4. Materials and Methods
Proton (1H) and carbon (13C) NMR spectra were recorded on Bruker Avance (III)-500 MHz spectrometer. Chemical shifts are reported in ppm relative to Me4Si (TMS, δ 0.00 ppm), or residual solvent peaks as an internal standard set to δ 7.26 and 77.16 ppm (CDCl3), or δ 3.31 and 49.00 ppm (CD3OD), or δ 2.50 and 39.52 ppm (d6-DMSO) or δ 7.01 and 20.43 ppm (toluene-d8). Electrospray ionization (ESI) mass spectrometry (MS) experiments were performed on a Waters QTOF Premier mass spectrometer (Micromass, UK) under normal conditions. Sodium formate solution was used as calibrant for high resolution mass spectra (HRMS) measurements. Reactions were monitored by thin layer chromatography (TLC) using 0.2 mm silica gel (Merck Kieselgel 60 F254) precoated aluminum plates, using UV light, ammonium molybdate or potassium permanganate staining solution to visualize. Silver nitrate-impregnated TLC plates were prepared by dipping silica gel precoated aluminum plates in a 20% silver nitrate solution in acetonitrile and drying these in an oven at 120 °C. Silver nitrate-impregnated silica gel was prepared by dispersing silica gel in a solution of the corresponding amount of silver nitrate in acetonitrile and concentrating this mixture to dryness. Flash column chromatography was performed on Davisil® silica gel (60, particle size 40–63 μm), or using Reveleris® silica or C18 reversed phase flash cartridges on a Grace Reveleris® automated flash system with continuous gradient facility. Solvents for reactions and chromatography were analytical grade and were used as supplied unless otherwise stated. Crystal structures were collected on an Agilent SuperNova diffractometer fitted with an EOS S2 detector, using CuKα radiation (1.54184 Å) at 120 K.
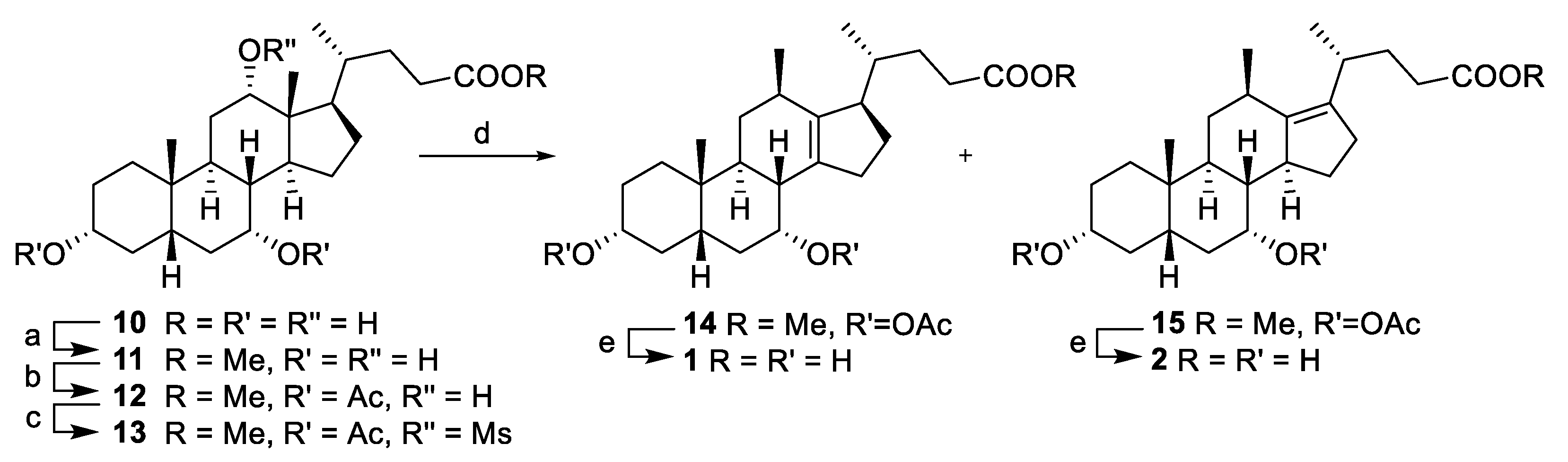
Methyl 3α,7α-diacetoxy-12α-hydroxy-5β-cholan-24-oate (
12) [
44]. To a solution of
11 [
45] (535.3 g, 1.27 mol) in ethyl acetate (2.5 L) was added 4-(dimethylamino)pyridine (7.80 g, 63 mmol) and pyridine (290 mL, 3.55 mol). The mixture was cooled to 3 °C before the dropwise addition of acetic anhydride (270 mL, 2.78 mol) and the reaction mixture was warmed to r.t. (48 h). Water (1.8 L) was added and the organic layer was washed with water (1 L), 10% HCl (2 × 600 mL), brine (600 mL), dried (MgSO
4) and concentrated. The product was purified via fractional precipitation from a mixture of ethyl acetate/petroleum ether (1:1.6) to afford the title compound
12 (283.4 g, 44%).
