1. Introduction
During development, the brain is exposed to a myriad of endocrine disrupting chemicals (EDCs) which can, even at low doses, interact with multiple cell signaling pathways and disrupt many biological processes [
1,
2,
3]. Thereby, exposure to environmental EDCs is suspected to be involved in the growing incidence of neurodevelopmental disorders. These conditions, which include sensorimotor, cognitive and learning deficits, attention deficit disorders with or without hyperactivity (ADHD), and autism spectrum disorders (ASD), affect more than 10% of children worldwide and constitute a major public health concern [
4]. According to the WHO, the incidence of neurodevelopmental disorders has dramatically increased over the past decades. For example, the incidence of ASD, estimated at 4 or 5 cases per 10,000 children in the 1970s, was reassessed at 1 per 110 in the 2000s [
5]. As these pathologies are complex and multifactorial, the contribution of EDCs is difficult to demonstrate and quantify. In humans, the first correlations have been established between in utero exposure to organophosphate pesticides and ADHD prevalence, and between exposure to polychlorinated biphenyls and reduced intellectual capacity [
5]. Recently, several studies have correlated exposure to bisphenol A (BPA) with increased cognitive and behavioral disorders [
6,
7]. BPA has been extensively used in industry, mainly to produce polycarbonate plastic and epoxy resins. Despite restriction of its use adopted in many countries, BPA is still ubiquitous in the environment and, consequently, in human biological fluids. In 2017, BPA was classified among the “substances of very high concern” in the REACH regulation [
8].
Although EDCs are likely to be involved in the etiology of neurodevelopmental disorders, so far, their toxicity has been poorly evaluated in the immature brain [
9]. In fact, in epidemiological and in vivo studies, intellectual, cognitive, emotional, and social behaviors are difficult to assess and quantify, making causal relationships difficult to establish. Thereby, it seems relevant to identify cellular and molecular mechanisms of EDC toxicity to define target processes and molecular markers that could ultimately allow a better evaluation of these compounds. Given the central role of neurons in the central nervous system (CNS) function and the fact that neuronal connectivity is impaired in many neurodevelopmental disorders, most studies have focused on EDCs’ impacts towards neurogenesis and synaptic plasticity [
9]. Conversely, few studies have analyzed the impact of EDCs on glial cells, and in particular, on oligodendrocytes, even though they carry out myelination, a fundamental step of CNS development [
9]. These cells arise later than neurons, from the third trimester of pregnancy. During this period, oligodendrocyte precursor cells (OPCs) proliferate, migrate, and differentiate into immature, and then mature, oligodendrocytes which myelinate the CNS by wrapping myelin sheaths around axons. In addition to their protective role, myelin sheaths increase the propagation rate of action potentials by insulating nerve fibers. Abnormal myelination contributes to neurological deficits. In particular, white matter alterations have been associated with ADHD and ASD [
10,
11]. By disrupting myelination, EDCs could contribute to the development of such disorders. Indeed, some studies have indicated that EDCs, especially BPA, can disrupt oligodendrocyte differentiation and myelination, but the evidence is still scarce [
3]. It must be emphasized that lipids represent 70% of myelin dry weight and, therefore, are, by far, the main myelin constituents [
12]. Thus, lipids are a valuable molecular target to assess oligodendrocyte differentiation and its disruption.
