
Clay, Zeolite and Oxide Minerals: Natural Catalytic Materials for the Ozonation of Organic Pollutants



Abstract
:1. Introduction
2. Ozonation
- Generation of BOD (biochemical oxygen demand) from DOC (dissolved organic carbon), minimizing ozone reactions with intermediate compounds. This would be the case of using ozone as a pretreatment to a biological process. In this case, the ozone concentration should be low and the process controlled by the absorption step.
- Elimination of DOC. In this case, mineralization is required and higher O3 concentrations are needed.
3. Catalytic Ozonation
3.1. Catalytic Ozonation Advantages
3.2. Catalytic Ozonation Mechanism
- chemisorption of ozone on the catalyst surface leading to the formation of active species which react with non-chemisorbed organic molecules;
- chemisorption of organic molecules (associative or dissociative) on the catalytic surface and their subsequent reaction with ozone;
- chemisorption of both ozone and organic molecules and the subsequent interaction between chemisorbed species.
- (i)
- Adsorption of the target pollutant and reaction intermediates: The target pollutant or intermediate reaction products may be adsorbed on the catalyst surface, generating an additional decay in the concentration of the compound, TOC (total organic carbon) or DOC, which could be misunderstood as catalytic activity. Therefore, the nature of the model molecule and its affinity towards the catalyst surface is of the utmost importance when evaluating catalytic activity. In addition, the adsorption of organic or inorganic species could block the actives sites. For example, Qi et al. [78] studied the catalyzed ozonation of 2-methylisoborneol (MIB), using different aluminum oxides: γ-AlOOH (HAO) and γ-Al2O3 (RAO). Both HAO and RAO could enhance ozone decomposition to generate hydroxyl radicals in the absence of MIB. However, the MIB adsorption capability of HAO was higher than that of RAO. Then, the adsorption of MIB on surface hydroxyl groups in HAO reduced the number of active sites participating in the ozonation reaction, inhibiting its catalytic effect.
- (ii)
- pH evolution during the reaction: Higher pH values promote O3 decomposition and generation of hydroxyl radicals. For example, Nawrocki and Fijołek [79] studied the catalytic activity of alumina and observed that sodium, the main contaminant in alumina, causes a pH increase after the introduction of the oxide in water. Then, ozone decomposition was attributed to the increase in pH and not to the true catalytic activity of the oxide.
- (iii)
- Leaching of active species and their contribution to the reaction: Yang et al. [80] determined the metal leaching from several solid catalysts (copper- and silver-oxide-based catalysts) and investigated the influence of the leached ions on the mineralization of two model compounds (oxalate and nitrobenzene). The homogeneous catalytic effect was found to be the dominant mechanism for the degradation of the model compounds under the chosen experimental conditions. The study aimed to draw attention to this important issue, since the homogeneous catalytic contribution could occur at very low concentrations, and just reporting the metal load over the solid catalyst before and after the reaction is insufficient. Moreover, the study presented by Inchaurrondo et al. [81] reported that the activity of the natural aluminosilicate, Montanit300®, was associated to the leached Mn, promoted by the interaction between the catalyst surface and carboxylic acids in solution. It is important to highlight that the homogeneous contribution was observed even at very low Mn concentrations (0.0074–0.066 mg/L). Then, the small traces of Mn present in the solid catalyst were key to the outstanding activity observed.
3.3. Main Operating Parameters in Catalytic Ozonation
3.3.1. Ozone Dose
3.3.2. Water pH
3.3.3. Water Matrix
3.3.4. Catalyst Dose
3.3.5. Temperature
3.3.6. Costs
4. Natural Mineral Catalysts
4.1. Clays
4.1.1. Definition
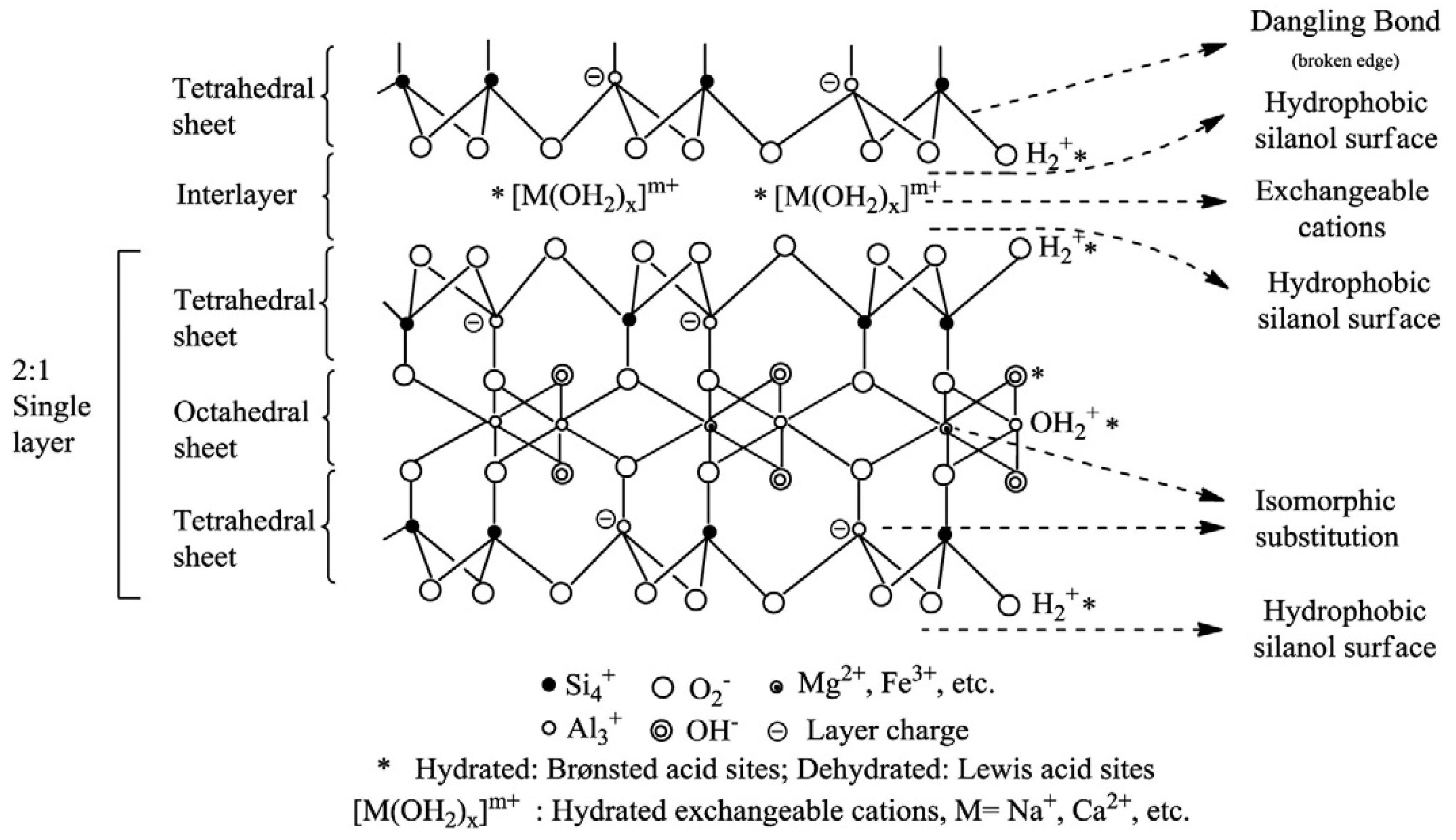
4.1.2. Structure and Properties

- Neutral siloxane surface: The siloxane surface is very unreactive due to the strong bond between atoms of silicon and oxygen. However, it presents hydrophobic characteristics with interesting adsorption properties.
- Isomorphic substitution sites: As mentioned before, the charge deficit originated from isomorphic substitution (usually Al for Si) is compensated by the presence of exchangeable cations. The location of the isomorphic substitution sites (octahedral or tetrahedral) influences the degree of polar or charged compounds’ adsorption. Isomorphic substitution in octahedral sheets creates soft Lewis base sites and in tetrahedral sheets, harder Lewis base sites. When the extent of Al for Si substitution increases, the hydrogen bonding to the charged surface by polar molecules increases.
- Metal cations occupying cation exchange sites: Organic solutes do not replace the exchangeable metal cation, but rather coordinate directly with the cation occupying the isomorphic substitution sites.
- 4.
- Hydrophobic sites: Through the sorption of organic molecules onto the clay’s surface. For example, the exchange of inorganic cations for alkyl ammonium cations on montmorillonite.
- 5.
- Water molecules surrounding exchangeable cations: The polarization of water molecules surrounding exchangeable cations or coordinated cations at broken edges (see point 6) generates Brönsted acid sites (proton donors). The strength of acidity depends on the nature of the exchangeable cation and amount of water. The presence of cations with higher charge and reduced size results in higher acidity.
- 6.
- Broken edge sites and exposed surface silanol and aluminol groups: These are surface hydroxyl groups located on the broken edges of clay minerals, which can form inner-sphere complexes with metal species, hydrogen bond to molecules accumulated at the interface, undergo replacement reactions with deuterium, tritium, and F− or be influenced by inorganic or organic cations through electrostatic interactions. These structural hydroxyl groups are among the most abundant and reactive active sites found on particles in soil. As the particle size decreases, the contribution of these sites to the overall reactivity increases.
4.1.3. Modification of Natural Clays
- (i)
- Ion-exchange reaction: The cation exchange capacity of clays is the most important parameter, which depends on the layer charge density. Cations present in the interlayer space of clays can be exchanged for the desired catalytic ion. Moreover, hydrophilic clays can be changed into hydrophobic clays, for example, with the exchange of inorganic cations for alkyl ammonium cations.
- (ii)
- Pillared interlayered clays (PILC): The interlamelar space in clays is generally not accessible to all substrates due to the strong electrostatic interaction between sheets and charge balancing cations. Therefore, modification through the introduction of large cations (“pillars”) between the sheets followed by calcination (the polycations are converted into the corresponding metal oxide clusters), increases porosity and surface area. In addition, the pillars made by a combination of Al with other metals (i.e., iron) introduce or constitute additional catalytic sites [121].
- (iii)
- Acid activation: The treatment with acids causes ion exchange reactions with H+, breakdown of layers causing more broken edge bonds and a reduction in particle size, and a selective leaching of central atoms in tetrahedral and octahedral sheets. In general, the acid treatment causes an increment in the amount of acid sites and surface area of the materials [21]. In addition, drastic acid treatments may cause dealumination (higher Si:Al ratio), increasing the hydrophobic characteristics of the material.
- (iv)
- Calcination: Purification of the materials is often performed through controlled calcination up to 400–500 °C, in order to remove carbonates and organic impurities.