1H and
13C NMR spectra matched that previously reported [
44].
Methyl 3α,7α-diacetoxy-12α-[(methylsulfonyl)oxy]-5β-cholan-24-oate (
13) [
46]. A solution of
12 (283.4 g, 559 mmol) in pyridine (1.7 L) was concentrated to approx. 1.4 L then cooled to 3 °C before the addition of methanesulfonyl chloride (94 mL, 1.19 mol). After stirring at r.t. (20 h) the reaction was quenched with water (100 mL) and ethyl acetate (600 mL), then concentrated. Water (1 L) and ethyl acetate (1 L) were added and the organic layer was washed with water (800 mL), 10% HCl (600 mL) and brine (800 mL). The organic layer was concentrated to dryness to afford the title compound
13 (319.8 g, 98%).
1H and
13C NMR spectra matched that previously reported [
46].
Methyl 3α,7α-diacetoxy-12β-methyl-18-nor-5β-chol-13(14)-en-24-oate (14) and methyl 3α,7β-diacetoxy-12β-methyl-18-nor-5β-chol-13(17)-en-24-oate (15). To a suspension of mesylate 13 (319.8 g, 547 mmol) in acetic acid (320 mL) at 100 °C was added sodium acetate (136.8 g, 1.65 mol) and stirred (18 h) before concentrating from toluene (2 × 800 mL) then ethyl acetate (800 mL). Ethyl acetate (1 L) and water (600 mL) were added and the organic layer was washed with water (500 mL), brine (500 mL), dried (MgSO4) and concentrated to afford a crude sample containing the title compounds 14 and 15 as a ~1:2 mixture of isomers (respectively), which were used in the next step.
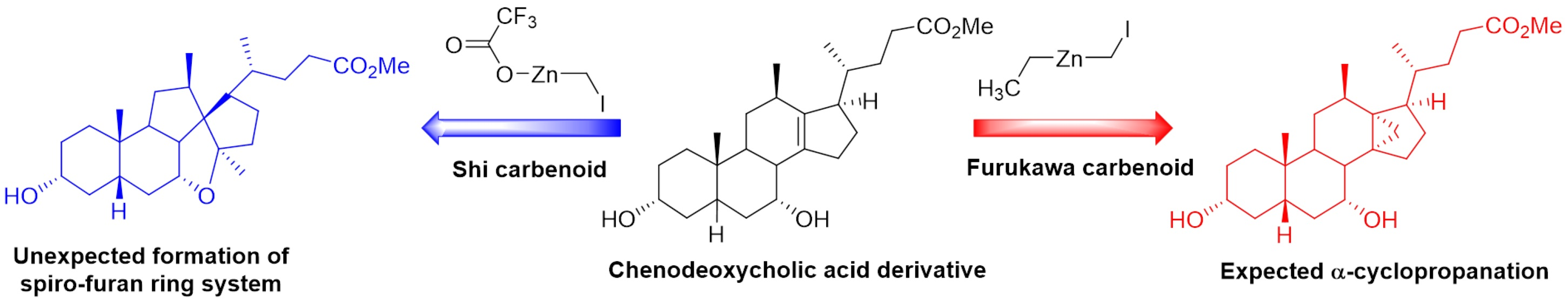
3α,7α-Dihydroxy-12β-methyl-18-nor-5β-chol-13(17)-en-24-oic acid (
1)
and 3α,7α-dihydroxy-12β-methyl-18-nor-5β-chol-13(14)-en-24-oic acid (
2) [
16]. To a solution of alkenes
14 and
15 in methanol (300 mL) was added NaOH (aq) (2M, 2.973 L, 5.95 mol) and the reaction mixture was stirred at 70 °C (18 h). The reaction was quenched with a solution of HCl (aq) (2M, 4.460 L, 8.92 mol) and the alkenes
1 (14.53 g, 7% from
13) and
2 (27.85 g, 13% from
13) were obtained separately by fractional precipitation from a mixture of methanol/ethyl acetate.
1H and
13C NMR spectra for each compound matched that previously reported [
16].
