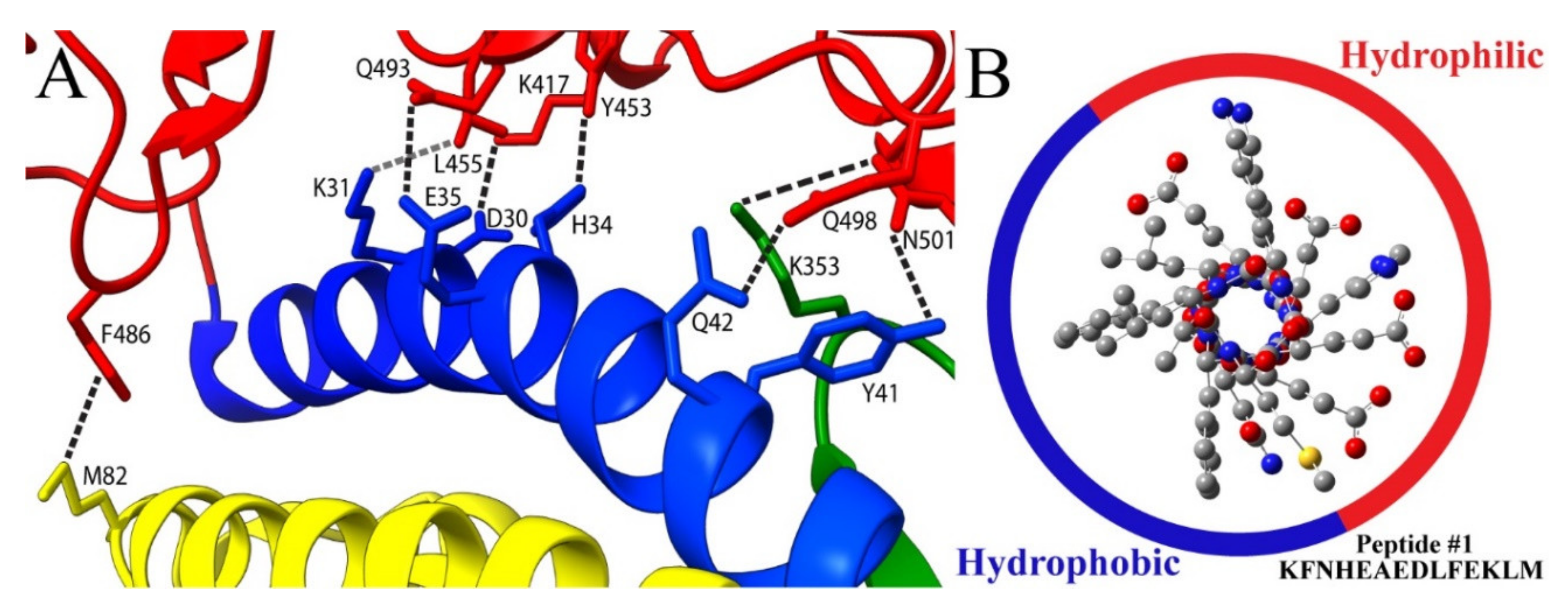{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Sequence—Head to Tail (N to C) | Isoelectric Point (pI) |
|---|---|---|
| 1 | KFNHEAEDLFEKLM-OH | 4.6 |
| 2 | KFNHEAEDLFEKLM-NH2 | 5.4 |
| 3 | KFNHEAEDLFEKLM (head-to-tail cycle) | 4.6 |
| 4 | Suc-KFNHEAEDLFEKLM-NH2 | 5.4 |
| 5 | Ac-KFNHEAEDLFEKLM-OH | 4.2 |
| 6 | CKFNHEAEDLFEKLMC-NH2 (S=S bridge) | 4.5 |
| 7 | Suc-KFNHEAEDLFEKLFEELFEDM-NH2 | 4.2 |
| 8 | GLFKEALEELWEA-NH2 | 4.3 |
| 9 | Suc-LLEKLLEWLE-NH2 | 4.6 |
| 10 | KFNDEAEDLFKLFEELFEDM-NH2 | 3.9 |
| 11 | MIEEQAKFLDKFNHEAEDLFK-NH2 | 4.8 |
| Peptide Number | Bulk Concentration (mM) | Binding (Yes/No) | Ka (M−1 s−1) | Kd (M−1) | KD (mM) |
|---|---|---|---|---|---|
| 1 | 5.0 | Yes | 4.62 ± 0.45 | (1.33 ± 0.27) × 10−2 | 2.84 ± 0.30 |
| 2 | 5.0 | Yes | 1.15 ± 0.08 | (2.58 ± 0.80) × 10−2 | 23.1 ± 0.84 |
| 3 | 1.25 | Yes | 15.1 ± 3.8 | (7.53 ± 0.87) × 10−3 | 0.518 ± 0.073 |
| 4 | 5.0 | Yes | 13.9 ± 3.6 | (1.87 ± 0.39) × 10−2 | 1.37 ± 0.07 |
| 5 | 5.0 | Yes | 0.64 ± 0.09 | (3.38 ± 0.06) × 10−2 | 42.9 ± 2.4 |
| 6 | 2.5 | Yes | 6.84 ± 2.46 | (1.12 ± 0.08) × 10−2 | 1.83 ± 0.54 |
| 7 | 1.25 | Yes | 4.29 ± 0.52 | (2.09 ± 0.39) × 10−2 | 5.05 ± 0.15 |
| 8 | 2.0 | Yes | 5.39 ± 1.31 | (1.19 ± 0.01) × 10−2 | 2.53 ± 0.58 |
| 9 | 2.5 | Yes | 5.83 ± 2.92 | (1.39 ± 0.16) × 10−2 | 3.37 ± 1.9 |
| 10 | 1.0 | Yes | 6.81 ± 2.41 | (1.75 ± 0.46) × 10−2 | 2.66 ± 0.28 |