4.1.4. Application of Clays in Ozonation Processes

{kind=link}
{kind=link}
{kind=link}
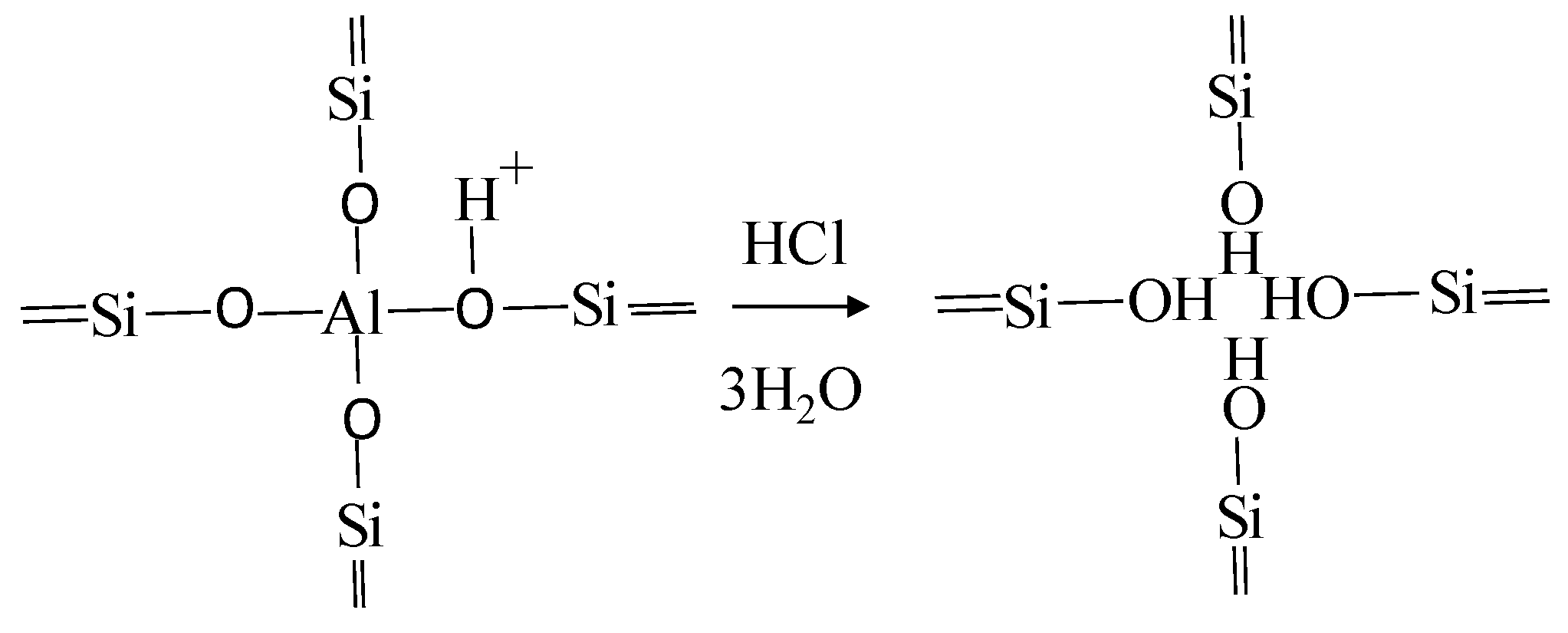{kind=link}
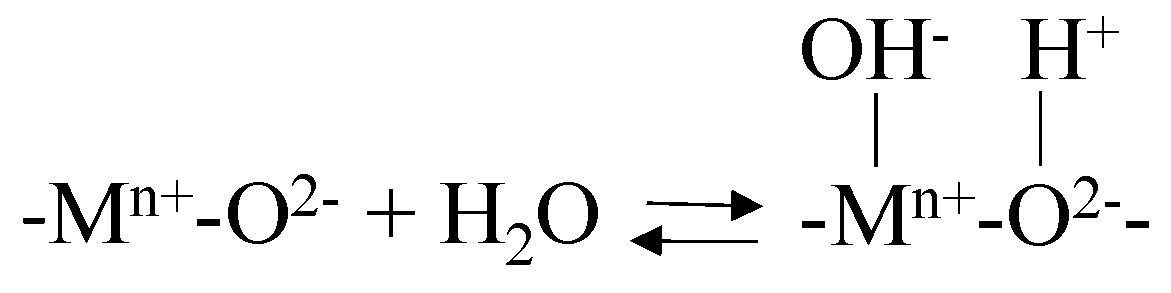{kind=link}
| Catalyst | Pollutant | Operating Conditions | Reaction Outcomes (X: Percentage Conversion) | Ref. |
|---|---|---|---|---|
| Crude bentonite (B), acid activated montmorillonite (HMt) and ion exchanged montmorillonite (NaMt, Fe(II)Mt) | Cationic dyes (methylene blue and methyl green) and Anionic dyes (methyl orange and methyl-thymol blue) | Semibatch reactor, O3 dose 600 mg/h, 22 °C, 20 mL of 10−4 M dye, catalyst load 40 mg, 5 min. | Dye conversion yields: B: 94–100%, NaMt: 72–96%, Fe(II)Mt: 95–96%, HMt-1:99–100%. The catalyst addition accelerated changes in relative absorbances of most UV-vis bands below 350 nm. | [122] |
| Thermal treated clay from tidal flat sediments | Perchloroethylene (PCE) | Batch reactor, O3 saturated water (5.8 ppm), 20 °C, 50 mL of 0.03 mM PCE, 0.8 wt.% clay, 10 min. | PCE Conversion: O3 alone: 60.6%, O3 + clay treated at 700 °C: 93.9%. | [63] |
| Kaolin plus MnO2 | Methylene blue (MB) | Bubbled column semibatch reactor, O3 dose 2.5 g/h, room T, 0.2 L of 0.3 g/L MB, 10 g kaolin and 5 g MnO2, 5 min, pH0 11. | COD Conversion: MnO2 + O3: 20%, kaolin+O3: 40%, MnO2 + O3 + kaolin: 80%. | [58] |
| CuFe2O4 nanoparticles supported on sepiolite (SEP) | Quinoline (QN) | Semibatch reactor, gas flow rate 1 L/min, room T, 0.5 L of 50 mg/L QN, catalyst load 1 g/L, 30 min, pH0 6.8. | TOC Conversion: O3 alone: 16.8%, SEP + O3: 44.1%, CuFe2O4 + O3: 55.8%, CuFe2O4/SEP + O3: 90.3%. Homogeneous contribution: 23.7%. Leaching: 0.41 mg/L Cu and 5.2 µg/L Fe. Negligible adsorption. | [123] |
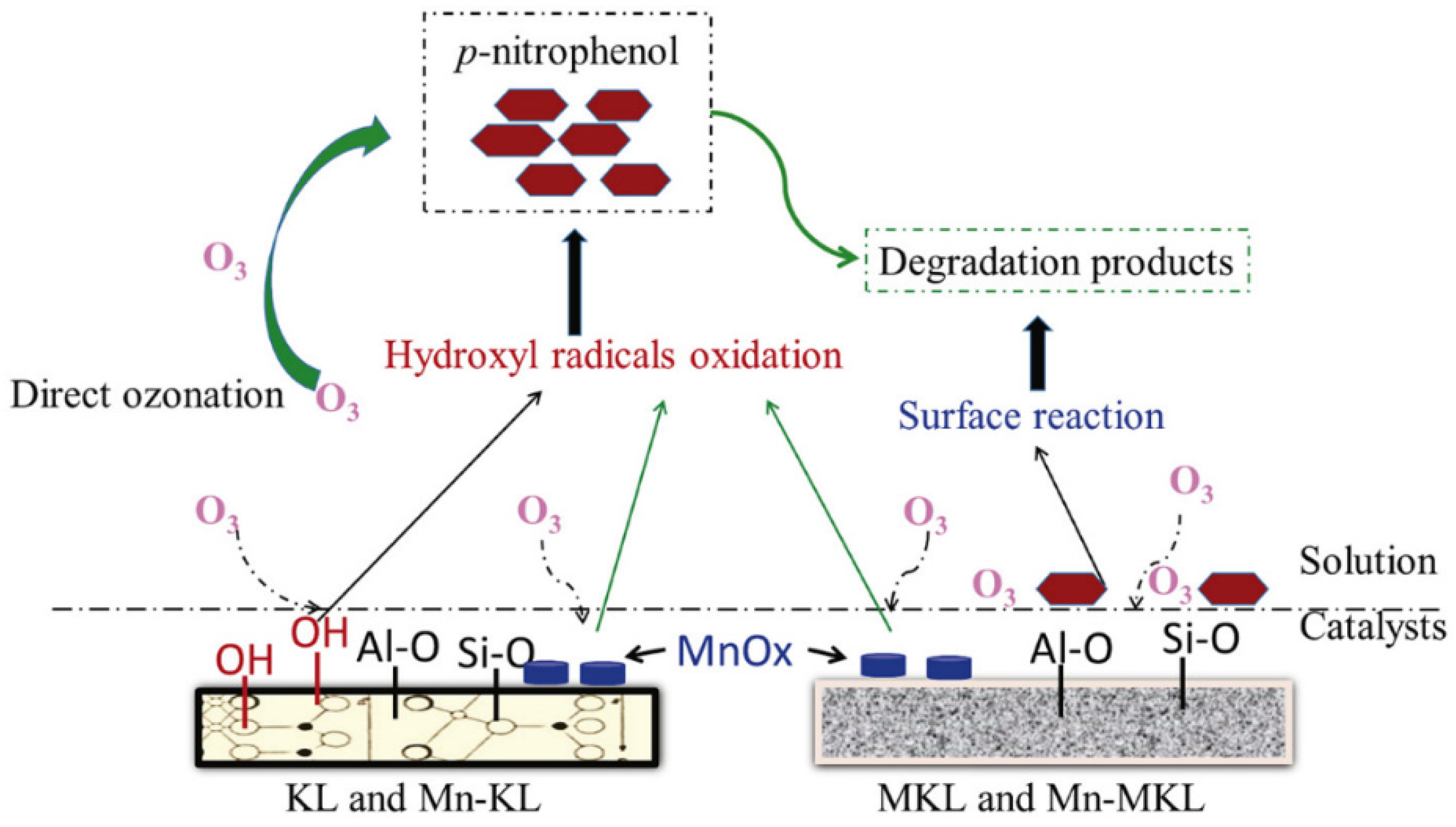
| Kaolinite (KL), Metakaolinite (MKL) and Mn oxides supported on KL (Mn-KL and Mn-MKL, 3% Mn loading) | p-Nitrophenol (NP) | Semibatch column reactor 0.25 L, gas flow rate 0.5 L/min, 30 °C, 0.1 L of 300 mg/L NP, catalyst load 0.5 g, 30 min, pH0 5.2. | TOC Conversion: Adsorption: 1–3%, O3 alone: 32%, KL+O3: 46%, MnKL + O3: 66%, MKL + O3: 41%, MnMKL + O3: 54%. Manganese leaching: 5.8% MnKL and 6.5% MnMKL. | [124] |
| Ion exchanged montmorillonite (NaMt, FeMt; CoMt; NiMt; CuMt) | Oxalic acid (OA) | Semibatch reactor, O2+O3 mixture throughput 5 mg/min, 25 °C, 0.5 L of 2.5 × 10−3 M OA, catalyst load 1 g, 180 min. | OA Conversion: O3 alone: 22.5%, Crude Mt + O3: 69%, NaMt + O3: 70.4%, CuMt + O3: 78.6%, CoMt + O3: 80.1%, NiMt+O3: 83.1%, FeMt + O3: 95.2%. | [52] |
| Ion exchanged montmorillonite (NaMt, FeMt; CoMt; NiMt; CuMt) | Oxalic acid (OA) | Semibatch reactor, O3 stream 6 mg/min, 25 °C, 10−3 M OA, catalyst load 2 g/L, pH 2.8, 60 min. | OA conversion: O3 alone: 1–2%, Crude Mt + O3: 5–6%, NaMt+O3: 10%, Fe(III)Mt + O3: 48%, Cu(II)Mt + O3: 70%, Co(II)Mt + O3: 95%, Ni(II)Mt + O3: 83%, Fe(II)Mt + O3: 100%. | [95] |
| Ion exchanged montmorillonite (NaMt, FeMt; CoMt; NiMt; CuMt) | Sulfamethoxazole (SMX) | Semibatch reactor, O3 feed 6 mg/min, 3 × 10−4 M SMX, catalyst load 1.91 g/L, pH 2.88, 20 min. | COD Conversion: NaMt + O3: 84%, FeMt + O3: 98%, CuMt + O3: 92%, CoMt + O3: 97%, NiMt + O3: 95%. | [120] |
| Fe-pillared clay | Indigo carmine (IC) | Semibatch bubble column reactor, gas flow rate 0.045 L/min, average O3 production 0.005 g/L, 1000 mg/L IC, 19 °C, catalyst load 0.1% w/w, pH 3, 10 min. | IC conversion: Adsorption: 2.9%, O3 alone: 25%, bentonite + O3: 30%, Fe particles: 40%, Fe-pillared clay + O3: 98%. | [126] |
| Montmorillonite (K10) and Al-Fe modified Montmorillonite (AFK 10) | Methylene blue (MB) and Malachite green (MG) | Semibatch reactor 0.06 L, O3 dose 0.5 g/h, 30 °C, 5 × 10−5 M dye, catalyst load 5 mg/L, 40 s. | Dye conversion: Adsorption: MB/K10: 78%, MG/K10: 51%, MB/AFK10: 24%, MG/AFK10: 56%. O3 alone: MG: 46%, MB: 17%. Catalytic ozonation: MB/K10: 59%, MG/K10: 70%, MB/AFK10: 99%, MG/AFK10: 88%. | [127] |
| Sepiolite (SEP) and Zero valent iron on sepiolite (ZVI/SEP) | Caffeine | Semibatch reactor 0.25 L, gas flow rate 6 mg/min ([O3]aqueous = 4 mg/L at t = 0), 25 °C, 10 mg/L caffeine, catalyst load 1 g/L, 60 min, pH 6. | TOC Conversion: O3 alone: 17.5%, SEP+O3: 30%, ZVI/SEP + O3: 41.1%. Caffeine adsorption 76%. | [128] |
| Kaolinite (KAO) from sulfide flotation wastewater | O-isopropyl-N-ethylthionocarbamate (IPETC) | Semibatch reactor 2.5 L, gas flow rate 1.67 L/min, O3 dose 2.065 mg/(min L), 20 °C, 2 L of 100 mg/L IPETC, catalyst load 0.5 g/L, 180 min, pH0 10. | IPETC Conversion (60 min): O3 alone: 84.81%, KAO + O3: 98.31%, adsorption: 4.96%. TOC Conversion (180 min): O3 alone: 12.47%, KAO + O3: 29.01%, adsorption: 2.58%. | [129] |
| Nanovermiculite loaded with Fe2+ | Sanitary landfill leachate | Semibatch rotating bed reactor 1.6 L, rotation speed 915 rpm, gas flow rate 3.9 L/min, O3 generator capacity: 34 g/h at 5 L/min, 23 °C, 0.2 L of 860 mg/L COD, catalyst load 0.05 g, 60 min, pH 5.8. | COD Conversion: Catalytic ozonation: 15.9%, adsorption: 1.7%. Color removal: 42.4%. | [130] |
| Raw sepiolite (SEP), Modified sepiolite (MOD-SEP), Mn-loaded modified sepiolite (Mn-SEP) | Regenerated papermaking wastewater | Semibatch reactor 1.6 L, gas flow rate 0.4 L/min, O3 20 mg/L in gas phase, room T, 1 L of 140–200 mg/L COD, catalyst load 0.8 g, 30 min, pH0 8. | COD Conversion: O3 alone: 34.8%, SEP + O3: 50.3%, MOD-SEP + O3: 62.7%, Mn-SEP + O3: 73.4%. Mn leaching < 0.2 mg/L. | [131] |
4.2. Zeolites
4.2.1. Definition
“A zeolite mineral is a crystalline substance with a structure characterized by a framework of linked tetrahedra, each consisting of four O atoms surrounding a cation. This framework contains open cavities in the form of channels and cages. These are usually occupied by H2O molecules and extra-framework cations that are commonly exchangeable. The channels are large enough to allow the passage of guest species. In the hydrated phases, dehydration occurs at temperatures mostly below about 400 °C and is largely reversible. The framework may be interrupted by (OH,F) groups; these occupy a tetrahedron apex that is not shared with adjacent tetrahedral [132]”.