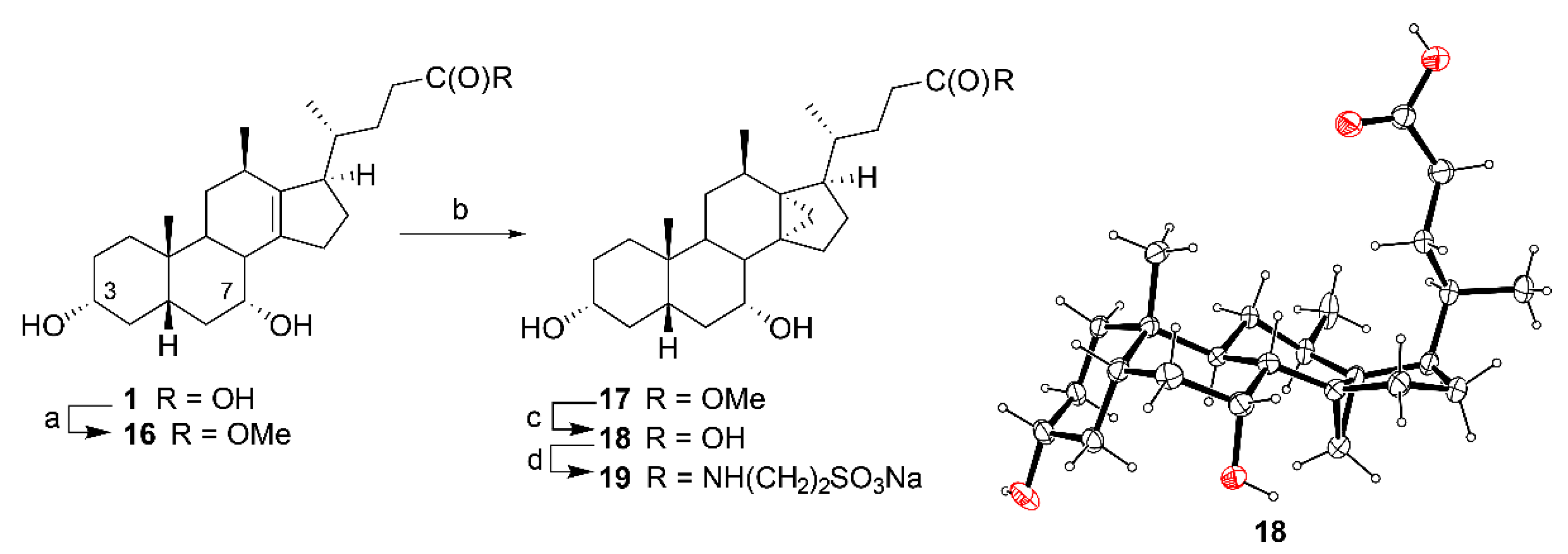
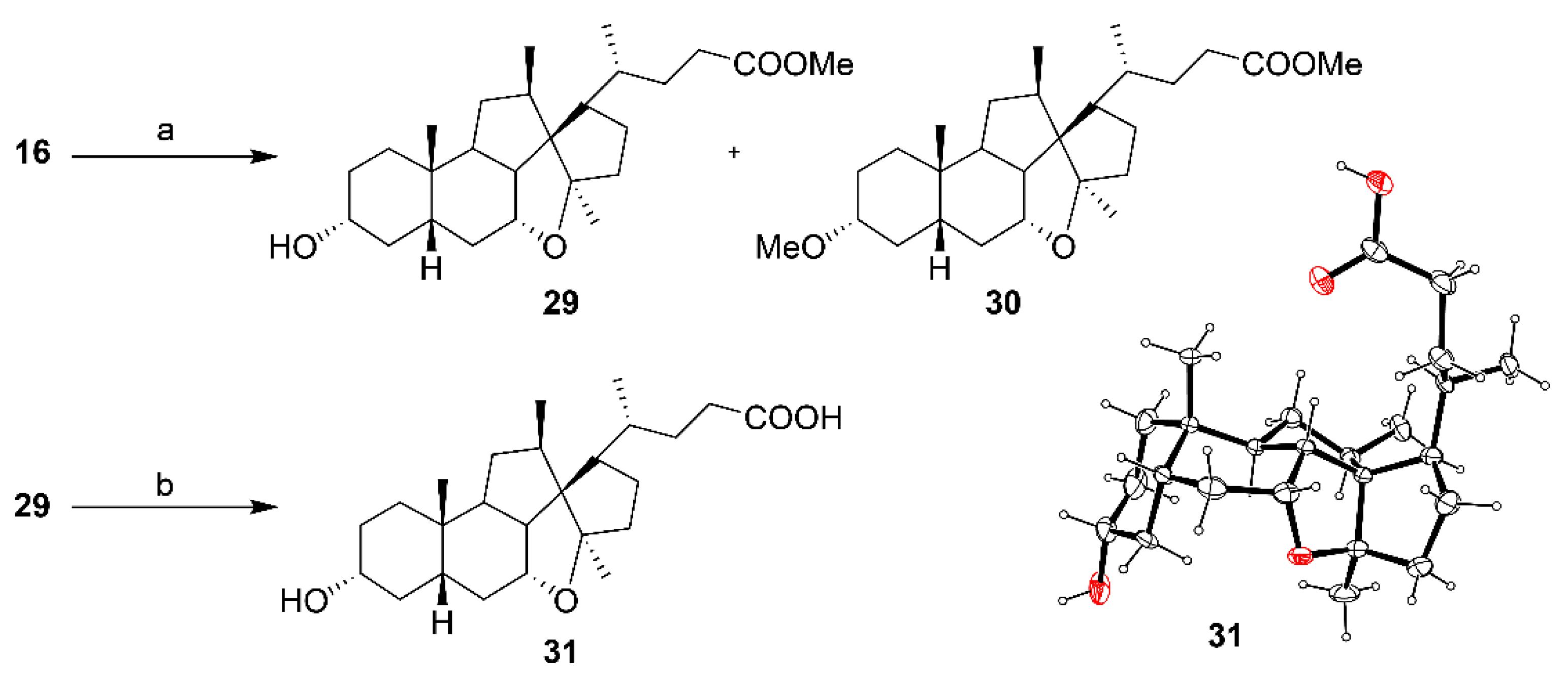
Methyl 3α,7α-dihydroxy-12β-methyl-18-nor-5β-chol-13(14)-en-24-oate (16). An ethereal solution of diazomethane was prepared by distilling a solution of Diazald (1.00 g, 2.56 mmol) in a mixture of Et2O (15 mL) and 1.8M KOH (aq) (2:8 mL water/EtOH, 17.8 mmol). The freshly distilled diazomethane was added slowly to a suspension of alkene 1 (1.00 g, 2.5 mmol) in ethyl acetate (100 mL) at r.t. until complete conversion to its methyl ester. Excess diazomethane was quenched with acetic acid and the solution was concentrated from toluene (50 mL) followed by methanol (50 mL) to afford the title compound 16 (0.83 g, 80%). 1H NMR (500 MHz, CDCl3) δ 4.08 (q, J = 2.9 Hz, 1H), 3.63 (s, 3H), 3.43 (tt, J = 11.1, 4.4 Hz, 1H), 2.62–2.56 (broad signal, 1H), 2.37–2.05 (m, 9H), 2.03–1.84 (m, 4H), 1.83–1.72 (m, 2H), 1.72–1.60 (m, 4H), 1.52 (dt, J = 14.0, 2.2 Hz, 1H), 1.44–1.16 (m, 3H), 1.05–0.93 (overlapping signals: m, 2H and 1.03, d, J = 7.1 Hz, 3H), 0.92 (d, J = 6.8 Hz, 3H), 0.87 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 174.5, 142.7, 137.9, 71.8, 68.1, 53.5, 51.4, 41.5, 41.0, 39.9, 35.6, 35.2, 34.9, 34.1, 33.6, 33.0, 32.7, 31.9, 31.3, 30.7, 25.2, 24.7, 22.3, 21.1, 18.9; HRMS(ESI) m/z calcd. for C25H40O4Na [M + Na]+: 427.2824, found 427.2825.
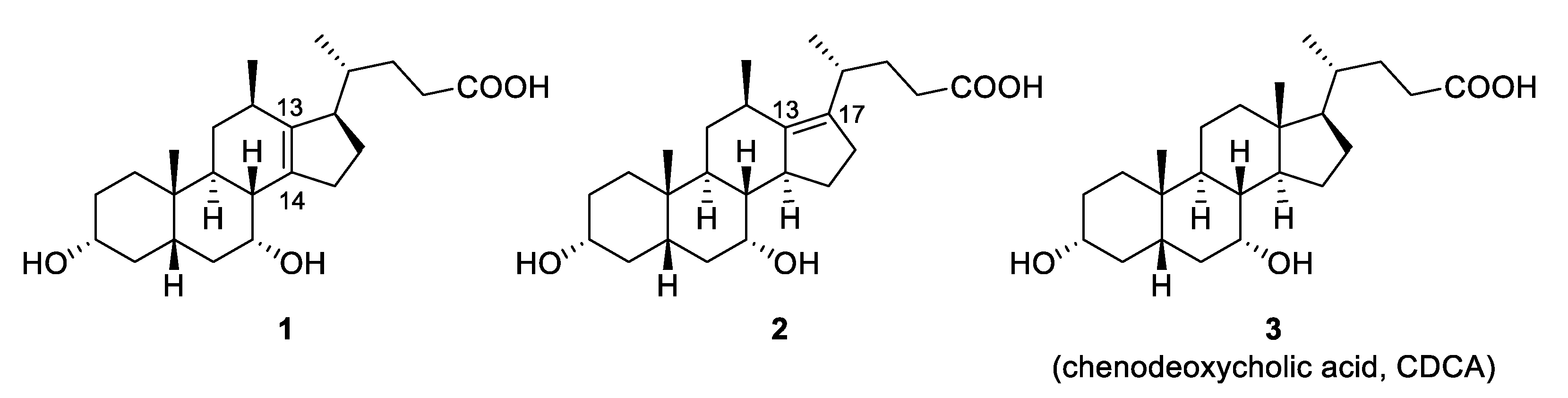