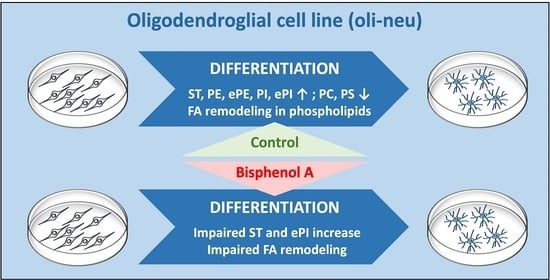
The aim of the present study was to assess the impact of BPA on oligodendrocyte differentiation in vitro through a comprehensive analysis of cellular lipid contents. For this purpose, we used the oligodendroglial cell line Oli-neu. Oli-neu are immortalized mouse OPCs transformed with activated
ErbB2 gene [
13]. These cells can undergo differentiation especially using the pharmacological agent PD174265 [
14,
15]. In proliferating conditions, Oli-neu cells exhibit a bipolar morphology typical of OPCs, while during differentiation, they become increasingly ramified and display an immature oligodendrocyte shape after 72 h (
Figure 1A). Moreover, proliferating Oli-neu cells express several markers of late OPCs including O4 and 2′,3′-cyclic nucleotide 3′ phosphodiesterase (CNPase) [
15]. During differentiation, CNPase and markers of mature oligodendrocytes accumulate, including myelin basic protein (MBP) (
Figure 1A) [
15]. Therefore, the Oli-neu cell line was chosen to model early oligodendrocyte differentiation. To investigate the impact of BPA on the lipidome of differentiating Oli-neu, the cells were exposed to several non-cytotoxic BPA concentrations for 72 h before the onset of differentiation and another 72 h during differentiation (
Figure 1B). Changes in lipidome accompanying Oli-neu differentiation were primarily characterized. As expected, Oli-neu differentiation led to a clear enrichment in several characteristic myelin lipid subclasses including ethanolamine plasmalogens (ePE) and sulfatides (ST). Importantly, the increase in ST was impaired by BPA, which could indicate a direct effect of this compound on oligodendrocyte early differentiation.
2. Material and Methods
2.1. Oli-Neu Culture, Differentiation, and BPA Treatment
Oli-neu cells were maintained in DMEM, high glucose GlutaMAXTM Supplement, pyruvate (Gibco reference 31966047, Thermo Fisher Scientific, Courtaboeuf, France) supplemented with 1% N2 (Gibco reference 17502001, Thermo Fisher Scientific, Courtaboeuf, France), 1% heat inactivated horse serum (Gibco reference 26050070, Thermo Fisher Scientific, Courtaboeuf, France), 1% penicillin-streptomycin (10,000 U/mL) (Gibco reference 15140122, Thermo Fisher Scientific, Courtaboeuf, France), 10 µg/mL insulin from bovine pancreas (Sigma I5500, Saint Quentin Fallavier, France), 0.4 µg/mL L-Thyroxine sodium salt pentahydrate (T4) (Sigma T0397, Saint Quentin Fallavier, France,), and 0.34 µg/mL 3,3′,5-Triiodo-L-thyronine sodium salt (T3) (Sigma T6397 Saint Quentin Fallavier, France).
Flasks and wells were coated with poly-L-lysine (Sigma P4707, Saint Quentin Fallavier, France) diluted at 20 µg/mL. Cells were exposed to BPA (Sigma 239658, Saint Quentin Fallavier, France) during 72 h of proliferation and an additional 72 h of differentiation induced by 1 μM PD174265 in DMSO (Sigma 513040-M, Saint Quentin Fallavier, France). The medium was changed after 48 h of differentiation. Oli-neu cells were exposed to 0.01, 0.1, 1, 10, 20, 50, and 100 µM BPA in DMSO. The final concentration of DMSO was 0.2%.
2.2. Cell Viability
Oli-neu cells were treated with BPA (0.01, 0.1, 1, 10, 20, 50, and 100 μM) in triplicate, in 96-well plates, during proliferation (72 h) followed by differentiation (72 h). Thiazolyl blue tetrazolium bromide (MTT, Merck M5655, Saint Quentin Fallavier, France) was extemporaneously diluted to prepare a 0.5 mg/mL solution in culture medium. At the end of the BPA treatment, 100 µL of MTT solution was directly added to the medium culture and cells were incubated for 3 h at 37 °C. After incubation, the medium was removed, cells were lysed with DMSO (100 μL), and homogenized (RT, 15 min) on a shaker plate. The optical density was read at 570 nm on a SpectraMax paradigm spectrophotometer (Molecular devices, Villepinte, France). The means were calculated from 5 independent experiments.