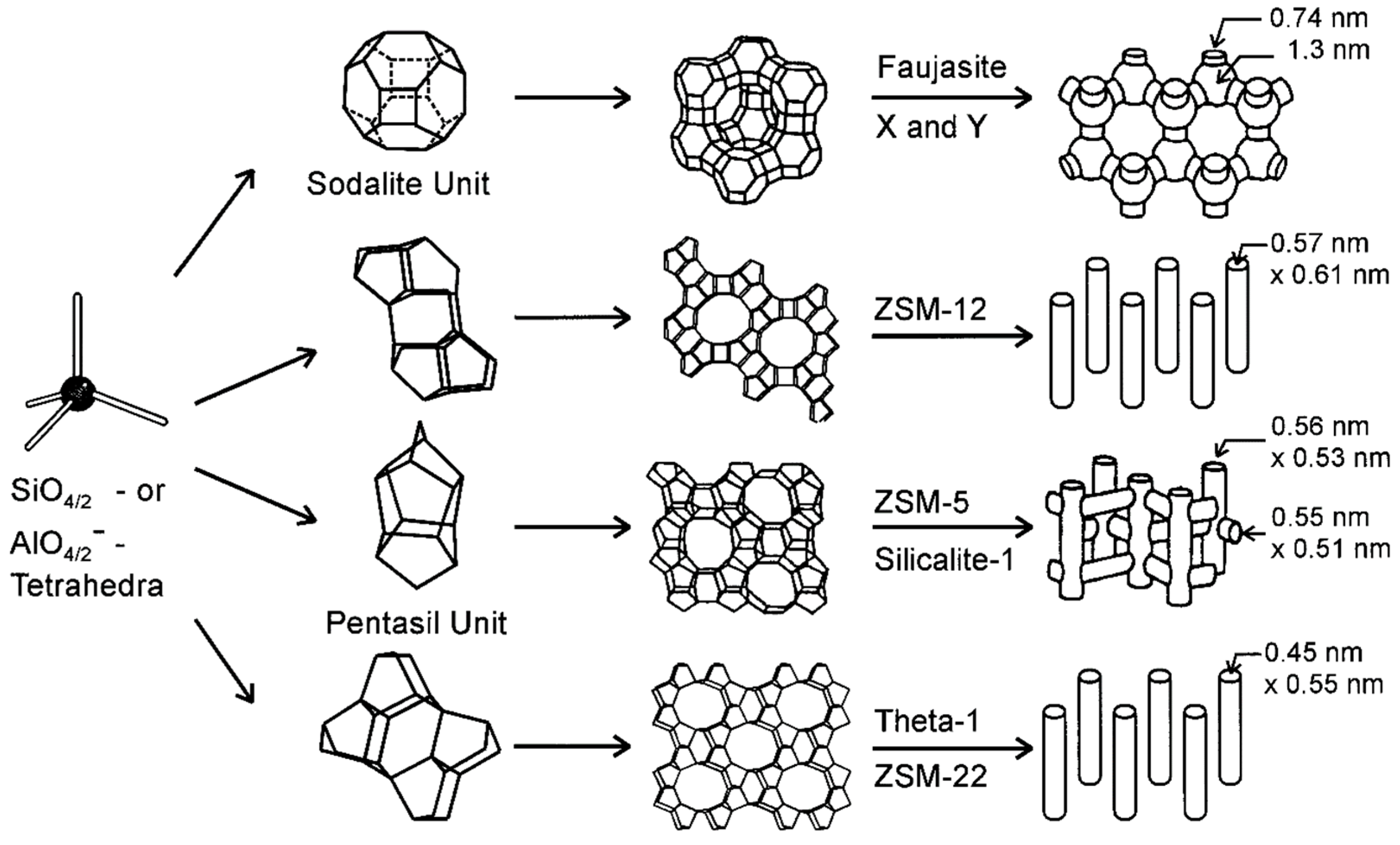
4.2.2. Structure and Properties
4.2.3. Modification of Natural Zeolites
- (i)
- Purification: Simple acid washing can remove impurities. In addition, purification usually involves available mineral processing equipment such as jaw crushers, crushing rolls, hydrocyclones, classifiers and shaking tables [140].
- (ii)
- Thermal Reduction: Is usually during the preparation of bifunctional catalysts. The zeolite is exchanged with the cationic form of the metal that will be used as the catalyst, and afterward is reduced in H2 atmosphere [140].
- (iii)
- Acid treatment with mineral acids: The acid treatment may remove the impurities that are blocking pores in the structure, leading to an increase in the specific surface area and microporosity of the material. In addition, cations are exchanged for H+, obtaining a proton-exchanged zeolite. Depending on the aggressiveness of the treatment (acid strength, temperature), it can lead to a progressive dealumination of the material, which causes a reduction in the cation exchange capacity and under drastic conditions, to the collapse of the structure [133]. On the other hand, a higher Si/Al ratio increases the hydrophobic character of the material, which is interesting in applications related to the adsorption/separation of non-polar molecules [133].
- (iv)
- Ammonium exchange: Another modification procedure to obtain a proton-exchanged zeolite is the ammonium exchange followed by calcination, leading to desorption of ammonia and the bonding of a hydroxyl proton at the bridging oxygen of an Si–O–Al arrangement [138]. Ammonium exchange can maintain the structure while acid treatment generally results in dealumination and reduction in thermal stability [133].
- (v)
- Modification by surfactants: Cationic (organic) surfactants are used to extend the adsorption capacity of zeolites to a broader spectrum of pollutants (for example, organic molecules and anionic compounds) [141]. Surfactants form a monolayer or “hemicelle” at the solid–aqueous interface via strong ionic bonds at surfactant concentrations below the critical micelle concentration (CMC). The hydrophobic tails of the surfactant molecules associate to form a bilayer when the concentration exceeds the CMC. These different coverings are favorable for the adsorption of different compounds. It has been reported that the removal of hydrocarbons is optimal at the point of full monolayer coverage [141]. On the other hand, anionic adsorption occurs in the bilayer configuration [141].
- (vi)
- Heat treatment (T ≥ 773 K) or steaming of acidic zeolites: This causes dihydroxylation of Brønsted acid sites and formation of Lewis acid sites. When framework dealumination occurs, the formation of Lewis acids sites can be attributed to the appearance of extra-framework aluminum species of octa-, penta-, or tetrahedral coordination [138].
- (vii)
- Hydrothermal Treatments (zeolite synthesis): Treatment with highly basic sodium or potassium hydroxide solutions causes amorphization and change in chemical composition; hence this treatment produces a highly active aluminosilicate gel, very useful for succeeding zeolite synthesis using as raw material the amorphous solid phase [140]. Synthetic zeolites are more expensive compared to the similar natural counterparts; however, they present a higher cation exchange capacity and improved tunable acidity and pore size (for example in hierarchical zeolites).
- (viii)
- Generation of a hierarchical structure: The presence of only microporous channels in zeolites may restrict diffusion, limit the selectivity range of desired products and promote deactivation by coking. Therefore, different synthesis or modification strategies have been studied to obtain hierarchical structured zeolites with not only their inherent microporosity, but also mesoporosity or even macroporosity [139,142]. In general, these methods can be subdivided in two main groups: (I) the top-down, where mesopores can be created in available zeolites by etching away a part of it, and in some cases recrystallizing again and (II) the bottom-up methods, where a hierarchical system is created during the zeolite synthesis [143]. From the top-down methods, dealumination is the oldest technique, which can be performed by means of steam, acid, or heat treatment [143]. However, there are some important disadvantages such as partial amorphization of the zeolite, wide mesopores size distribution and the existence of cavities and mesopores not connected to the surface. Another alternative is desilication, where the zeolite is contacted with an alkaline solution. The Si-O-Si bonds are selectively hydrolyzed causing the preferential removal of Si from the zeolite framework and the formation of mesopores. However, when using too concentrated alkaline solutions, a lot of zeolite material can be lost and microporosity could decrease drastically [143].
4.2.4. Application of Natural Zeolites in Ozonation Processes
| Catalyst | Pollutant | Operating Conditions | Reaction Outcomes | Ref. |
|---|---|---|---|---|
| Iron-coated zeolite (clinoptilolite) (ICZ) | Humic acid (HA) | Semibatch reactor 2 L, gas flow rate 2 L/min, O3 10 mg/L, 24 °C, 30 mg/L HA, catalyst load 0.75 g/L, 60 min, pH0 6.5. | COD Conversion: Adsorption: 7.15%. O3 alone: 21.39%. Catalytic ozonation with IZC: 62%. Catalytic ozonation with natural zeolite: 22.31%. Fe leached 54.59µg/L | [144] |
| Cerium-loaded natural zeolite (CZ) | Penicillin G (PG) | Semibatch reactor 1 L, gas flow rate 0.6 L/min, O3 dose 6 mg/min, 25 °C, 50 mg/L PG, catalyst load 2 g/L, 120 min, pH0 4.5. | TOC Conversion: Adsorption: 4%. O3 alone: 9%. Catalytic ozonation: 22%. Ce leached in cycles of 20 min: 0.057 mg/L (1st cycle), 0.246 mg/L (5th cycle). | [145] |
| Natural zeolite (Chilean mining company “Minera Formas) | O3 decay | Differential circular flow reactor: after reaching dissolved O3 saturation (125 µM, 20 °C) the gas flow (2 L/min) is closed and begins liquid recirculation (1.5 L/min, 1 L) through the fixed bed (19 mL, 15 g/L). | Apparent first order ozone decay constants (×103 s−1): O3 alone: 0.5 (pH 2), 0.6 (pH 7), 3 (pH 8). Catalytic ozonation: 0.9 (pH 2), 1 (pH 7), 3.6 (pH 8). | [89] |
| Acid-treated natural zeolite (53% clinoptilolite, 40% mordenite, 7% quartz) (AZ) | Methylene blue | Differential circular flow reactor: after reaching dissolved O3 saturation (125 µM, 20 °C) the gas flow (2 L/min) is closed, MB is added (94 × 10−6 M) and begins liquid recirculation (1.5 L/min, 1 L) through the fixed bed (19 mL, 15 g/L), pH0 6. | Pseudo first order rate constants of MB removal (×103 s−1): O3 alone: 0.5, O3 + AZ: 5, AZ:1.9. | [146] |
| Montanit300® (M, natural volcanic stone, rich with zeolite clinoptilolite) and acid treated Montanit300® (5MH45 with HCl and 80MS with H2SO4) | Orange II (OII) | Semibatch reactor 1 L, O3 dose 14 mg/(L min), 24 °C, 0.5 L of 100 mg/L OII, catalyst load 1 g/L, 240 min, pH0 6. | TOC Conversion: Adsorption: Negligible. O3 alone: 66%. Zeolite + O3: 91% (M), 65% (5MH45), 88% (80MS). Fe leached (mg/L): 0.95 (M), 0.5 (5MH45), 0.8 (80MS). Mn leached (mg/L): 0.066 (M), 0.0037 (5MH45), 0.0074 (80MS). | [81] |
| Natural zeolite Faujasite | Landfill leachate | Semibatch reactor, O3 dose 27 mg/m3, <15 °C, 0.5 L of 2500 mg/L COD, catalyst load 160 g/L, 100 min, pH0 5–9. | COD Conversion: Adsorption: 66% (pH 8.2). O3 alone: 45% (pH 9), 30% (pH 5). Zeolite + O3: 65% (pH 11), 50% (pH 5). Amonia Conversion: Adsorption: 57% (pH 8.2). O3 alone: 28% (pH 9), 10% (pH 5). Zeolite + O3: 20% (pH 11), 35% (pH 5). | [147] |
4.3. Oxides
4.3.1. Structure and Properties
4.3.2. Aluminum Oxides
4.3.3. Iron Oxides
4.3.4. Manganese Oxides
| Catalyst | Pollutant | Operating Conditions | Reaction Outcomes | Ref. |
|---|---|---|---|---|
| Fe and Mn modified bauxite (IMB and MMB) | 2,4,6-Trichloroanisole (TCA) | Batch reactor 1 L, O3 dissolved concentration 0.62 mg/L, room T, 28.2 µg/L TCA, catalyst load 0.5 g/L, 60 min, pH 6.5. | TCA Conversion: O3 alone: 56%, Bauxite + O3: 72.4%, MMB + O3: 85.5%, IMB+O3: 99%. Adsorption < 10%. Leaching: Mn 0.065 mg/L, Fe 0.02 mg/L (30 min). | [153] |
| Raw bauxite | 2,4,6-Trichloroanisole (TCA) | Semibatch reactor 3 L, O3 dissolved concentration 0.5 mg/L, 20 °C, 100 ng/L TCA, catalyst load 0.2 g/L, 10 min, pH 6. | TCA Conversion: O3 alone: 34.6%, Bauxite + O3: 86%, γ-AlOOH+O3: 77%, γ-Al2O3 + O3: 60%. Adsorption 10%. Al leaching < 0.05 mg/L. | [46] |
| Raw (RBO) and calcined (CBO) bauxite ores | p-Nitrophenol (NP) | Semibatch reactor, O3 2.46 mg/min, 0.1 L of 300 mg/L NP, catalyst load 5 g/L, 10 min, pH0 5. | COD Conversion: O3 alone: 48%, RBO + O3: 58%, COB + O3: 73.5 Adsorption negligible. | [76] |
| Magnetite ore | Reactive Red-120 (RR-120) | Semibatch reactor, O3 1 mg/min, 25 °C, 0.1 L of 100 mg/L RR-120, catalyst load 0.2 g, 10 min, pH 11. | RR-120 Conversion: O3 alone: 40%, raw magnetite + O3: 66%, calcined magnetite + O3: 84.5%. Adsorption 13%. | [157] |