Methyl 3α,7α-dihydroxy-12β-methyl-13α,14α-methylene-18-nor-5β-cholan-24-oate (17). To Et2Zn (1M in hexanes, 2.5 mL, 0.25 mmol) in dichloromethane (10 mL) was added a solution of CH2I2 (0.23 mL, 2.8 mmol) in dichloromethane (5 mL), followed by addition of 16 (0.100 g, 0.247 mmol) in dichloromethane (5 mL) and the reaction mixture was stirred at 80 °C. After 4 h, the reaction was quenched with NH4Cl (20 mL), extracted with ethyl acetate (20 mL) and washed with brine (20 mL), dried (MgSO4) and concentrated. The residue was purified by flash chromatography on silica gel (ethyl acetate/petroleum ether, 0:10 to 8:2) to afford the title compound 17 (0.081 g, 78%). 1H NMR (500 MHz, CDCl3) δ 4.09 (q, J = 3.2 Hz, 1H), 3.64 (s, 3H), 3.46 (tt, J = 11.1 Hz, 4.5 Hz, 1H) 2.39 (ddd, J = 15.0, 10.4, 5.5 Hz, 1H), 2.24–2.10 (m, 4H), 1.99–1.92 (ddd, J = 14.8 Hz, 5.5 Hz, 3.9 Hz, 1H), 1.85–1.80 (m, 2H), 1.79–1.61 (m, 8H), 1.52–1.45 (m, 1H), 1.44–1.24 (m, 5H), 1.22 (d, J = 7.5 Hz, 3H), 1.20–1.08 (m, 2H), 1.01 (td, J = 14.3, 3.4 Hz, 1H), 0.87 (s, 3H), 0.85 (d, J = 6.6 Hz, 3H), 0.81–0.74 (m, 1H), 0.65 (d, J = 3.9 Hz, 1H), 0.35 (d, J = 3.9 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 174.2, 71.9, 69.0, 51.5, 50.5, 43.0, 41.0, 39.8, 39.5, 35.4, 34.8, 34.0, 33.7, 33.04, 32.99, 32.9, 32.6, 30.6, 30.1, 29.9, 27.1, 23.8, 22.8, 22.4, 20.9, 18.7; HRMS(ESI) m/z calcd. for C26H43O4 [M + H]+: 419.3161, found 419.3157.