| 11 | 1.0 | No | − | − | − |
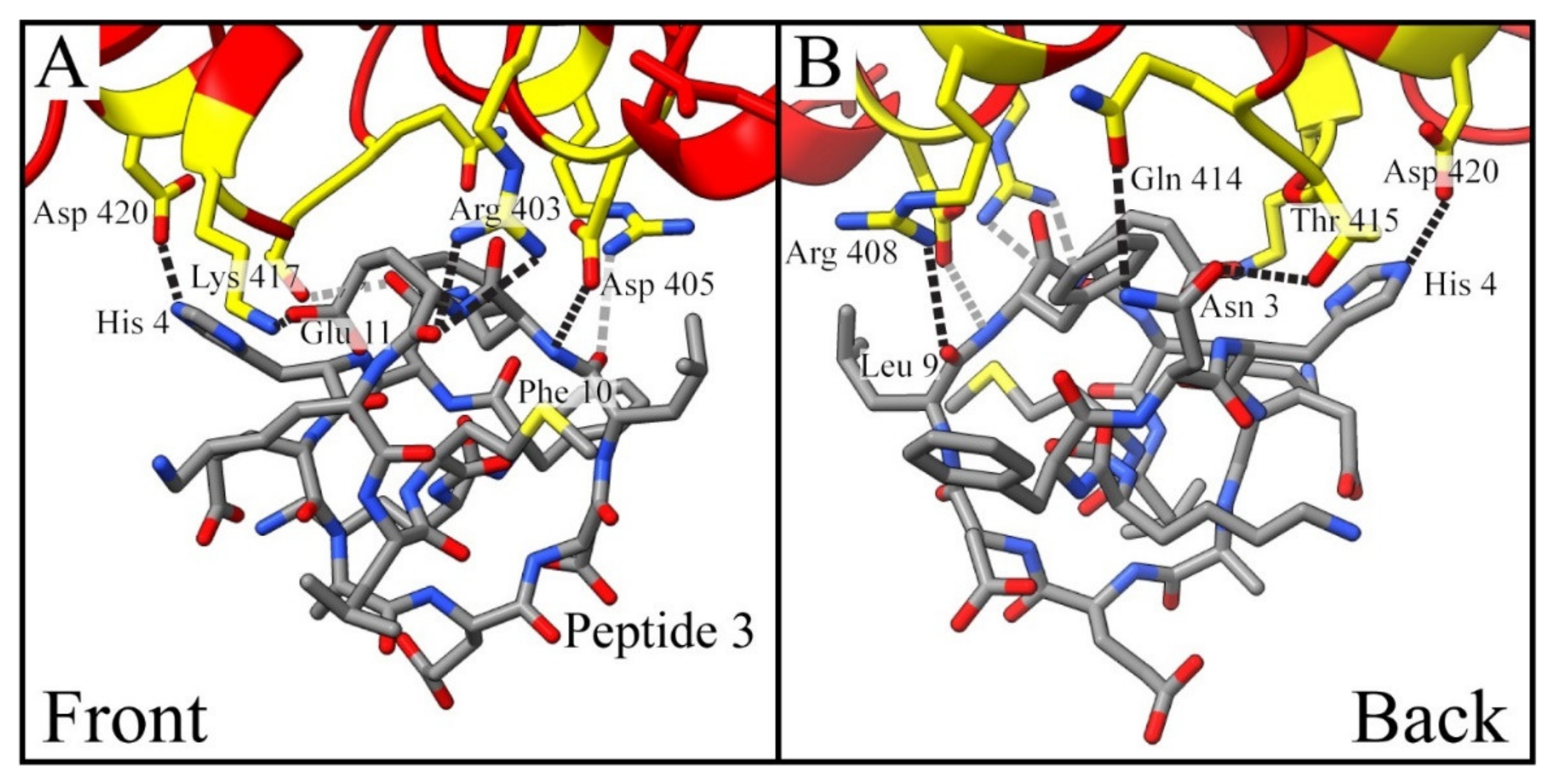
| Peptide 3 | Peptide 4 | ||
|---|---|---|---|
| Residue Interaction | Distance (Å) | Residue Interaction | Distance (Å) |
| Arg 403 → Glu 11 * | 2.9 | Arg 403 → Glu 7 | 3.5 |
| Arg 403 → Glu 11 * | 3.5 | Asp 405→ Met 14-NH2 | 3.1 |
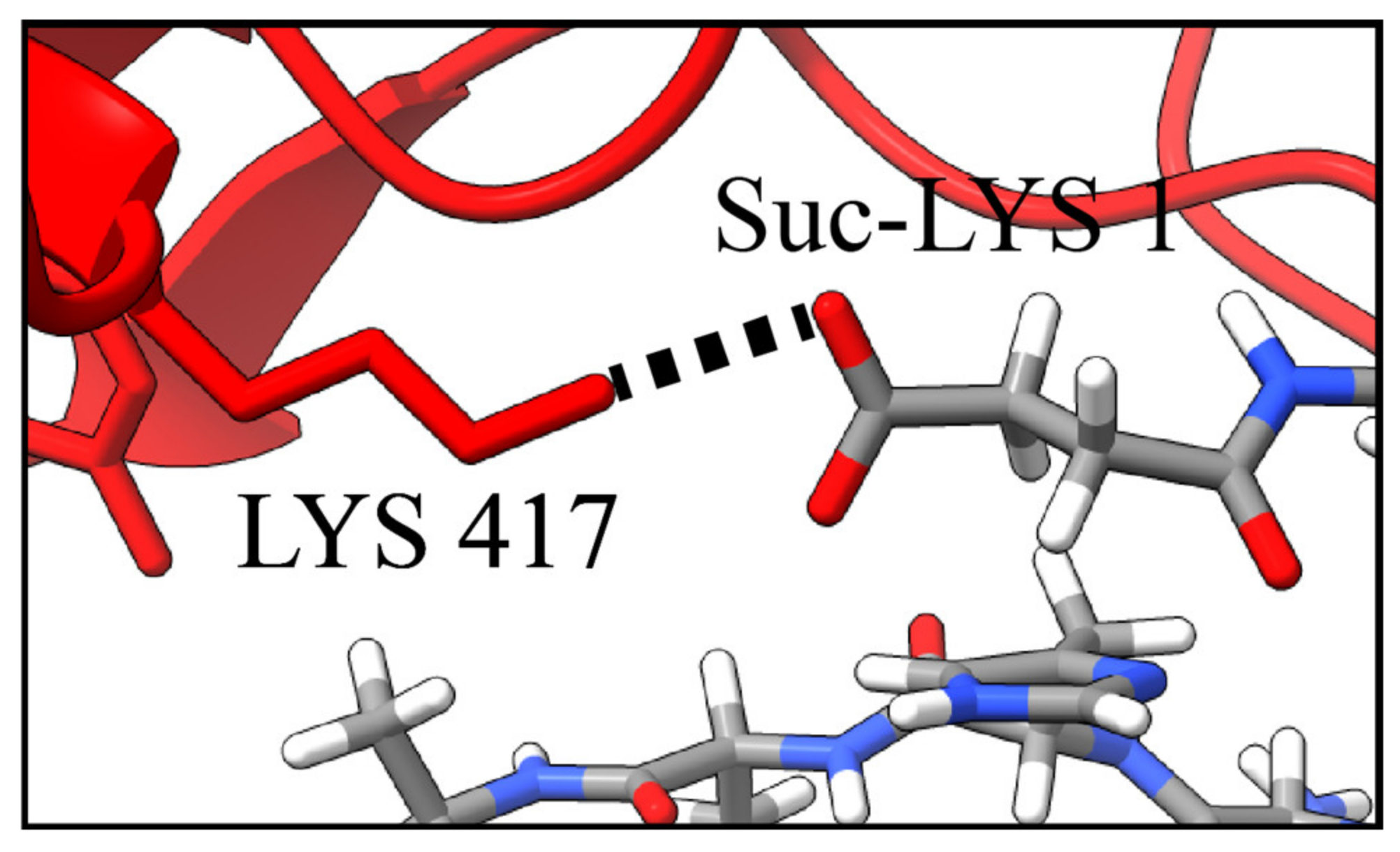
| Asp 405 → Phe 10 * | 2.6 | Lys 417 → Suc-Lys 1 | 2.8 |
| Arg 408 → Leu 9 * | 3.2 | Gly 496 * → Asp 8 | 3.2 |
| Gln 414 → Asn 3 | 2.6 | Asn 501 → Leu 9 * | 3.2 |
| Thr 415 → Asn 3 | 2.9 | Val 503 → Leu 13 * | 3.1 |
| Lys 417 → Glu 11 | 3.2 | Tyr 505 → Glu 7 | 2.9 |
| Asp 420 → His 4 | 3.7 | Tyr 505 → Glu 7 | 2.9 |
| Variant (Pango Lineage) | Spike Mutation | Residue Charge Change | Net Charge Change |
|---|---|---|---|
| Alpha (B.1.1.7) Overall charge change: Positive | D178H | (−) to neutral | (+) |
| E484K | (−) to (+) | (++) | |
| N501Y | neutral to neutral | no change | |
| A570D | neutral to (−) | (−) | |
| P681H | (+) to neutral | (−) | |
| T716I | neutral to neutral | no change | |
| S982A | neutral to neutral | no change | |
| D1118H | (−) to neutral | (+) | |
| Beta (B.1.351) Overall charge change: Positive | L18F | (+) to neutral | (−) |
| D80A | (−) to neutral | (+) | |
| D215G | (−) to neutral | (+) | |
| R246I | (+) to neutral | (−) | |
| K417N | (+) to neutral | (−) | |
| E484K | (−) to (+) | (++) | |
| N501Y | neutral to neutral | no change | |