| Natural magnetite modified by oxygen and argon glow discharge plasma | Oxazine dye Basic Blue 3 (BB3) | Batch reactor, O3 dissolved concentration 1.2 mg/L, 0.25 L of 90 mg/L BB3, catalyst load 0.6 g/L, 15 min, pH0 6.7. | BB3 Conversion: O3 alone: 51.02%, raw magnetite + O3: 63.78%, modified magnetite + O3: 93.47%. Adsorption < 7%. Fe leaching 0.2 mg/L (O3 0.3 mg/L and 20 min reaction). | [158] |
| Plasma-treated goethite nanoparticles: natural (NG), using N2 (PTG-N2) and using Ar (PTG-Ar) | Sulfasalazine antibiotic (SSZ) | Semibatch reactor, gas flow rate 1 L/h, O3 5 mg/L in gas phase, 25 °C, 10 mg/L SSZ, catalyst load 1.5 g/L, 40 min, pH 7. | SSZ Conversion: O3 alone: 61.44%, O3 + NG: 75.64%, O3 + PTG-Ar: 93.47%, O3 +PTG-N2: 96.05%. TOC Conversion: O3 alone: 31.54%, O3 + NG: 38.41%, O3 + PTG-Ar: 52.32%, O3 + PTG-N2: 56.69%. Adsorption < 10%. Fe leached: 0.18 mg/L (NG), 0.079 mg/L (PTG-N2). | [159] |
| Goethite | Para-chlorobenzoic acid (pCBA) | Batch reactor, O3 dissolved concentration 3 mg/L, p-CBA 1.2 mg/L at pH 3 and 3 mg/L at pH 2, catalyst load 5 g/L. | p-CBA Conversion (pH 3, 1 min): O3 alone: 70%, goethite + O3: 86%. p-CBA Conversion (pH 2, 10 min): O3 alone < 1%, goethite + O3: 16%. | [160] |
| Limonite: raw (NL) and modified through plasma O2/Ar (PTL/O2/Ar) | Sulfasalazine antibiotic | Semibatch reactor, gas flow rate 1 L/h, O3 15 mg/L in gas phase, 0.1 mM SSZ, catalyst load 1.5 g/L, 50–120 min, pH0 7. | SSZ Conversion (50 min): O3 alone: 62.8%, O3 + NL: 74.9%, O3 + PTL/O2/Ar: 98.8%. TOC Conversion (120 min): O3 alone: 42.5%, O3 + NL: 54.1%, O3 + PTL/O2/Ar: 78.5%. Adsorption 7.4–10.6%. Fe leached (50 min): 0.221 mg/L (NL), 0.062 mg/L (PTL/O2/Ar). | [109] |
| Manganese sand ore | Aniline | Semibatch reactor, O3 1.76 mg/min, 25 °C, 0.1 L of 200 mg/L aniline, catalyst load 3 g/L, 10 min, pH0 7.2. | COD Conversion: O3 alone: 42.6%, raw manganese ore + O3: 61%, calcined manganese ore + O3: 67.6%. Adsorption < 10%. | [165] |
| Manganese ore | Landfill leachate | Semibatch column reactor, air flow rate 7 L/min, O3 2.882 g/h, 1 L of 3083 mg/L COD landfill leachate, catalyst load 0.6 g/L, 100 min, pH0 8. | COD Conversion: O3 alone: 41.37%, manganese ore + O3: 61%. | [166] |
| Silicate ore (SO) and manganese-modified silicate ore (MnSO) | Ciprofloxacin (CIP) | Semibatch reactor 0.5 L, gas flow rate 0.3 L/min, O3 0.4 mg/min, 20 °C, 0.4 L of 20 mg/L CIP, catalyst load 0.5 g/L, 30 min, pH0 7. | TOC Conversion: O3 alone: 28%, SO + O3: 40%, MnSO + O3: 49%, MnSO + O2: 10%, SO + O2: 15%. Mn leaching 0.069–0.097 mg/L. | [167] |
4.4. Others
| Catalyst | Pollutant | Operating Conditions | Reaction Outcomes | Ref. |
|---|---|---|---|---|
| Pumice | p-Chloronitrobenzene (p-CNB) | Semibatch reactor 1.2 L, total applied O3 0.6 mg/L, 23 °C, 1 L of 100 µg/L p-CNB, catalyst load 1 g/L, 20 min, pH0 6.86. | p-CNB Conversion: O3 alone: 54%, pumice + O3: 84.3%. Adsorption: 3.9%. Negligible leaching (Si, Al, Fe, Mg, Ti, K, Na). | [65] |
| Fe-pumice | p-Chloronitrobenzene (p-CNB) | Batch reactor 1 L, O3 0.9 mg/L in liquid phase, 25 °C, 1 L of 100 µg/L p-CNB, catalyst load 0.5 g/L, 15 min, pH 6. | p-CNB Conversion: O3 alone: 40%, pumice+O3: 76%, Fe-pumice + O3: 91%. Adsorption < 5.5%. Fe leaching 2.1 µg/L. | [171] |
| Iron silicate-loaded pumice | Diclofenac (DCF) | Semibatch reactor, gas flow rate 1 L/min, O3 dose 5.52 mg/L, 25 °C, 0.5 L of 29.6 mg/L DFC, catalyst load 0.8 g/L, 60 min, pH0 7. | TOC Conversion: O3 alone: 32.3%, O3 + cat.: 73.3%. DFC Conversion: O3 alone: 100%, O3+cat.: 100%, O2 + cat.: 7.3%. | [67] |
| Volcanic rocks P1 and P2 | Parabens: methyl (MP), ethyl (EP), propyl (PP), benzyl (BeP) and butylparaben (BuP) | Semibatch reactor 2 L, gas flow rate 0.2 L/min, transferred O3 dose 20 mg/L, 25 °C, parabens 10 mg/L, catalyst load 0.5 g/L, pH0 3.5. | O3 alone Conversion: 28% (MP), 26% (PP), 44% (BeP), 26% (EP), 41% (BuP). P1+O3 Conversion: 72% (MP), 72% (PP), 85% (BeP), 68% (EP), 70% (BuP). P2+O3 Conversion: 62% (MP), 58% (PP), 70% (BeP), 58% (EP), 64% (BuP). Adsorption negligible. Leaching: 0.35 Al, 0.45 Fe, 0.15 Na and 0.69 mg/L Mg. | [172] |
| Volcanic rock | Parabens: methyl (MP), ethyl (EP), propyl (PP), benzyl (BeP) and butylparaben (BuP) | Semibatch reactor 2 L, gas flow rate 0.2 L/min, 25 °C, 2 L of mixture of parabens 10 mg/L each (total COD 90 mg/L), catalyst load 0.5 g/L, pH0 5.2. | COD Conversion: O3 alone <15%, O3 + cat.: 37% (with transferred O3 dose of 55 mg/L). Adsorption negligible. Leaching: 0.13 Al, 0.21 Fe, 1.12 Na, 3.18 Ca and 0.2 mg/L Mg. | [173] |
| Sepiolite, volcanic rock and iron shavings (zerovalent iron (ZVI)) | Simulated olive mill wastewater | Semibatch reactor 0.5 L, gas flow rate 0.5 L/min, O3 20 g/Nm3 in gas phase, 20 °C, 0.5 L of 1211 mg/L COD, catalyst load 1 g/L, 120 min, pH 3. | COD Conversion: O3 alone: 29%, sepiolite+O3: 31%, Volcanic rock + O3: 37%, ZVI + O3: 60%. Adsorption negligible. Fe leaching: 400 mg/L (ZVI). | [174] |
| Volcanic sand (VS) | Benzothiazole (BT) | Differential circular flow reactor (20 °C): after reaching dissolved O3 saturation (125 µM, 20 °C) the gas flow (120 L/h) is closed, BT is added (222 µM) and begins liquid recirculation (1 L/min, 1 L) through the fixed bed (10 g/L cat., 19 mL). | BT Conversion at pH 2, 10 min: O3 alone: 35%, VS + O3: 70%, VS: 5%. BT Conversion at pH 7, 10 min: O3 alone: 74%, VS+O3: 93%. | [88] |
| Soil: Sand containing organic matter (S), baked sand (BS) and goethite (G) | p-Chlorobenzoic acid (p-CBA) | Batch reactor, O3 dosage 3 mg/L, 25 °C, 2.56 × 10−3 mM pCBA, catalyst load 50 g/L (S, BS) and 5 g/L (G), pH 5.6, 420 s. | p-CBA Conversion: BS+O3: 73.2%, S + O3: 86%, G+O3: 88%. | [175] |
| Natural Mackinawite | N,N-dimethylacetamide (DMAC) | Semibatch reactor 0.25 L, 30 °C, gas flow rate 0.3 L/min, O3 50 mg/L in gas phase, catalyst load 3.5 g/L, 20 min, pH0 6.8. | DMAC Conversion: O3 alone: 9.2%, catalytic O3: 96.6%. Adsorption 3%. Fe leaching: 209.2 mg/L. | [49] |
| Polonite® (POL), Wollastonite (WOLL), zeolite (ZeoCat) | Contaminants of emerging concern (CECs: Atrazine (ATZ), ibuprofen (IBP), naproxen (NPX), and gemfibrozil (GBZ)) in Milli-Q (MQW) and simulated synthetic wastewater (SWW) | Semibatch reactor 1 L, gas flow rate 1 L/min, O3 8 g/Nm3 in gas phase, room T, 0.8 L of SWW (pH 7.6) or MQW (pH 5–6) with 150 µg of CECs, catalyst load 25 g/L. Disinfection tests: SWW spiked with E. coli 2 × 105 MPN/mL. | ATZ Conversion with a total ozone dosage (TOD) of 18 mg/L, in MQW: O3 alone: 50%, O3+POL: 100%, O3+WOLL: 44%, O3+ZeoCat: 46%. Adsorption < 13%. ATZ Conversion with TOD of 40 mg/L, in SWW: O3 alone: 87%, O3+POL: 79%, O3 + WOLL: 65%, O3 + ZeoCat: 79%. TOD to reach disinfection criteria in SWW: POL or ZeoCat + O3: 34–39 mg/L, O3 alone: 38–49 mg/L. | [176] |
| Tourmaline (TOU) | Atrazine (ATZ) | Batch reactor 0.25 L, O3 3 mg/L in liquid phase, 5 °C, 5 µM ATZ, catalyst load 1 g/L, 30 min, pH0 7. | ATZ Conversion: O3 alone: 28.8%, O3 + TOU: 100%. Adsorption < 3%. Leaching: Fe (3.2–4.6 µg/L) and Al (6.8–5.4 µg/L) | [177] |
| Calcined Zeolite, Talc and Kaolin | p-Chlorobenzoic acid (p-CBA) | Batch reactor, O3 2 mg/L in liquid phase, 23 °C, 4 µM p-CBA, catalyst load 0.5 g/L, 2 min, pH0 7. | p-CBA Conversion: O3 alone: 94.5%, O3 + Zeolite: 99.5%, O3 + Kaolin: 95%, O3 + Talc: 98.7%. Adsorption negligible. | [178] |
| Brucite (Mg(OH)) | Azo dye active brilliant red (X-3B) | Semibatch reactor, O3 flow rate 0.3 mg/min, 20 °C, 0.05 L of 500 mg/L X-3B, catalyst load 0.5 g, 15 min. | COD Conversion: O3 alone: 9%, Mg(OH) + O3: 33%. X-3B Conversion: O3 alone: 47%, Mg(OH) + O3: 89%. Adsorption 4%. | [179] |
| Brucite (Mg(OH)) and Magnesia (MgO) | Phenol (Ph) | Semibatch reactor, gas flow rate 5 mL/min, O3 0.36 mg/min, 25 °C, 0.1 L of 100 mg/L Ph, catalyst load 5 g/L, 60 min, pH0 = 6.35 (O3), 10.18 (Mg(OH)), 10.8 (MgO). | COD Conversion: O3 alone: 38%, Mg(OH) + O3: 58%, MgO + O3: 90%. Adsorption negligible | [180] |
| Galena (PbS) | O-isopropyl-ethylthionocarbamate (IPETC) | Semibatch reactor, O3 dosage 2.065 mg/(min L), 2 L of 100 mg/L IPETC, catalyst load 0.75 g/L, 20 min, pH 10. | IPETC Conversion: O3 alone: 50%, PbS + O3: 90%. Adsorption ≈ 10% Leached Pb 14.9–23.2 mg/L (180 min) and ≈1 mg/L (20 min). | [181] |
| CuS | C. I. Reactive Blue 5 (RB-5) | Semibatch reactor, O3 1 wt.%, gas flow rate 1.23 SLPM, 21–23 °C, 0.25 L of 1 g/L RB-5, catalyst load 1.2 g/L, 10 min. | Color Conversion: O3 alone: 57%, CuS + O3: 90%. | [182] |
| CuS | Acid Red-151 (AR-151), Remazol Brilliant Blue-R (RBBR), Reactive Black-5 (RB-5) | Semibatch reactor, O3 dosage 115 mg/(min L), 100 mg/L dye, catalyst load 0.1 g/L, 80 min, pH 3, 7, 10. | TOC Conversion pH 3: O3 alone: 25% (AR-151), 18% (RBBR), 24% (RB-5); CuS + O3: 40% (AR-151), 38% (RBBR), 40% (RB-5). Cu leached 27.7 mg/L TOC Conversion pH 7: O3 alone: 54% (AR-151), 48% (RBBR), 43% (RB-5); CuS+O3: 86% (AR-151), 83% (RBBR), 75% (RB-5). Cu leached 7 mg/L TOC Conversion pH 10: O3 alone: 63% (AR-151), 66% (RBBR), 79% (RB-5); CuS + O3: 95% (AR-151), 93% (RBBR), 86% (RB-5). Cu leached 4 mg/L. | [183] |
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Water Assessment Programme (WWAP). The United Nations World Water Development Report 2019: Leaving No One Behind; UNESCO: Paris, France, 2019; Available online: https://unesdoc.unesco.org/ark:/48223/pf0000367306 (accessed on 22 March 2022).