3α,7α-Dihydroxy-12β-methyl-13α,14α-methylene-18-nor-5β-cholan-24-oic Acid (18). To a solution of 17 (0.045 g, 0.11 mmol) in MeOH (2 mL) was added NaOH (aq) (2M, 0.5 mL, 1 mmol). The reaction was stirred at 70 °C (2 h) before being quenched with a solution of HCl (aq) (2M, 1 mL, 2 mmol). The product was extracted with ethyl acetate (15 mL), washed with NaHCO3 (10 mL), brine (10 mL), dried (MgSO4) and concentrated to afford the title compound 18 (0.040 g, 92%). 1H NMR (500 MHz, CDCl3) δ 4.11 (q, J = 2.9 Hz, 1H), 3.48 (tt, J = 11.1, 4.4 Hz, 1H), 2.44 (ddd, J = 15.8, 10.5, 5.5 Hz, 1H), 2.27–2.13 (m, 4H), 2.00–1.93 (m, 1H), 1.86–1.62 (m, 9H), 1.52–1.45 (m, 1H), 1.45–1.25 (m, 5H), 1.23 (d, J = 7.5 Hz, 3H), 1.21–1.11 (m, 2H), 1.01 (td, J = 14.4, 3.6 Hz, 1H), 0.88–0.85 (overlapping signals: 0.86, d; 0.86, s, 6H), 0.84–0.76 (m, 1H), 0.66 (d, J = 4.1 Hz, 1H), 0.37 (d, J = 4.1 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 178.5, 72.0, 69.1, 50.5, 43.0, 41.1, 39.7, 39.5, 35.5, 34.8, 34.1, 33.8, 33.00, 32.95, 32.76, 32.67, 30.6, 30.1, 30.0, 26.9, 23.8, 22.8, 22.4, 20.9, 18.7; HRMS(ESI) m/z calcd for C25H41O4 [M + H]+: 405.3005, found 405.3005. Crystal data for C25H40O4 (M = 404.57 g/mol): orthorhombic, space group P212121, a = 11.4831(2) Å, b = 12.4753(2) Å, c = 19.9429(3) Å, V = 2856.92(8) Å3, Z = 4, T = 120(2) K, μ(CuKα) = 0.489 mm−1, 17,623 reflections measured (4.18° ≤ Θ ≤ 67.684°), 5577 unique (Rint = 0.0392), which were used in all calculations. The final R1 was 0.0434 (I > 2σ(I)) and wR2 was 0.1393 (all data).