| D614G | (−) to neutral | (+) | |
| A701V | neutral to neutral | no change | |
| Gamma (P.1) Overall charge change: Positive | L18F | neutral to neutral | no change |
| T20N | neutral to neutral | no change | |
| P26S | neutral to neutral | no change | |
| D138Y | (−) to neutral | (+) | |
| R190S | (+) to neutral | (−) | |
| K417T | (+) to neutral | (−) | |
| E484K | (−) to (+) | (++) | |
| N501Y | neutral to neutral | no change | |
| D614G | (−) to neutral | (+) | |
| H655Y | neutral to neutral | no change | |
| T1027I | neutral to neutral | no change | |
| Delta (B.1.617.2) Overall charge change: Positive | T19R | neutral to (+) | (+) |
| V70F | neutral to neutral | no change | |
| T95I | neutral to neutral | no change | |
| G142D | neutral to (−) | (−) | |
| E156- | remove (−) | (+) | |
| F157- | remove neutral | no change | |
| R158G | (+) to neutral | (−) | |
| A222V | neutral to neutral | no change | |
| W258L | neutral to (+) | (+) | |
| K417N | (+) to neutral | (−) | |
| L452R | neutral to (+) | (+) | |
| T478K | neutral to (+) | (+) | |
| D614G | (−) to neutral | (+) | |
| P681R | neutral to (+) | (+) | |
| D950N | (−) to neutral | (+) | |
| Omicron (B.1.1.529) Overall charge change: positive | A67V | neutral to neutral | no change |
| T95I | neutral to neutral | no change | |
| G142D | neutral to (−) | (−) | |
| L212I | neutral to neutral | no change | |
| G339D | neutral to (−) | (−) | |