- Jury, W.A.; Vaux, H.J. The emerging global water crisis: Managing scarcity and conflict between water users. Adv. Agron. 2007, 95, 1–76. [Google Scholar] [CrossRef]
- IPCC. Climate Change 2021: The Physical Science Basis; Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change; Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S.L., Péan, C., Berger, S., Caud, N., Chen, Y., Goldfarb, L., Gomis, M.I., et al., Eds.; Cambridge University Press: Cambridge, UK, 2021. [Google Scholar]
- Russell, D.L. Practical Wastewater Treatment; Wiley-Interscience: Hoboken, NJ, USA, 2006. [Google Scholar]
- Carbajo, J.B.; Rodríguez, A.; Rosal, R.; Letón, P.; García-Calvo, E. Uso de Ozono: Ozono, Ozono/H2O2. In Tecnologías de Tratamiento de Aguas para su Reutilización; 2012. Available online: http://www.consolider-tragua.com/documentos/tecnologias_tratamiento_agua.pdf (accessed on 22 March 2022).
- Ghuge, S.P.; Saroha, A.K. Catalytic ozonation for the treatment of synthetic and industrial effluents—Application of mesoporous materials: A review. J. Environ. Manag. 2018, 211, 83–102. [Google Scholar] [CrossRef] [PubMed]
- Gogate, P.R.; Pandit, A.B. A review of imperative technologies for wastewater treatment II: Hybrid methods. Adv. Environ. Res. 2004, 8, 553–597. [Google Scholar] [CrossRef]
- Garrido-Ramírez, E.G.; Theng, B.K.G.; Mora, M.L. Clays and oxide minerals as catalysts and nanocatalysts in Fenton-like reactions—A review. Appl. Clay Sci. 2010, 47, 182–192. [Google Scholar] [CrossRef]
- Oller, I.; Malato, S.; Sánchez-Pérez, J.A. Combination of Advanced Oxidation Processes and biological treatments for wastewater decontamination—A review. Sci. Total Environ. 2011, 409, 4141–4166. [Google Scholar] [CrossRef]
- Tian, X.; Song, Y.; Shen, Z.; Zhou, Y.; Wang, K.; Jin, X.; Han, Z.; Liu, T. A comprehensive review on toxic petrochemical wastewater pretreatment and advanced treatment. J. Clean. Prod. 2020, 245, 118692. [Google Scholar] [CrossRef]
- Gottschalk, C.; Libra, J.A.; Saupe, A. Ozonation of Water and Wastewater; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009. [Google Scholar] [CrossRef]
- Nawrocki, J.; Kasprzyk-Hordern, B. The efficiency and mechanisms of catalytic ozonation. Appl. Catal. B 2010, 99, 27–42. [Google Scholar] [CrossRef]
- Wang, J.; Chen, H. Catalytic ozonation for water and wastewater treatment: Recent advances and perspective. Sci. Total Environ. 2020, 704, 135249. [Google Scholar] [CrossRef] [PubMed]
- Hagen, J. Industrial Catalysis: A Practical Approach, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Borsatto, F.; Inglezakis, V.J. Natural zeolite markets and strategic considerations. In Handbook of Natural Zeolites; Bentham Science Publishers: Bacau, Romania, 2012. [Google Scholar]
- Martínez, F.; Molina, R.; Pariente, M.I.; Siles, J.A.; Melero, J.A. Low-cost Fe/SiO2 catalysts for continuous Fenton processes. Catal. Today 2017, 280, 176–183. [Google Scholar] [CrossRef]
- Airi, A.; Signorile, M.; Bonino, F.; Quagliotto, P.; Bordiga, S.; Martens, J.A.; Crocellà, V. Insights on a hierarchical MFI zeolite: A combined spectroscopic and catalytic approach for exploring the multilevel porous system down to the active sites. ACS Appl. Mater. Interfaces 2021, 13, 49114–49127. [Google Scholar] [CrossRef]
- Doustkhah, E.; Ide, Y. Microporous layered silicates: Old but new microporous materials. New J. Chem. 2020, 44, 9957–9968. [Google Scholar] [CrossRef]
- Korde, A.; Min, B.; Kapaca, E.; Knio, O.; Nezam, I.; Wang, Z.; Leisen, J.; Yin, X.; Zhang, X.; Sholl, D.S.; et al. Single-walled zeolitic nanotubes. Science 2022, 375, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Rahim Pouran, S.; Abdul Raman, A.A.; Wan Daud, W.M.A. Review on the application of modified iron oxides as heterogeneous catalysts in Fenton reactions. J. Clean. Prod. 2014, 64, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.H. An overview on strategies towards clay-based designer catalysts for green and sustainable catalysis. Appl. Clay Sci. 2011, 53, 87–96. [Google Scholar] [CrossRef]
- Kasprzyk-Hordern, B.; Ziółek, M.; Nawrocki, J. Catalytic ozonation and methods of enhancing molecular ozone reactions in water treatment. Appl. Catal. B 2003, 46, 639–669. [Google Scholar] [CrossRef]
- Von Sonntag, C.; von Gunten, U. Chemistry of Ozone in Water and Wastewater Treatment: From Basic Principles to Applications; IWA Publishing: London, Britain, 2012. [Google Scholar] [CrossRef]
- Maldonado, M.; Malato, S.; Perezestrada, L.; Gernjak, W.; Oller, I.; Domenech, X.; Peral, J. Partial degradation of five pesticides and an industrial pollutant by ozonation in a pilot-plant scale reactor. J. Hazard. Mater. 2006, 138, 363–369. [Google Scholar] [CrossRef]
- Mendret, J.; Azais, A.; Favier, T.; Brosillon, S. Urban wastewater reuse using a coupling between nanofiltration and ozonation: Techno-economic assessment. Chem. Eng. Res. Des. 2019, 145, 19–28. [Google Scholar] [CrossRef]
- Mansas, C.; Mendret, J.; Brosillon, S.; Ayral, A. Coupling catalytic ozonation and membrane separation: A review. Sep. Purif. Technol. 2020, 236, 116221. [Google Scholar] [CrossRef]
- Guillossou, R.; Le Roux, J.; Brosillon, S.; Mailler, R.; Vulliet, E.; Morlay, C.; Nauleau, F.; Rocher, V.; Gaspéri, J. Benefits of ozonation before activated carbon adsorption for the removal of organic micropollutants from wastewater effluents. Chemosphere 2020, 245, 125530. [Google Scholar] [CrossRef]
- Derco, J.; Gotvajn, A.Ž.; Čižmárová, O.; Dudáš, J.; Sumegová, L.; Šimovičová, K. Removal of micropollutants by ozone-based processes. Processes 2021, 9, 1013. [Google Scholar] [CrossRef]
- Guo, Y.; Zhao, E.; Wang, J.; Zhang, X.; Huang, H.; Yu, G.; Wang, Y. Comparison of emerging contaminant abatement by conventional ozonation, catalytic ozonation, O3/H2O2 and electro-peroxone processes. J. Hazard. Mater. 2020, 389, 121829. [Google Scholar] [CrossRef] [PubMed]
- Moussavi, G.; Khavanin, A.; Alizadeh, R. The investigation of catalytic ozonation and integrated catalytic ozonation/biological processes for the removal of phenol from saline wastewaters. J. Hazard. Mater. 2009, 171, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Pocostales, P.; Álvarez, P.; Beltrán, F.J. Catalytic ozonation promoted by alumina-based catalysts for the removal of some pharmaceutical compounds from water. Chem. Eng. J. 2011, 168, 1289–1295. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, C.; Wang, Y.; Hu, C. Inhibition of nano-metal oxides on bromate formation during ozonation process. Ozone Sci. Eng. 2014, 36, 549–559. [Google Scholar] [CrossRef]
- Nie, Y.; Li, N.; Hu, C. Enhanced inhibition of bromate formation in catalytic ozonation of organic pollutants over Fe–AlLDH/Al2O3. Sep. Purif. Technol. 2015, 151, 256–261. [Google Scholar] [CrossRef]
- Ibn Abdul Hamid, K.; Scales, P.J.; Allard, S.; Croue, J.-P.; Muthukumaran, S.; Duke, M. Ozone combined with ceramic membranes for water treatment: Impact on HO radical formation and mitigation of bromate. J. Environ. Manag. 2020, 253, 109655. [Google Scholar] [CrossRef]
- Tang, X.; Zhang, Y.; Li, W.; Geng, J.; Ren, H.; Xu, K. Mechanism and toxicity evaluation of catalytic ozonation over Cu/Ce–Al2O3 system aiming at degradation of humic acid in real wastewater. Sci. Rep. 2021, 11, 8748. [Google Scholar] [CrossRef]
- Fei, J.; Lin, X.; Li, X.; Huang, Y.; Ma, L. Effect of Fe-based catalytic ozonation and sole ozonation on the characteristics and conversion of organic fractions in bio-treated industrial wastewater. Sci. Total Environ. 2021, 774, 145821. [Google Scholar] [CrossRef]
- Rivas, J.; Rodríguez, E.; Beltrán, F.J.; García-Araya, J.F.; Alvarez, P. Homogeneous catalyzed ozonation of simazine. Effect of Mn(II) and Fe(II). J. Environ. Sci. Health 2001, 36, 317–330. [Google Scholar] [CrossRef]
- Andreozzi, R.; Caprio, V.; Marotta, R.; Tufano, V. Kinetic modeling of pyruvic acid ozonation in aqueous solutions catalyzed by Mn(II) and Mn(IV) ions. Water Res. 2001, 35, 109–120. [Google Scholar] [CrossRef]
- Ni, C.-H.; Chen, J.-N.; Yang, P.-Y. Catalytic ozonation of 2-dichlorophenol by metallic ions. Water Sci. Technol. 2003, 47, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Kuo, C.-Y.; Chang, C.-L. Homogeneous catalytic ozonation of C.I. Reactive Red 2 by metallic ions in a bubble column reactor. J. Hazard. Mater. 2008, 154, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Psaltou, S.; Karapatis, A.; Mitrakas, M.; Zouboulis, A. The role of metal ions on p-CBA degradation by catalytic ozonation. J. Environ. Chem. Eng. 2019, 7, 103324. [Google Scholar] [CrossRef]
- Legube, B. Catalytic ozonation: A promising advanced oxidation technology for water treatment. Catal. Today 1999, 53, 61–72. [Google Scholar] [CrossRef]
- Ma, J.; Graham, N.J.D. Degradation of atrazine by manganese-catalysed ozonation—Influence of radical scavengers. Water Res. 2000, 34, 3822–3828. [Google Scholar] [CrossRef]
- Beltrán, F.J.; Rivas, F.J.; Montero-de-Espinosa, R. Ozone-enhanced oxidation of oxalic acid in water with cobalt catalysts. 2. Heterogeneous catalytic ozonation. Ind. Eng. Chem. Res. 2003, 42, 3218–3224. [Google Scholar] [CrossRef]
- Ernst, M.; Lurot, F.; Schrotter, J.-C. Catalytic ozonation of refractory organic model compounds in aqueous solution by aluminum oxide. Appl. Catal. B 2004, 47, 15–25. [Google Scholar] [CrossRef]
- Qi, F.; Xu, B.; Chen, Z.; Ma, J.; Sun, D.; Zhang, L.; Wu, F. Ozonation catalyzed by the raw bauxite for the degradation of 2,4,6-trichloroanisole in drinking water. J. Hazard. Mater. 2009, 168, 246–252. [Google Scholar] [CrossRef]
- Vittenet, J.; Aboussaoud, W.; Mendret, J.; Pic, J.-S.; Debellefontaine, H.; Lesage, N.; Faucher, K.; Manero, M.-H.; Thibault-Starzyk, F.; Leclerc, H.; et al. Catalytic ozonation with γ-Al2O3 to enhance the degradation of refractory organics in water. Appl. Catal. A-Gen. 2015, 504, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, F.; Cao, H.; Xie, Y.; Xiao, J.; Chen, Y.; Ghazi, Z.A. Selection of active phase of MnO2 for catalytic ozonation of 4-nitrophenol. Chemosphere 2017, 168, 1457–1466. [Google Scholar] [CrossRef]
- Peng, J.; Yan, J.; Chen, Q.; Jiang, X.; Yao, G.; Lai, B. Natural mackinawite catalytic ozonation for N, N-dimethylacetamide (DMAC) degradation in aqueous solution: Kinetic, performance, biotoxicity and mechanism. Chemosphere 2018, 210, 831–842. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, Y.; Yu, G.; Wang, Y. Revisiting the role of reactive oxygen species for pollutant abatement during catalytic ozonation: The probe approach versus the scavenger approach. Appl. Catal. B 2021, 280, 119418. [Google Scholar] [CrossRef]
- Andreozzi, R.; Insola, A.; Caprio, V.; Marotta, R.; Tufano, V. The use of manganese dioxide as a heterogeneous catalyst for oxalic acid ozonation in aqueous solution. Appl. Catal. A-Gen. 1996, 138, 75–81. [Google Scholar] [CrossRef]