3α,7α-Dihydroxy-12β-methyl-13,14-methylene-18-nor-N-(2-sulfoethyl)-5β-cholan-24-amide (19). Cyclopropane 18 (0.190 g, 0.470 mmol) was concentrated from triethylamine (0.16 mL, 1.15 mmol) then dissolved in THF (9 mL), cooled to −10 °C then isobutyl chloroformate (0.07 mL, 0.5 mmol) was added. After 1 h, triethylamine (0.16 mL, 1.15 mmol) was added followed by an aqueous solution of taurine (0.145 g, 1.14 mmol in 0.92 mL water) and the reaction was allowed to warm to r.t. (16 h). The mixture was concentrated, the residue was dissolved in MeOH (5 mL) and NaOH (2M, 1 mL, 2 mmol) was added. After 2 min, the mixture was concentrated then purified by flash chromatography on silica gel (chloroform/MeOH, 10:0 to 1:1) followed by chromatography on RP-C18 (water/MeOH, 20:1 to 1:4) to afford the title compound 19 (0.102 g, 40%). 1H NMR (500 MHz, CD3OD) δ 4.11 (q, J = 2.9 Hz, 1H), 3.60 (td, J = 6.9, 2.2 Hz, 2H), 3.45–3.37, (m, 1H), 2.98 (td, J = 7.1, 1.2, Hz, 2H), 2.36–2.27 (m, 1H), 2.27–2.19 (m, 3H), 2.12–2.00 (m, 2H), 1.94–1.86 (m, 2H), 1.85– 1.72 (m, 4H), 1.71–1.66 (m, 1H), 1.65–1.58 (m, 2H), 1.58–1.51 (m, 1H), 1.51–1.44 (m, 1H), 1.43–1.31 (m, 3H),1.29 (d, J = 7.5 Hz, 3H), 1.27–1.16 (m, 2H), 1.05 (td, J = 14.2, 3.4 Hz, 1H), 0.95 (s, 3H), 0.91 (d. J = 6.5 Hz), 0.89–0.81 (m, 1H), 0.70 (d, J = 4.3 Hz, 1H), 0.42 (d, J = 4.5 Hz, 1H); 13C NMR (126 MHz, MeOD) δ 176.3, 72.8, 70.1, 52.1, 51.6, 44.6, 42.8, 41.1, 40.5, 36.8, 36.7, 36.2, 36.1, 35.3, 35.1, 35.0, 34.3, 34.3, 31.8, 31.4, 31.3, 29.3, 25.7, 24.0, 23.4, 21.4, 19.2; HRMS(ESI) m/z calcd. for C27H44NNaO6S [M + Na]+: 533.2787, found 533.3123.
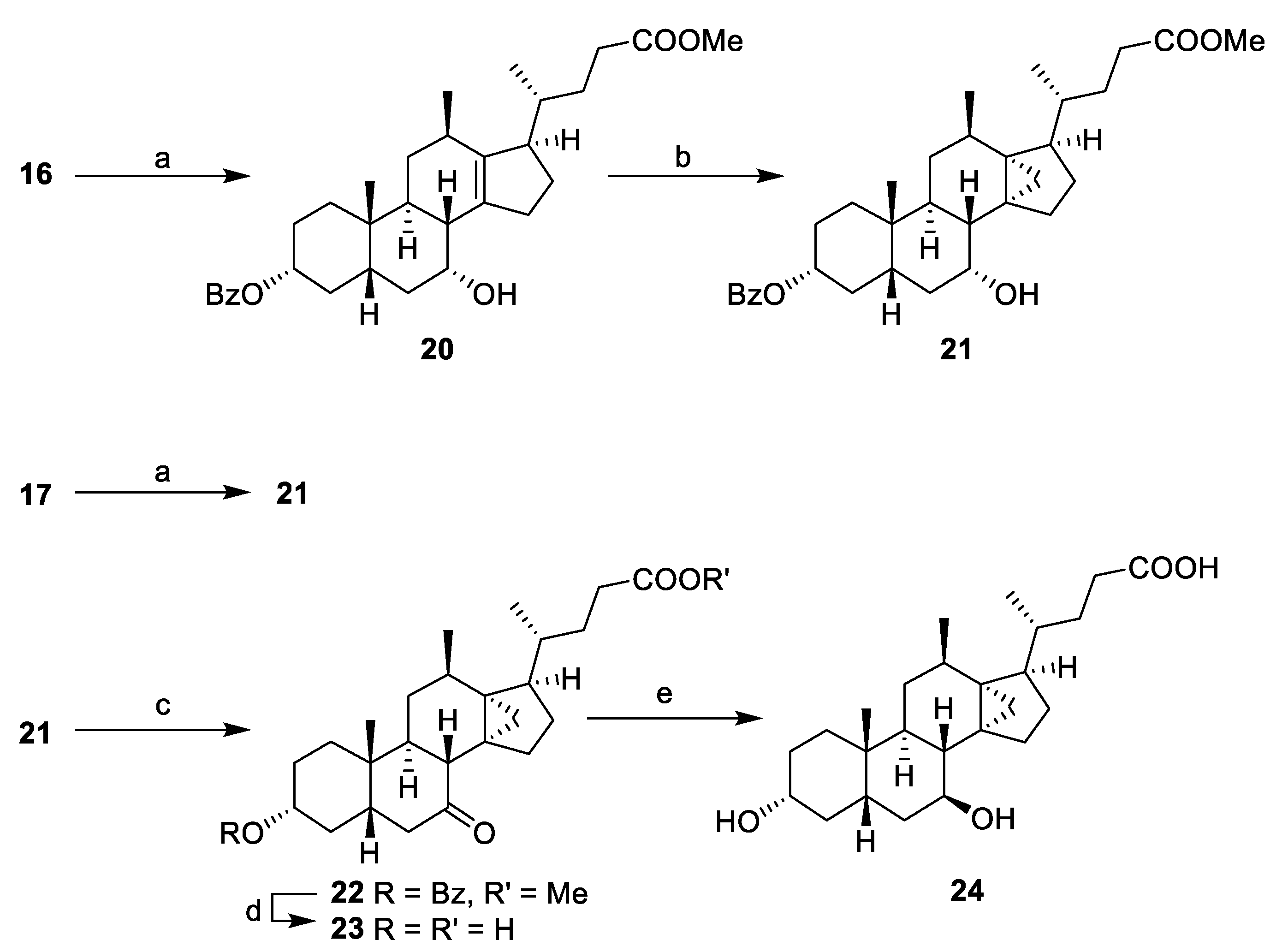
Methyl 3α-benzoyl-7α-hydroxyl-12β-methyl-18-nor-5β-chol-13(14)-en-24-oate (20). To a solution of 16 (1.00 g, 2.47 mmol) in a mixture of toluene (11 mL) and pyridine (12 mL) was added benzoyl chloride (0.44 mL, 3.8 mmol) and the mixture was stirred at r.t. (2 h). The reaction was quenched with water (20 mL), extracted with ethyl acetate (15 mL) and the organic layer was washed with NaOH (1M, 20 mL), water (20 mL) and brine (20 mL), dried (MgSO4), concentrated and the resulting residue was purified by flash chromatography on silica gel (ethyl acetate/ petroleum ether, 0:10 to 3:7) to afford 20 (1.172 g, 93%). 