| S371L | neutral to neutral | no change | |
| S373P | neutral to neutral | no change | |
| S375F | neutral to neutral | no change | |
| K417N | (+) to neutral | (−) | |
| N440K | neutral to (+) | (+) | |
| G446S | neutral to neutral | no change | |
| S477N | neutral to neutral | no change | |
| T478K | neutral to (+) | (+) | |
| E484A | (−) to neutral | (+) | |
| Q493R | neutral to (+) | (+) | |
| G496S | neutral to neutral | no change | |
| Q498R | neutral to (+) | (+) | |
| N501Y | neutral to neutral | no change | |
| Y505H | neutral to neutral | no change | |
| T547K | neutral to (+) | (+) | |
| D614G | (−) to neutral | (+) | |
| H655Y | neutral to neutral | no change | |
| N679K | neutral to (+) | (+) | |
| P681H | neutral to neutral | no change | |
| N764K | neutral to (+) | (+) | |
| D796Y | (−) to neutral | (+) | |
| N856K | neutral to (+) | (+) | |
| Q954H | neutral to neutral | no change | |
| N969K | neutral to (+) | (+) | |
| L981F | neutral to neutral | no change |
| Peptide | Calculated MW (g/mol) | Parent Ion (m/z) * | Purity by HPLC (%) |
|---|---|---|---|
| 1 | 1750.97 | 876.35 [2+] | 97.17 |
| 2 | 1749.98 | 876.00 [2+] | 97.51 |
| 3 | 1732.97 | 865.40 [2−] | 95.59 |
| 4 | 1850.07 | 925.95 [2+] | 97.19 |
| 5 | 1793.00 | 897.40 [2+] | 97.07 |
| 6 | 1954.27 | 978.10 [2+] | 96.49 |
| 7 | 2759.99 | 1380.90 [2+] | 95.86 |
| 8 | 1533.76 | 1534.65 [1+] | 96.62 |