2.3. Western Blotting
Frozen cell pellets were homogenized in cold RIPA buffer (Sigma R0278, Saint Quentin Fallavier, France) containing protease inhibitors (cOmplete
TM Tablets, Roche 4693159001, Merck, Saint Quentin Fallavier, France) for 1 h. An equal amount of 10 µg protein per sample was used. Western Blot migration, transfer, incubation, revelation, and quantification were performed as previously described using mouse anti-β-actin (1:20,000, Sigma A5316, Saint Quentin Fallavier, France), mouse anti-CNPase (1:500, Merck-Millipore MAB326, Guyancourt, France), and HRP-conjugate goat anti-mouse IgG (1:2000, Sigma 12-349, Saint Quentin Fallavier, France) antibodies [
16]. Actin was used as loading control.
2.4. Immunocytofluorescence
Oli-neu cells were plated on poly-L-lysine-coated Ibidi 8-well chamber slides (BioValley, Marne la Vallee, France) and exposed to BPA. Then, cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% triton and saturated with 3.5% donkey serum in PBS. Immunocytofluorescence staining was performed using mouse anti-MBP (1:200, Merck-Millipore MAB382, Guyancourt, France) and mouse anti-CNPase (1:250, Merck-Millipore MAB326, Guyancourt, France). Cells were incubated overnight with primary antibodies at 4 °C, washed three times, and incubated with donkey anti-mouse secondary antibody conjugated to Alexa fluor 488 (1:500, Invitrogen A21202, Villebon-sur-Yvette, France) for 1 h. DAPI was used to counterstain nuclei (Sigma D9542, Saint Quentin Fallavier, France). Images were acquired using an Eclipse Ti-E inverted fluorescence microscope and the NIS-Elements software (Nikon, Champigny sur Marne, France).
2.5. RT-PCR
Cells (
N = 6 to 9 independent experiments per group) were lysed and total RNA was extracted with a Nucleospin RNA XS Plus kit (Macherey Nagel, Hoerdt, France). Then, RNA was dosed with a Qubit broad range kit using QFX DeNovix Fluorometer (Proteigene, Saint-Marcel, France), and 1 µg RNA was subjected to retrotranscription using an iScript
TM cDNA synthesis kit (Bio-Rad, Marnes-La-Coquette, France). Then, qPCRs were achieved for each sample in duplicates using SYBR
® Green Supermix (Bio-Rad, Marnes-La-Coquette, France) in a CFX96 Real-Time PCR machine (Bio-Rad, Marnes-La-Coquette, France), with a 3-step program (5 s of denaturation at 95 °C, 10 s of annealing at 60 °C, and 10 s of elongation at 72 °C). Specific primers (Eurogentec, Angers, France) were designed with the NCBI primer design tool. Sequences are given in
Supplementary Table S1.
Rpl13a (ribosomal protein L13a) was chosen to normalize the quantitative experiments based on reference gene suitability testing. The results are expressed as relative expression to D0 samples.
2.6. Lipidomic Analysis
2.6.1. Cell Lysis and Lipid Extraction
Cell pellets of 2.5 million cells (N = 6 independent experiments per group) were stored at −80 °C until the start of the analytical process. Pellets were resuspended in 1 mL of ultra-pure water and sonicated for 5 min. Then, suspensions were spiked with 15 µL of a mixture of internal standards (15 µM concentration each) and extracted with 7.25 mL of a chloroform/methanol/water mixture (2:2:1.8 v/v/v) containing 3,5-di-tert-4-butylhydroxytoluene 0.01% (w/v) as antioxidant agent. Then, samples were centrifuged at 3000 rpm for 10 min. Organic phases were collected, evaporated to dryness at 45 °C under reduced pressure, and the lipid extracts were dissolved in 90 µL of acetonitrile/isopropanol/chloroform/water (35:35:20:10) mixture.