1H NMR (500 MHz, CDCl3) δ 8.02–7.97 (m, 2H), 7.52–7.47 (m, 1H), 7.42–7.35 (m, 2H), 4.85 (tt, J = 11.4, 4.6 Hz, 1H), 4.13 (q, J = 2.9 Hz, 1H), 3.64 (s, 3H), 2.66–2.60 (m, 1H), 2.59–2.48 (m, 1H), 2.43–2.32 (m, 3H), 2.32–2.23 (m, 2H), 2.19–2.10 (m, 2H), 2.05–1.97 (m, 2H), 1.96–1.88 (m, 1H), 1.88–1.76 (m, 4H), 1.75–1.66 (m, 2H), 1.66–1.59 (m, 1H), 1.58–1.52 (m, 2H), 1.32–1.21 (m, 2H), 1.17 (td, J = 14.4, 3.4 Hz, 1H), 1.09–0.96 (overlapping signals: m, 1H; 1.05, d, J = 6.8 Hz, 3H), 0.96–0.89 (overlapping signals: d, J = 6.8 Hz, 3H; 0.93, s, 3H); 13C NMR (126 MHz, CDCl3) δ 174.5, 166.1, 142.9, 137.9, 132.5, 130.9, 129.5, 128.1, 74.8, 68.1, 53.5, 51.4, 41.5, 40.7, 35.6, 35.4, 35.0, 34.9, 34.1, 33.6, 33.0, 32.7, 31.9, 31.4, 26.9, 25.2, 24.8, 22.3, 21.1, 18.9; HRMS(ESI) m/z calcd. for C32H44O5Na [M + Na]+: 531.3086, found 531.3090.
Methyl 3α-benzoyl-7α-hydroxy-12β-methyl-13α,14α-methylene-18-nor-5β-cholan-24-oate (21). From 17: To a solution of 17 (0.418 g, 0.999 mmol) in toluene (5 mL) and pyridine (10 mL) was added benzoyl chloride (0.2 mL, 2 mmol) and the mixture was stirred at r.t. (1 h). Water (15 mL) and ethyl acetate (15 mL) were added and the organic layer was washed with NaOH (1M, 20 mL), water (20 mL) and brine (20 mL). The crude mixture was concentrated from toluene and MeOH, treated with Amberlyst® A26 resin (hydroxide form) then purified by flash chromatography on silica gel (ethyl acetate/petroleum ether, 0:10 to 3:7) to afford compound 21 (0.493 g, 94%). From 20: To Et2Zn (1M in hexanes, 2.5 mL, 2.5 mmol) in dichloromethane (15 mL) was added a solution of CH2I2 (0.57 mL, 6.9 mmol) in dichloromethane (10 mL), followed by the dropwise addition of 20 (0.590 g, 1.16 mmol) in dichloromethane (15 mL) and the reaction mixture was stirred at 80 °C (18 h). The reaction was quenched with NH4Cl (100 mL), extracted with ethyl acetate (2 × 50 mL) and the combined organic layers were washed with NaHCO3 (50 mL), brine (100 mL), dried (MgSO4) and concentrated. The residue was purified by flash chromatography on silica gel (ethyl acetate/petroleum ether, 0:10 to 3:7) to afford the title compound 21 (0.462 g, 76%). 1H NMR (500 MHz, CDCl3) δ 8.02 (d, J = 7.3 Hz, 2H), 7.50 (t, J = 7.3 Hz, 1H), 7.39 (t, J = 7.7 Hz, 2H), 4.83 (tt, J = 11.5, 4.4 Hz, 1H), 4.11 (broad signal, 1H), 3.65 (s, 3H), 2.49–2.33 (m, 2H), 2.27–2.21 (m, 1H), 2.20–2.04 (m, 3H), 2.04–1.95 (m, 1H), 1.95–1.71 (m, 8H), 1.70–1.62 (m, 1H), 1.60–1.45 (m, 3H), 1.45–1.31 (m, 2H), 1.23 (d, J = 7.4 Hz, 3H), 1.21–1.09 (m, 3H), 0.92 (s, 3H), 0.89–0.77 (overlapping signals: m, 1H; 0.85, d, J = 6.5 Hz, 3H), 0.69–0.64 (m, 1H), 0.45–0.38 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 174.2, 166.1, 132.5, 131.0, 129.5, 128.1, 75.0, 69.0, 51.4, 50.4, 43.0, 40.9, 39.5, 35.3, 35.2, 34.9, 34.1, 33.7, 33.0, 32.9, 32.8, 32.6, 30.1, 29.9, 27.1, 27.0, 23.8, 22.8, 22.4, 20.8, 18.7; HRMS(ESI) m/z calcd. for C33H46O5Na [M + Na]+: 545.3243, found 545.3248.