| 9 | 1384.61 | 1385.60 [1+] | 96.40 |
| 10 | 2508.75 | 1256.35 [2+] | 96.62 |
| 11 | 2581.89 | 1291.85 [2+] | 96.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mackin, R.T.; Edwards, J.V.; Atuk, E.B.; Beltrami, N.; Condon, B.D.; Jayawickramarajah, J.; French, A.D. Structure/Function Analysis of Truncated Amino-Terminal ACE2 Peptide Analogs That Bind to SARS-CoV-2 Spike Glycoprotein. Molecules 2022, 27, 2070. https://doi.org/10.3390/molecules27072070
Mackin RT, Edwards JV, Atuk EB, Beltrami N, Condon BD, Jayawickramarajah J, French AD. Structure/Function Analysis of Truncated Amino-Terminal ACE2 Peptide Analogs That Bind to SARS-CoV-2 Spike Glycoprotein. Molecules. 2022; 27(7):2070. https://doi.org/10.3390/molecules27072070
Chicago/Turabian StyleMackin, Robert T., J. Vincent Edwards, E. Berk Atuk, Noah Beltrami, Brian D. Condon, Janarthanan Jayawickramarajah, and Alfred D. French. 2022. "Structure/Function Analysis of Truncated Amino-Terminal ACE2 Peptide Analogs That Bind to SARS-CoV-2 Spike Glycoprotein" Molecules 27, no. 7: 2070. https://doi.org/10.3390/molecules27072070
APA StyleMackin, R. T., Edwards, J. V., Atuk, E. B., Beltrami, N., Condon, B. D., Jayawickramarajah, J., & French, A. D. (2022). Structure/Function Analysis of Truncated Amino-Terminal ACE2 Peptide Analogs That Bind to SARS-CoV-2 Spike Glycoprotein. Molecules, 27(7), 2070. https://doi.org/10.3390/molecules27072070