2.6.2. Sample Injection and Data Analysis
Sample injection was achieved using an ultra-high-performance liquid chromatography high-resolution mass spectrometry (UHPLC-HRMS) device consisting of a UPLC
® system (Waters, Manchester, UK) hyphenated to a Synapt
® G2 (Q-TOF) mass spectrometer (Waters, Manchester, UK). Chromatographic separation was achieved on an Acquity
® CSH C18 column (100 mm × 2.1 mm, 1.7 μm). UHPLC-HRMS data acquisition and analysis were adapted from a previously described protocol [
17]. Data dependent acquisition was performed to provide tandem mass spectra of the five most intense ions detected on a first mass spectrum. Lipid annotation included polar head group identification in glycerophospholipids, sphingoid base characterization in sphingolipids, and fatty acyl side chains. Moreover,
sn-1 and
sn-2 locations of fatty acid side chains in phospholipids were determined based on the relative intensity of carboxylate product ions displayed on the MS/MS spectra. The regioisomer mentioned for each lipid species identified corresponded to the major regioisomer (
Supplementary Table S2). However, the presence of a minor amount of the other regioisomer cannot be excluded. Concentration of each lipid species was determined using the corresponding internal standard. Lipid content was expressed as percentage of total fatty acid (mol%), which grants the advantage of comparing lipid distribution between samples without bias from their individual total lipid amount. The heatmap was built using statistical analysis (one factor) in Metaboanalyst 5.0 software (Montreal, Canada) with the default parameters [
18].
2.7. Statistical Analysis
Principal component analysis score plots were generated using the SIMCA-P+ software version 13.0.3 (Umetrics, Umeå, Sweden). Statistical analyses such as t-tests were performed on GraphPad Prism 8.0 (Graph-Pad Software, San Diego, CA, USA). Mol% of lipid subclasses and species were compared by a multiple t-test. MTT viability and gene expression data were analyzed by one-way ANOVA and Dunnett multiple comparison post-test after validation of normal distribution by a Shapiro–Wilk test. D0 was used as control group. The value of p < 0.05 was considered to be statistically significant.
3. Results
A MTT viability assay was achieved by exposing Oli-neu cells to BPA concentrations ranging from 0 to 100 µM during 72 h of proliferation followed by 72 h of differentiation (
Figure 1C). Cytotoxicity appeared from 20 µM BPA concentration (74.3 ± 6.1% viable cells,
p < 0.001), and cell viability gradually decreased to 1.9 ± 1.4% viable cells (
p < 0.001) at 100 µM of BPA. The subcytotoxic doses of BPA (up to 10 µM) were, thus, selected to perform lipidomic and gene expression analyses in Oli-neu cells (
Figure 1B). Four groups were finally considered: proliferating cells (P0), differentiated control cells (D0), differentiated cells exposed to BPA 1 µM (D1), and differentiated cells exposed to BPA 10 µM (D10).
To validate the differentiation protocol and address the effects of BPA through well-known markers of oligodendrocyte differentiation, we first assessed the expression of five genes coding myelin proteins, i.e., myelin basic protein (
Mbp), myelin-associated glycoprotein (
Mag), 2′,3′-cyclic nucleotide 3′ phosphodiesterase (
Cnp), myelin oligodendrocyte glycoprotein (
Mog), and proteolipid protein 1 (
Plp1) (
Figure 2A). Expression of these myelin genes experienced a dramatic increase between P0 and D0.
Plp1,
Mbp,
Cnp,
Mog, and
Mag expressions increased by 9.4 (
p < 0.001), 9.7 (
p < 0.001), 12.5 (
p < 0.001), 98.0 (
p < 0.001), and 104.2 folds (
p < 0.001), respectively. In differentiated Oli-neu, BPA exposure at 10 µM decreased the expression of
Plp1,
Cnp,
Mog, and
Mag genes by 0.75 (
p < 0.001), 0.69 (
p < 0.01), 0.63 (
p < 0.001), and 0.62 (
p < 0.001), respectively. Moreover, we investigated the levels of CNPase protein, an early expressed myelin-associated enzyme [
15]. Western blot analysis revealed low levels of CNPase in proliferating cells, while a strong CNPase signal was evidenced in differentiated cells, with comparable intensity among D0, D1 and D10 (
Figure 2B). In addition, CNPase was clearly detected by immunocytofluorescence in differentiated cells (
Figure 2C). As observed in Western blot, the intensity of CNPase staining appeared similar between control D0 cells and cells exposed to BPA. Finally, on the one hand, the efficiency of differentiation was confirmed by the upregulated expression of several genes coding for myelin proteins and CNPase accumulation. On the other hand, 10 µM BPA partially counteracted the increase in the expression of several genes coding for myelin proteins, including
Cnp, without affecting CNPase immunoreactivity.