- Azzouz, A.; Kotbi, A.; Niquette, P.; Sajin, T.; Ursu, A.V.; Rami, A.; Monette, F.; Hausler, R. Ozonation of oxalic acid catalyzed by ion-exchanged montmorillonite in moderately acidic media. Reac. Kinet. Mech. Cat. 2010, 99, 289–302. [Google Scholar] [CrossRef]
- Yan, H.; Chen, W.; Liao, G.; Li, X.; Ma, S.; Li, L. Activity assessment of direct synthesized Fe-SBA-15 for catalytic ozonation of oxalic acid. Sep. Purif. Technol. 2016, 159, 1–6. [Google Scholar] [CrossRef]
- Kasprzyk-Hordern, B.; Raczyk-Stanisławiak, U.; Świetlik, J.; Nawrocki, J. Catalytic ozonation of natural organic matter on alumina. Appl. Catal. B 2006, 62, 345–358. [Google Scholar] [CrossRef]
- Amin, N.A.S.; Akhtar, J.; Rai, H.K. Screening of combined zeolite-ozone system for phenol and COD removal. Chem. Eng. J. 2010, 158, 520–527. [Google Scholar] [CrossRef]
- Ikhlaq, D.R.; Brown, B. Kasprzyk-Hordern, Catalytic ozonation for the removal of organic contaminants in water on ZSM-5 zeolites. Appl. Catal. B 2014, 154–155, 110–122. [Google Scholar] [CrossRef]
- Gao, G.; Kang, J.; Shen, J.; Chen, Z.; Chu, W. Catalytic ozonation of sulfamethoxazole by composite iron-manganese silicate oxide: Cooperation mechanism between adsorption and catalytic reaction. Environ. Sci. Pollut. Res. 2016, 23, 21360–21368. [Google Scholar] [CrossRef]
- Gao, L.; Zhai, Y.; Ma, H.; Wang, B. Degradation of cationic dye methylene blue by ozonation assisted with kaolin. Appl. Clay Sci. 2009, 46, 226–229. [Google Scholar] [CrossRef]
- Wang, J.; Bai, Z. Fe-based catalysts for heterogeneous catalytic ozonation of emerging contaminants in water and wastewater. Chem. Eng. J. 2017, 312, 79–98. [Google Scholar] [CrossRef]
- Lv, A.; Hu, C.; Nie, Y.; Qu, J. Catalytic ozonation of toxic pollutants over magnetic cobalt-doped Fe3O4 suspensions. App. Catal. B 2012, 117–118, 246–252. [Google Scholar] [CrossRef]
- Yang, L.; Hu, C.; Nie, Y.; Qu, J. Surface acidity and reactivity of β-FeOOH/Al2O3 for pharmaceuticals degradation with ozone: In situ ATR-FTIR studies. Appl. Catal. B 2010, 97, 340–346. [Google Scholar] [CrossRef]
- Yan, L.; Bing, J.; Wu, H. The behavior of ozone on different iron oxides surface sites in water. Sci. Rep. 2019, 9, 14752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.H.; Jung, J.; Yoon, J.H.; Lee, M.J. Catalytic ozonation of PCE by clay from tidal flat sediments. Catal. Lett. 2002, 78, 77–79. [Google Scholar] [CrossRef]
- Sui, M.; Sheng, L.; Lu, K.; Tian, F. FeOOH catalytic ozonation of oxalic acid and the effect of phosphate binding on its catalytic activity. App. Catal. B 2010, 96, 94–100. [Google Scholar] [CrossRef]
- Yuan, L.; Shen, J.; Chen, Z.; Liu, Y. Pumice-catalyzed ozonation degradation of p-chloronitrobenzene in aqueous solution. Appl. Catal. B 2012, 117–118, 414–419. [Google Scholar] [CrossRef]
- Bing, J.; Hu, C.; Nie, Y.; Yang, M.; Qu, J. Mechanism of catalytic ozonation in Fe2O3/Al2O3 @SBA-15 aqueous suspension for destruction of Ibuprofen. Environ. Sci. Technol. 2015, 49, 1690–1697. [Google Scholar] [CrossRef]
- Gao, G.; Shen, J.; Chu, W.; Chen, Z.; Yuan, L. Mechanism of enhanced diclofenac mineralization by catalytic ozonation over iron silicate-loaded pumice. Sep. Purif. Technol. 2017, 173, 55–62. [Google Scholar] [CrossRef]
- Chen, W.; Li, X.; Pan, Z.; Ma, S.; Li, L. Effective mineralization of Diclofenac by catalytic ozonation using Fe-MCM-41 catalyst. Chem. Eng. J. 2016, 304, 594–601. [Google Scholar] [CrossRef]
- Camarasa, E.; Vial, C.; Poncin, S.; Wild, G.; Midoux, N.; Bouillard, J. Influence of coalescence behaviour of the liquid and of gas sparging on hydrodynamics and bubble characteristics in a bubble column. Chem. Eng. Process. Process Intensif. 1999, 38, 329–344. [Google Scholar] [CrossRef]
- López-López, A.; Pic, J.-S.; Benbelkacem, H.; Debellefontaine, H. Influence of t-butanol and of pH on hydrodynamic and mass transfer parameters in an ozonation process. Chem. Eng. Process. Process Intensif. 2007, 46, 649–655. [Google Scholar] [CrossRef]
- Tizaoui, C.; Grima, N.M.; Derdar, M.Z. Effect of the radical scavenger t-butanol on gas–liquid mass transfer. Chem. Eng. Sci. 2009, 64, 4375–4382. [Google Scholar] [CrossRef]
- Pi, Y.; Schumacher, J.; Jekel, M. The use of para-chlorobenzoic acid (pCBA) as an ozone/hydroxyl radical probe compound. Ozone Sci. Eng. 2005, 27, 431–436. [Google Scholar] [CrossRef]
- Sánchez-Polo, M.; Leyva-Ramos, R.; Rivera-Utrilla, J. Kinetics of 1,3,6-naphthalenetrisulphonic acid ozonation in presence of activated carbon. Carbon 2005, 43, 962–969. [Google Scholar] [CrossRef]
- Kuosa, M.; Kallas, J.; Häkkinen, A. Ozonation of p-nitrophenol at different pH values of water and the influence of radicals at acidic conditions. J. Environ. Chem. Eng. 2015, 3, 325–332. [Google Scholar] [CrossRef]
- Huang, X.; Li, X.; Pan, B.; Li, H.; Zhang, Y.; Xie, B. Self-enhanced ozonation of benzoic acid at acidic pHs. Water Res. 2015, 73, 9–16. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Yu, J.; Han, C.; Yan, G.; Guo, S. p-Nitrophenol removal by bauxite ore assisted ozonation and its catalytic potential: P-nitrophenol removal by bauxite ore. Clean Soil Air Water 2015, 43, 1010–1017. [Google Scholar] [CrossRef]
- Haber, J.; Block, J.H.; Delmon, B. Methods and Procedures for Catalyst Characterization. In Handbook of Heterogeneous Catalysis; Ertl, G., Knozinger, H., Schuth, F., Weitkamp, J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; p. hetcat0066. [Google Scholar] [CrossRef]
- Qi, F.; Xu, B.; Chen, Z.; Zhang, L.; Zhang, P.; Sun, D. Mechanism investigation of catalyzed ozonation of 2-methylisoborneol in drinking water over aluminum (hydroxyl) oxides: Role of surface hydroxyl group. Chem. Eng. J. 2010, 165, 490–499. [Google Scholar] [CrossRef]
- Nawrocki, J.; Fijołek, L. Effect of aluminium oxide contaminants on the process of ozone decomposition in water. Appl. Catal. B 2013, 142–143, 533–537. [Google Scholar] [CrossRef]
- Yang, W.; Vogler, B.; Lei, Y.; Wu, T. Metallic ion leaching from heterogeneous catalysts: An overlooked effect in the study of catalytic ozonation processes. Environ. Sci. Water Res. Technol. 2017, 3, 1143–1151. [Google Scholar] [CrossRef]
- Inchaurrondo, N.; di Luca, C.; Žerjav, G.; Grau, J.M.; Pintar, A.; Haure, P. Catalytic ozonation of an azo-dye using a natural aluminosilicate. Catal. Today 2021, 361, 24–29. [Google Scholar] [CrossRef]
- Aghaeinejad-Meybodi, A.; Ebadi, A.; Shafiei, S.; Khataee, A.; Kiadehi, A.D. Degradation of Fluoxetine using catalytic ozonation in aqueous media in the presence of nano-γ-alumina catalyst: Experimental, modeling and optimization study. Sep. Purif. Technol. 2019, 211, 551–563. [Google Scholar] [CrossRef]
- Tizaoui, C.; Grima, N. Kinetics of the ozone oxidation of Reactive Orange 16 azo-dye in aqueous solution. Chem. Eng. J. 2011, 173, 463–473. [Google Scholar] [CrossRef]
- Saeid, S.; Tolvanen, P.; Kumar, N.; Eränen, K.; Peltonen, J.; Peurla, M.; Mikkola, J.-P.; Franz, A.; Salmi, T. Advanced oxidation process for the removal of ibuprofen from aqueous solution: A non-catalytic and catalytic ozonation study in a semi-batch reactor. Appl. Catal. B 2018, 230, 77–90. [Google Scholar] [CrossRef]
- Lin, J.; Kawai, A.; Nakajima, T. Effective catalysts for decomposition of aqueous ozone. Appl. Catal. B 2002, 39, 157–165. [Google Scholar] [CrossRef]
- Antoniou, M.G.; Hey, G.; Rodríguez Vega, S.; Spiliotopoulou, A.; Fick, J.; Tysklind, M.; la Cour Jansen, J.; Andersen, H.R. Required ozone doses for removing pharmaceuticals from wastewater effluents. Sci. Total Environ. 2013, 456–457, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Vel Leitner, N.K.; Delouane, B.; Legube, B.; Luck, F. Effects of catalysts during ozonation of salicylic acid, peptides and humic substances in aqueous solution. Ozone Sci. Eng. 1999, 21, 261–276. [Google Scholar] [CrossRef]
- Valdes, H.; Murillo, F.; Manoli, J.; Zaror, C. Heterogeneous catalytic ozonation of benzothiazole aqueous solution promoted by volcanic sand. J. Hazard. Mater. 2008, 153, 1036–1042. [Google Scholar] [CrossRef]
- Valdés, H.; Farfán, V.J.; Manoli, J.A.; Zaror, C.A. Catalytic ozone aqueous decomposition promoted by natural zeolite and volcanic sand. J. Hazard. Mater. 2009, 165, 915–922. [Google Scholar] [CrossRef]
- Geelhoed, J.S.; Hiemstra, T.; Van Riemsdijk, W.H. Phosphate and sulfate adsorption on goethite: Single anion and competitive adsorption. Geochim. Cosmochim. Acta 1997, 61, 2389–2396. [Google Scholar] [CrossRef]
- Elovitz, M.S.; von Gunten, U.; Kaiser, H.-P. Hydroxyl radical/ozone ratios during ozonation processes. II. The effect of temperature, pH, alkalinity, and DOM properties. Ozone Sci. Eng. 2000, 22, 123–150. [Google Scholar] [CrossRef]
- Li, B.; Li, C.; Qu, R.; Wu, N.; Qi, Y.; Sun, C.; Zhou, D.; Wang, Z. Effects of common inorganic anions on the ozonation of polychlorinated diphenyl sulfides on silica gel: Kinetics, mechanisms, and theoretical calculations. Water Res. 2020, 186, 116358. [Google Scholar] [CrossRef] [PubMed]
- Katsoyiannis, I.A.; Canonica, S.; von Gunten, U. Efficiency and energy requirements for the transformation of organic micropollutants by ozone, O3/H2O2 and UV/H2O2. Water Res. 2011, 45, 3811–3822. [Google Scholar] [CrossRef]
- Nawrocki, J. Catalytic ozonation in water: Controversies and questions. Discussion paper. Appl. Catal. B 2013, 142–143, 465–471. [Google Scholar] [CrossRef]
- Shahidi, D.; Roy, R.; Azzouz, A. Total removal of oxalic acid via synergistic parameter interaction in montmorillonite catalyzed ozonation. J. Environ. Chem. Eng. 2014, 2, 20–30. [Google Scholar] [CrossRef]
- Roth, J.A.; Sullivan, D.E. Solubility of ozone in water. Ind. Eng. Chem. Fund. 1981, 20, 137–140. [Google Scholar] [CrossRef]
- Andreozzi, R.; Caprio, V.; Ermellino, I.; Insola, A.; Tufano, V. Ozone solubility in phosphate-buffered aqueous solutions: Effect of temperature, tert-butyl alcohol, and pH. Ind. Eng. Chem. Res. 1996, 35, 1467–1471. [Google Scholar] [CrossRef]
- Biń, K. Ozone solubility in liquids. Ozone Sci. Eng. 2006, 28, 67–75. [Google Scholar] [CrossRef]
- Hoigné, J.; Bader, H. Rate constants of reactions of ozone with organic and inorganic compounds in water—I. Water Res. 1983, 17, 173–183. [Google Scholar] [CrossRef]
- Kaiser, H.-P.; Köster, O.; Gresch, M.; Périsset, P.M.J.; Jäggi, P.; Salhi, E.; von Gunten, U. Process control for ozonation systems: A novel real-time approach. Ozone Sci. Eng. 2013, 35, 168–185. [Google Scholar] [CrossRef]
- Yong, E.L.; Lin, Y.-P. Effects of pH value and temperature on the initiation, promotion, inhibition and direct reaction rate constants of natural organic matter in ozonation. RSC Adv. 2016, 6, 18587–18595. [Google Scholar] [CrossRef]
- EPA. Alternative Disinfectants and Oxidants Guidance Manual; 1999. Available online: https://nepis.epa.gov/Exe/ZyPDF.cgi/2000229L.PDF?Dockey=2000229L.PDF (accessed on 22 March 2022).