A comprehensive lipidomic analysis was then performed by UHPLC-HRMS to assess changes accompanying Oli-neu differentiation and the impacts of BPA. Principal component analysis (PCA) models comparing lipid distribution (mol%) of P0, D0, D1, and D10 showed clear separation between P0 and a cluster including D0, D1, and D10 (
Supplementary Figure S1). Nevertheless, a clear clustering was observed when comparing D0 to P0, D0 to D1, and D0 to D10 (
Figure 3A). It is noteworthy that no separation was exhibited between D0 and differentiated cells exposed to lower concentrations of BPA, 0.01 µM and 0.1 µM (data not shown).
Regarding differentiation of unexposed cells, comparison of P0 to D0 revealed significant lipid distribution changes (
Figure 3B,
Supplementary Tables S3 and S4). A marked increase was observed from P0 to D0 for both phosphatidylethanolamine (PE) and their ether analogs (ePE), respectively, from 3.1 ± 0.27 mol% to 4.8 ± 0.21 mol% and from 3.1 ± 0.23 mol% to 7.3 ± 0.72 mol% (
p < 0.001). Regarding phospholipids, additional changes included an increase in phosphatidylinositol (PI) content from 5.3 ± 1.7 mol% to 10.0 ± 3.3 mol% (
p < 0.05). It was concomitant to a dramatic decrease in phosphatidylcholine (PC) abundance (from 72.0 ± 4.8 mol% to 59.6 ± 5.1 mol%,
p < 0.01) and, to a lesser extent, in phosphatidylserine (PS) (from 0.89 ± 0.17 mol% to 0.62 ± 0.13 mol%,
p < 0.05). Regarding sphingolipids, differentiation was accompanied by an increase in ceramide (Cer), sulfatide (ST), hydroxylated sulfatide (STOH), and sphingomyelin (SM) amounts, respectively, from 0.13 ± 0.02 mol% to 0.20 ± 0.03 mol% (
p < 0.01), from 0.46 ± 0.09 mol% to 1.69 ± 0.38 mol% (
p < 0.001), from 0.09 ± 0.03 mol% to 0.29 ± 0.16 mol% (
p < 0.05), and from 1.9 ± 0.32 mol% to 2.6 ± 0.17 mol% (
p < 0.01). Moreover, diglyceride (DG), free fatty acid (FA), and lyso-phosphatidylcholine (LPC) contents also increased between P0 and D0, from 0.20 ± 0.02 mol% to 0.25 ± 0.03 mol% (
p < 0.05), from 1.7 ± 0.33 mol% to 2.4 ± 0.45 mol% (
p < 0.01), and from 0.35 ± 0.08 mol% to 0.47 ± 0.09 mol% (
p < 0.05), respectively. The global composition in fatty acyl side chains was also determined in lipid subclasses (
Supplementary Table S2). In phospholipids, from P0 to D0, the global content in saturated FA (SFA) and monounsaturated FA (MUFA) decreased from 14.2 ± 2.9% to 8.6 ± 1.3% (
p < 0.01) and from 60.4 ± 3.6% to 48.3 ± 1.1% (
p < 0.001), respectively (
Figure 3C). Conversely, the content in polyunsaturated FA (PUFA) increased from 25.4 ± 1.8% in P0 to 43.1 ± 2.1% in D0 (
p < 0.001).