- Plumlee, M.H.; Stanford, B.D.; Debroux, J.-F.; Hopkins, D.C.; Snyder, S.A. Costs of advanced treatment in water reclamation. Ozone: Sci. Eng. 2014, 36, 485–495. [Google Scholar] [CrossRef]
- Mahamuni, N.N.; Adewuyi, Y.G. Advanced oxidation processes (AOPs) involving ultrasound for waste water treatment: A review with emphasis on cost estimation. Ultrason. Sonochem. 2010, 17, 990–1003. [Google Scholar] [CrossRef] [PubMed]
- Fast, S.A.; Gude, V.G.; Truax, D.D.; Martin, J.; Magbanua, B.S. A critical evaluation of Advanced Oxidation Processes for emerging contaminants removal. Environ. Process. 2017, 4, 283–302. [Google Scholar] [CrossRef]
- Esplugas, S.; Giménez, J.; Contreras, S.; Pascual, E.; Rodríguez, M. Comparison of different advanced oxidation processes for phenol degradation. Water Res. 2002, 36, 1034–1042. [Google Scholar] [CrossRef]
- Ferre-Aracil, J.; Valcárcel, Y.; Negreira, N.; de Alda, M.L.; Barceló, D.; Cardona, S.C.; Navarro-Laboulais, J. Ozonation of hospital raw wastewaters for cytostatic compounds removal. Kinetic modelling and economic assessment of the process. Sci. Total Environ. 2016, 556, 70–79. [Google Scholar] [CrossRef]
- Yang, L.; Sheng, M.; Li, Y.; Xue, W.; Li, K.; Cao, G. A hybrid process of Fe-based catalytic ozonation and biodegradation for the treatment of industrial wastewater reverse osmosis concentrate. Chemosphere 2020, 238, 124639. [Google Scholar] [CrossRef]
- Heidari, Z.; Pelalak, R.; Eshaghi Malekshah, R.; Pishnamazi, M.; Rezakazemi, M.; Aminabhavi, T.M.; Shirazian, S. A new insight into catalytic ozonation of sulfasalazine antibiotic by plasma-treated limonite nanostructures: Experimental, modeling and mechanism. Chem. Eng. J. 2022, 428, 131230. [Google Scholar] [CrossRef]
- Krichevskaya, M.; Klauson, D.; Portjanskaja, E.; Preis, S. The cost evaluation of Advanced Oxidation Processes in laboratory and pilot-scale experiments. Ozone Sci. Eng. 2011, 33, 211–223. [Google Scholar] [CrossRef]
- Guggenheim, S. Definition of Clay and Clay Mineral: Joint Report of the AIPEA Nomenclature and CMS Nomenclature Committees. Clays Clay Miner. 1995, 43, 255–256. [Google Scholar] [CrossRef]
- Sirinakorn, T.; Imwiset, K.; Bureekaew, S.; Ogawa, M. Inorganic modification of layered silicates toward functional inorganic-inorganic hybrids. Appl. Clay Sci. 2018, 153, 187–197. [Google Scholar] [CrossRef]
- Ide, Y.; Ochi, N.; Ogawa, M. Effective and selective adsorption of Zn2+ from seawater on a layered silicate. Angew. Chem. 2011, 123, 680–682. [Google Scholar] [CrossRef]
- Mokhtar, M. Application of synthetic layered sodium silicate magadiite nanosheets for environmental remediation of Methylene Blue dye in water. Materials 2017, 10, 760. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Maroto, J.M.; Alonso-Azcárate, J. What is clay? A new definition of “clay” based on plasticity and its impact on the most widespread soil classification systems. Appl. Clay Sci. 2018, 161, 57–63. [Google Scholar] [CrossRef]
- Schulze, D.G. Clay Minerals. In Encyclopedia of Soils in the Environment; Elsevier: Amsterdam, The Netherlands, 2005; pp. 246–254. [Google Scholar]
- Yariv, S.; Cross, H. Colloid Geochemistry of Clay Minerals. In Geochemistry of Colloid Systems; Springer: Berlin/Heidelberg, Germany, 1979; pp. 287–333. [Google Scholar] [CrossRef]
- Zhou, C.H.; Keeling, J. Fundamental and applied research on clay minerals: From climate and environment to nanotechnology. Appl. Clay Sci. 2013, 74, 3–9. [Google Scholar] [CrossRef]
- Johnston, C.T. Sorption of organic compounds on clay minerals: A surface functional group approach. In CMS Workshop Lectures. In Organic Pollutants in the Environment; Sahwney, B., Ed.; The Clay Mineral Society: Boulder, CO, USA, 1996; pp. 1–44. [Google Scholar]
- Shahidi, D.; Moheb, A.; Abbas, R.; Larouk, S.; Roy, R.; Azzouz, A. Total mineralization of sulfamethoxazole and aromatic pollutants through Fe2+-montmorillonite catalyzed ozonation. J. Hazard. Mater. 2015, 298, 338–350. [Google Scholar] [CrossRef]
- Navalon, S.; Alvaro, M.; Garcia, H. Heterogeneous Fenton catalysts based on clays, silicas and zeolites. Appl. Catal. B 2010, 99, 1–26. [Google Scholar] [CrossRef]
- Boudissa, F.; Mirilà, D.; Arus, V.-A.; Terkmani, T.; Semaan, S.; Proulx, M.; Nistor, I.-D.; Roy, R.; Azzouz, A. Acid-treated clay catalysts for organic dye ozonation—Thorough mineralization through optimum catalyst basicity and hydrophilic character. J. Hazard. Mater. 2019, 364, 356–366. [Google Scholar] [CrossRef]
- Liu, D.; Wang, C.; Song, Y.; Wei, Y.; He, L.; Lan, B.; He, X.; Wang, J. Effective mineralization of quinoline and bio-treated coking wastewater by catalytic ozonation using CuFe2O4/Sepiolite catalyst: Efficiency and mechanism. Chemosphere 2019, 227, 647–656. [Google Scholar] [CrossRef]
- Ma, W.; Hu, J.; Yoza, B.A.; Wang, Q.; Zhang, X.; Li, Q.X.; Guo, S.; Chen, C. Kaolinite based catalysts for efficient ozonation of recalcitrant organic chemicals in water. Appl. Clay Sci. 2019, 175, 159–168. [Google Scholar] [CrossRef]
- Bing, J.; Li, L.; Lan, B.; Liao, G.; Zeng, J.; Zhang, Q.; Li, X. Synthesis of cerium-doped MCM-41 for ozonation of p-chlorobenzoic acid in aqueous solution. Appl. Catal. B 2012, 115–116, 16–24. [Google Scholar] [CrossRef]
- Bernal, M.; Romero, R.; Roa, G.; Barrera-Díaz, C.; Torres-Blancas, T.; Natividad, R. Ozonation of indigo carmine catalyzed with Fe-pillared clay. Int. J. Photoenergy 2013, 2013, 918025. [Google Scholar] [CrossRef]
- Mirilă, D.C.; Boudissa, F.; Beltrao-Nuñes, A.P.; Platon, N.; Didi, M.A.; Nistor, I.D.; Roy, R.; Azzouz, A. Organic dye ozonation catalyzed by chemically modified montmorillonite K10–Role of surface basicity and hydrophilic character. Ozone Sci. Eng. 2020, 42, 517–530. [Google Scholar] [CrossRef]
- Savun-Hekimoğlu, B.; Eren, Z.; Ince, N.H. Catalytic ozonation of caffeine with sepiolite: Effects of impregnation with zero-valent iron and ultrasound. Environ. Prog. Sustain. Energy 2021, 40, e13552. [Google Scholar] [CrossRef]
- Fu, P.; Lin, X.; Wang, L.; Ma, Y. Catalytic ozonation of refractory O-isopropyl-N-ethylthionocarbamate collector with coexisted kaolinite in sulfide flotation wastewaters. Appl. Clay Sci. 2020, 198, 105834. [Google Scholar] [CrossRef]
- Braga, W.L.M.; de Melo, D.H.A.; de Morais, D.; Samanamud, G.R.L.; França, A.B.; Finzi Quintão, C.M.; Loures, C.C.A.; de Urzedo, A.P.F.M.; Naves, L.L.R.; de Freitas Gomes, J.H.; et al. Optimization of the treatment of sanitary landfill by the ozonization catalysed by modified nanovermiculite in a rotating packed bed. J. Clean. Prod. 2020, 249, 119395. [Google Scholar] [CrossRef]
- Cheng, Z.; Yang, R.; Wang, Y. Mn/sepiolite as the heterogeneous ozonation catalysts applied to the advanced treatment of regenerated-papermaking wastewater. Water Sci. Technol. 2017, 75, 1025–1033. [Google Scholar] [CrossRef]
- Coombs, D.S.; Alberti, A.; Armbruster, T.; Artioli, G.; Colella, C.; Galli, E.; Grice, J.D.; Liebau, F.; Mandarino, J.A.; Minato, H.; et al. Recommended nomenclature for zeolite minerals: Report of the subcommittee on zeolites of the International Mineralogical Association, Commission on New Minerals and Mineral Names. Mineral. Mag. 1998, 62, 533–571. [Google Scholar] [CrossRef]
- Wang, S.; Peng, Y. Natural zeolites as effective adsorbents in water and wastewater treatment. Chem. Eng. J. 2010, 156, 11–24. [Google Scholar] [CrossRef]
- Weitkamp, J. Zeolites and catalysis. Solid State Ion 2000, 131, 175–188. [Google Scholar] [CrossRef]
- Moshoeshoe, M.; Nadiye-Tabbiruka, M.; Obuseng, V. A review of the chemistry, structure, properties and applications of zeolites. Am. J. Mater. Sci. 2017, 7, 196–221. [Google Scholar]
- Barrer, R.M. Zeolites and their synthesis. Zeolites 1981, 1, 130–140. [Google Scholar] [CrossRef]
- Kallo, D. Applications of natural zeolites in water and wastewater treatment. In Natural Zeolites Occurrence, Properties, Applications; Bish, D.L., Ming, D.W., Eds.; De Gruyter: Berlin, Germany, 2001. [Google Scholar]
- Hunger, M. Catalytically Active Sites: Generation and Characterization. In Zeolites and Catalysis; Čejka, J., Corma, A., Zones, S., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 493–546. [Google Scholar] [CrossRef]
- Gackowski, M.; Datka, J. Acid properties of hierarchical zeolites Y. Molecules 2020, 25, 1044. [Google Scholar] [CrossRef] [Green Version]
- Roque-Malherbe, R. Applications of natural zeolites in pollution abatement ad industry. In Handbook of Surfaces and Interfaces of Materials; Elsevier: Amsterdam, The Netherlands, 2001; pp. 495–522. [Google Scholar] [CrossRef]
- Inglezakis, V.J.; Zorpas, A.A. Handbook of Natural Zeolites; Bentham Science: Sharjah, United Arab Emirates, 2012. [Google Scholar]
- Jia, X.; Khan, W.; Wu, Z.; Choi, J.; Yip, A.C.K. Modern synthesis strategies for hierarchical zeolites: Bottom-up versus top-down strategies. Adv. Powder Technol. 2019, 30, 467–484. [Google Scholar] [CrossRef]
- Kerstens, D.; Smeyers, B.; Van Waeyenberg, J.; Zhang, Q.; Yu, J.; Sels, B.F. State of the art and perspectives of hierarchical zeolites: Practical overview of synthesis methods and use in catalysis. Adv. Mater. 2020, 32, 2004690. [Google Scholar] [CrossRef]
- Gümüş, D.; Akbal, F. A comparative study of ozonation, iron coated zeolite catalyzed ozonation and granular activated carbon catalyzed ozonation of humic acid. Chemosphere 2017, 174, 218–231. [Google Scholar] [CrossRef]
- Zhang, J.; Xiong, Z.; Wei, J.; Song, Y.; Ren, Y.; Xu, D.; Lai, B. Catalytic ozonation of penicillin G using cerium-loaded natural zeolite (CZ): Efficacy, mechanisms, pathways and toxicity assessment. Chem. Eng. J. 2020, 383, 123144. [Google Scholar] [CrossRef]
- Valdés, H.; Tardón, R.F.; Zaror, C.A. Role of surface hydroxyl groups of acid-treated natural zeolite on the heterogeneous catalytic ozonation of methylene blue contaminated waters. Chem. Eng. J. 2012, 211–212, 388–395. [Google Scholar] [CrossRef]
- AlGburi, H.R.; Aziz, H.A.; Zwain, H.M.; Noor, A.F.M. Treatment of Landfill Leachate by Heterogeneous Catalytic Ozonation with Granular Faujasite Zeolite. Environ. Eng. Sci. 2021, 38, 635–644. [Google Scholar] [CrossRef]
- Scheinost, A.C. Metal Oxides. In Encyclopedia of Soils in the Environment; Elsevier: Amsterdam, The Netherlands, 2005; pp. 428–438. [Google Scholar]
- Kasprzyk-Hordern, B. Chemistry of alumina, reactions in aqueous solution and its application in water treatment. Adv. Colloid Interface Sci. 2004, 110, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Kung, H.H. Transition Metal Oxides, Surface Chemistry and Catalysis; Elsevier: Amsterdam, The Netherlands, 1989. [Google Scholar]
- Ikhlaq, A.; Brown, D.R.; Kasprzyk-Hordern, B. Mechanisms of catalytic ozonation on alumina and zeolites in water: Formation of hydroxyl radicals. Appl. Catal. B 2012, 123–124, 94–106. [Google Scholar] [CrossRef]
- Di Luca, C.; Inchaurrondo, N.; Marcé, M.; Parra, R.; Esplugas, S.; Haure, P. On disclosing the role of mesoporous alumina in the ozonation of sulfamethoxazole: Adsorption vs. Catalysis. Chem. Eng. J. 2021, 412, 128579. [Google Scholar] [CrossRef]
- Qi, F.; Xu, B.; Zhao, L.; Chen, Z.; Zhang, L.; Sun, D.; Ma, J. Comparison of the efficiency and mechanism of catalytic ozonation of 2,4,6-trichloroanisole by iron and manganese modified bauxite. Appl. Catal. B 2012, 121–122, 171–181. [Google Scholar] [CrossRef]
- Siddiqui, S.I.; Chaudhry, S.A. Iron oxide and its modified forms as an adsorbent for arsenic removal: A comprehensive recent advancement. Process Saf. Environ. Prot. 2017, 111, 592–626. [Google Scholar] [CrossRef]
- Zhang, T.; Li, C.; Ma, J.; Tian, H.; Qiang, Z. Surface hydroxyl groups of synthetic α-FeOOH in promoting OH generation from aqueous ozone: Property and activity relationship. Appl. Catal. B 2008, 82, 131–137. [Google Scholar] [CrossRef]
- Larouk, S.; Ouargli, R.; Shahidi, D.; Olhund, L.; Shiao, T.C.; Chergui, N.; Sehili, T.; Roy, R.; Azzouz, A. Catalytic ozonation of Orange-G through highly interactive contributions of hematite and SBA-16—To better understand azo-dye oxidation in nature. Chemosphere 2017, 168, 1648–1657. [Google Scholar] [CrossRef]
- Moussavi, G.; Khosravi, R.; Omran, N.R. Development of an efficient catalyst from magnetite ore: Characterization and catalytic potential in the ozonation of water toxic contaminants. Appl. Catal. A-Gen. 2012, 445–446, 42–49. [Google Scholar] [CrossRef]
- Taseidifar, M.; Khataee, A.; Vahid, B.; Khorram, S.; Joo, S.W. Production of nanocatalyst from natural magnetite by glow discharge plasma for enhanced catalytic ozonation of an oxazine dye in aqueous solution. J. Mol. Catal. A-Chem. 2015, 404–405, 218–226. [Google Scholar] [CrossRef]
- Pelalak, R.; Alizadeh, R.; Ghareshabani, E. Enhanced heterogeneous catalytic ozonation of pharmaceutical pollutants using a novel nanostructure of iron-based mineral prepared via plasma technology: A comparative study. J. Hazard. Mater. 2020, 392, 122269. [Google Scholar] [CrossRef]
- Park, J.-S.; Choi, H.; Cho, J. Kinetic decomposition of ozone and para-chlorobenzoic acid (pCBA) during catalytic ozonation. Water Res. 2004, 38, 2285–2292. [Google Scholar] [CrossRef] [PubMed]
- Remucal, C.K.; Ginder-Vogel, M. A critical review of the reactivity of manganese oxides with organic contaminants. Environ. Sci. Processes Impacts 2014, 16, 1247. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Du, Z.; Li, X.; Zhang, Y.; He, Y.; Zhang, Y. Degradation of methylene blue by natural manganese oxides: Kinetics and transformation products. R. Soc. Open Sci. 2019, 6, 190351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Ma, J.; He, H. Recent advances in catalytic decomposition of ozone. J. Environ. Sci. 2020, 94, 14–31. [Google Scholar] [CrossRef]
- Tong, S.; Liu, W.; Leng, W.; Zhang, Q. Characteristics of MnO2 catalytic ozonation of sulfosalicylic acid and propionic acid in water. Chemosphere 2003, 50, 1359–1364. [Google Scholar] [CrossRef]
- Chen, C.; Yoza, B.A.; Chen, H.; Li, Q.X.; Guo, S. Manganese sand ore is an economical and effective catalyst for ozonation of organic contaminants in petrochemical wastewater. Water Air Soil Pollut. 2015, 226, 182. [Google Scholar] [CrossRef]
- Van, T.H.; Trinh, T.V. Application of regression analysis for ozone and catalytic ozonation of organic compounds from landfill leachate with ceramic raschig rings and natural manganese ore. Ozone Sci. Eng. 2016, 38, 133–142. [Google Scholar] [CrossRef]
- Luo, L.; Zou, D.; Lu, D.; Xin, B.; Zhou, M.; Zhai, X.; Ma, J. Heterogeneous catalytic ozonation of ciprofloxacin in aqueous solution using a manganese-modified silicate ore. RSC Adv. 2018, 8, 33534–33541. [Google Scholar] [CrossRef] [Green Version]
- Rachel, A.; Lavedrine, B.; Subrahmanyam, M.; Boule, P. Use of porous lavas as supports of photocatalysts. Catal. Commun. 2002, 3, 165–171. [Google Scholar] [CrossRef]
- Inchaurrondo, N.; Maestre, A.; Žerjav, G.; Pintar, A.; Ramos, C.; Haure, P. Screening of catalytic activity of natural iron-bearing materials towards the Catalytic Wet Peroxide Oxidation of Orange II. J. Environ. Chem. Eng. 2018, 6, 2027–2040. [Google Scholar] [CrossRef]
- Çifçi, D.İ.; Meriç, S. A review on pumice for water and wastewater treatment. Desalin. Water Treat. 2016, 57, 18131–18143. [Google Scholar] [CrossRef]
- Yuan, L.; Shen, J.; Chen, Z.; Guan, X. Role of Fe/pumice composition and structure in promoting ozonation reactions. Appl. Catal. B 2016, 180, 707–714. [Google Scholar] [CrossRef]
- Gomes, J.F.; Quinta-Ferreira, M.E.; Costa, R.; Quinta-Ferreira, R.M.; Martins, R.C. Paraben degradation using catalytic ozonation over volcanic rocks. Environ. Sci. Pollut. Res. 2018, 25, 7346–7357. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.F.; Frasson, D.; Pereira, J.L.; Gonçalves, F.J.M.; Castro, L.M.; Quinta-Ferreira, R.M.; Martins, R.C. Ecotoxicity variation through parabens degradation by single and catalytic ozonation using volcanic rock. Chem. Eng. J. 2019, 360, 30–37. [Google Scholar] [CrossRef]
- Martins, R.C.; Ramos, C.M.; Quinta-Ferreira, R.M. Low-cost catalysts to enhance ozone action on the depuration of olive mill wastewaters. Ind. Eng. Chem. Res. 2014, 53, 15357–15368. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.-N.; Choi, H.; Hwang, T.-M.; Kang, J.-W. Characterization of ozone decomposition in a soil slurry: Kinetics and mechanism. Water Res. 2002, 36, 219–229. [Google Scholar] [CrossRef]
- Kolosov, P.; Peyot, M.-L.; Yargeau, V. Novel materials for catalytic ozonation of wastewater for disinfection and removal of micropollutants. Sci. Total Environ. 2018, 644, 1207–1218. [Google Scholar] [CrossRef]
- Wang, D.; Xu, H.; Ma, J.; Lu, X.; Qi, J.; Song, S. Strong promoted catalytic ozonation of atrazine at low temperature using tourmaline as catalyst: Influencing factors, reaction mechanisms and pathways. Chem. Eng. J. 2018, 354, 113–125. [Google Scholar] [CrossRef]
- Psaltou, S.; Kaprara, E.; Kalaitzidou, K.; Mitrakas, M.; Zouboulis, A. The effect of thermal treatment on the physicochemical properties of minerals applied to heterogeneous catalytic ozonation. Sustainability 2020, 12, 10503. [Google Scholar] [CrossRef]
- Dong, Y.; He, K.; Zhao, B.; Yin, Y.; Yin, L.; Zhang, A. Catalytic ozonation of azo dye active brilliant red X-3B in water with natural mineral brucite. Catal. Commun. 2007, 8, 1599–1603. [Google Scholar] [CrossRef]
- He, K.; Dong, Y.M.; Li, Z.; Yin, L.; Zhang, A.M.; Zheng, Y.C. Catalytic ozonation of phenol in water with natural brucite and magnesia. J. Hazar. Mater. 2008, 159, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.; Wang, L.; Lin, X.; Ma, Y.; Hou, Z. Ozonation of recalcitrant O-isopropyl-N-ethylthionocarbamate catalyzed by galena in flotation effluents and its dissolution behaviors. Miner. Eng. 2021, 165, 106859. [Google Scholar] [CrossRef]
- Yong, K.; Wu, J.S. Andrews, Heterogeneous catalytic ozonation of aqueous reactive dye. Ozone Sci. Eng. 2005, 27, 257–263. [Google Scholar] [CrossRef]
- Pirgalıoğlu, S.; Özbelge, T.A. Comparison of non-catalytic and catalytic ozonation processes of three different aqueous single dye solutions with respect to powder copper sulfide catalyst. Appl. Catal. A-Gen. 2009, 363, 157–163. [Google Scholar] [CrossRef]
- Kolosov, P.; Yargeau, V. Impact of catalyst load, chemical oxygen demand and nitrite on disinfection and removal of contaminants during catalytic ozonation of wastewater. Sci. Total Environ. 2019, 651, 2139–2147. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inchaurrondo, N.S.; Font, J. Clay, Zeolite and Oxide Minerals: Natural Catalytic Materials for the Ozonation of Organic Pollutants. Molecules 2022, 27, 2151. https://doi.org/10.3390/molecules27072151
Inchaurrondo NS, Font J. Clay, Zeolite and Oxide Minerals: Natural Catalytic Materials for the Ozonation of Organic Pollutants. Molecules. 2022; 27(7):2151. https://doi.org/10.3390/molecules27072151
Chicago/Turabian StyleInchaurrondo, Natalia Soledad, and Josep Font. 2022. "Clay, Zeolite and Oxide Minerals: Natural Catalytic Materials for the Ozonation of Organic Pollutants" Molecules 27, no. 7: 2151. https://doi.org/10.3390/molecules27072151
APA StyleInchaurrondo, N. S., & Font, J. (2022). Clay, Zeolite and Oxide Minerals: Natural Catalytic Materials for the Ozonation of Organic Pollutants. Molecules, 27(7), 2151. https://doi.org/10.3390/molecules27072151