We next assessed the impact of BPA exposure on Oli-neu differentiation based on lipidomic analysis data. At both 1 and 10 μM doses, BPA decreased ST levels from 1.69 ± 0.38 mol% in D0 to 0.97 ± 0.13 (
p < 0.05) in D1 and 0.77 ± 0.09 (
p < 0.01) in D10 (
Figure 3B). Among the 10 identified ST species, which were all upregulated during differentiation (
p < 0.05), 5 species were significantly decreased by 10 µM BPA in differentiated cells (
Figure 3B, right panel). A decrease in ePI content was also measured in differentiated cells following exposure to 10 µM BPA (from 0.44 ± 0.08 mol% in D0 to 0.23 ± 0.02 mol% in D10,
p < 0.001). Regarding the global content of FA side chains in phospholipids, MUFA content was increased from 48.3 ± 1.1% in D0 to 51.0 ± 1.4% in D1 (
p < 0.01) and 52.3 ± 1.6% in D10 (
p < 0.001), while PUFA content was decreased from 43.1 ± 2.1% in D0 to 40.0 ± 1.4% in D1 (
p < 0.01) and 38.7 ± 0.75% in D10 (
p < 0.001) (
Figure 3C).
In addition to a lipidome analysis, we investigated lipid metabolism through gene expression by RT-qPCR. The lipidomic analysis revealing changes in lipid contents during Oli-neu differentiation and following BPA exposure, therefore, we assessed the gene expression of enzymes involved in corresponding lipid metabolic pathways (
Figure 4). Comparison of lipid contents both between P0 and D0 and between D0 and D1 or D10 groups especially highlighted changes in ST, thus, leading to explore the sphingolipid metabolism pathway (
Figure 4 and
Figure 5). Between P0 and D0, the expressions of
Gal3st1 coding for cerebroside sulfotransferase enzyme (CST),
Sptlc2 coding for serine palmitoyltransferase long chain base subunit 2 (SPTLC2), and
Ugt8a coding for ceramide UDP-galactosyltransferase (CGT) exhibited a respective fold change of 2.3 (
p < 0.001), 2.0 (
p < 0.001), and 6.2 (
p < 0.001) (
Figure 4). Gene expression of enzymes involved in sphingolipid catabolism, including
Galc coding for galactosylceramidase (GALC),
Gba coding for glucosylceramidase beta (GBA),
Gba2 coding for glucosylceramidase beta 2 (GBA2),
Smpd1 coding for sphingomyelin phosphodiesterase 1 (SMPD1), and
Smpd2 coding for sphingomyelin phosphodiesterase 2 (SMPD2), were approximately doubled between P0 and D0 (
p < 0.001). In differentiated Oli-neu, BPA altered the expression of several genes coding for enzymes of the sphingolipid metabolism pathway (
Figure 4 and
Figure 5). Among genes exhibiting an increased expression between P0 and D0, BPA induced a decrease in
Sptlc2 and
Ugt8a expressions, respectively, by 0.86 (
p < 0.05) and 0.78 (
p < 0.01) at 10 µM dose (D10) as compared with D0.
Gba expression, also increased during differentiation, was decreased by BPA exposure in both D1 (0.84,
p < 0.05) and D10 (0.75,
p < 0.001) groups. In addition,
Sgms2 coding for sphingomyelin synthase 2 (SGMS2) and
Ugcg coding for UDP-glucose ceramide glucosyltransferase (UGCG) were decreased in the D10 group by 0.79 (
p < 0.05) and 0.85 (
p < 0.05), respectively.
Since SFA, MUFA, and PUFA phospholipid contents also displayed changes between P0 and D0 and between D0 and D1 or D10 groups, fatty acid elongation was investigated through a gene expression analysis of
Elovl genes coding for elongase enzymes ELOVL1 to ELOVL7 (
Figure 4). Expression of
Elovl7 increased by 8-fold (
p < 0.001) between P0 and D0 groups, whereas
Elovl1,
Elovl4,
Elovl5, and
Elovl6 expressions were not modified. In differentiated Oli-neu, BPA exposure at 10 µM (D10 group) decreased the expression of
Elovl7 by 0.77 (
p < 0.01). Conversely, the expression of
Elovl1 was increased in both D1 and D10 groups by 2.1- (
p < 0.01) and 2.0-fold (
p < 0.05), respectively. (one-way ANOVA and Dunnett multiple comparison post-test, *
p < 0.05, **
p < 0.01, and ***
p < 0.001).



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