Methyl 3α-benzoyl-7-oxo-12β-methyl-13α,14α-methylene-18-nor-5β-cholan-24-oate (22). Pyridinium chlorochromate (PCC) method: To a solution of 21 (0.190 g, 0.36 mmol) in dichloromethane (5 mL) was added silica gel (0.300 g) then PCC (1.200 g, 5.6 mmol) and the mixture was stirred at r.t. (2 h). The crude mixture was filtered, washed with ethyl acetate (30 mL) and the filtrate was washed with water and brine until the aqueous washing were clear affording the title compound 22 (0.181 g, 95%). Dess–Martin method: To a solution of 21 (0.045 g, 0.086 mmol) in dichloromethane (5 mL) was added Dess–Martin periodinane (1.4 equiv.) and the reaction was stirred at r.t. (4 h). The mixture was quenched with isopropanol (0.5 mL), concentrated and the residue was purified by flash chromatography on silica gel (ethyl acetate/toluene, 0:10 to 3:7) to afford the title compound 22 (0.029 g, 65%). 1H NMR (500 MHz, CDCl3) δ 8.04–7.95 (m, 2H), 7.56–7.48 (m, 1H), 7.45–7.36 (m, 2H), 4.94 (tt, J = 11.3, 4.7 Hz, 1H), 3.67 (s, 3H), 2.93–2.86 (m, 1H), 2.66 (d, J = 12.3 Hz, 1H), 2.41 (ddd, J = 15.4, 10.2, 5.4 Hz, 1H), 2.23–2.08 (m, 3H), 2.07–1.97 (m, 3H), 1.97–1.91 (m, 1H), 1.90–1.83 (m, 3H), 1.83–1.75 (m, 1H), 1.75–1.66 (m, 1H), 1.53–1.40 (m, 3H), 1.36–1.26 (m, 3H), 1.25–1.22 (overlapping signals: m, 1H; d, J = 7.5 Hz, 3H), 1.21 (s, 3H), 1.17–1.09 (m, 1H), 1.09–1.00 (m, 1H), 0.87–0.81 (overlapping signals: m, 1H; 0.84, d, J = 6.5 Hz, 3H), 0.25–0.20 (m, 1H); 13C NMR (126 MHz, CDCl3) δ 210.5, 174.2, 166.0, 132.8, 130.5, 129.5, 128.2, 73.6, 52.3, 51.5, 50.2, 45.1, 44.9, 39.4, 39.0, 34.8, 34.1, 33.8, 33.7, 32.9, 31.0, 30.9, 27.1, 26.4, 24.5, 23.1, 22.4, 20.6, 18.7; HRMS(ESI) m/z calcd. for C33H45O5 [M + H]+: 521.3267, found 521.3271.
3α-Hydroxy-7-oxo-12β-methyl-13α,14α-methylene-18-nor-5β-cholan-24-oic acid (23). To a solution of 22 (0.043 g, 0.083 mmol) in MeOH (2 mL) was added NaOH (aq) (2M, 0.5 mL, 1 mmol). The reaction was stirred at 70 °C (2 h) before being quenched with a solution of HCl (aq) (2M, 1 mL, 2 mmol). The product was extracted with ethyl acetate (15 mL), washed with NaHCO3 (15 mL), brine (15 mL), dried (MgSO4), and purified via flash chromatography on silica gel (acetone/dichloromethane + 1% AcOH, 0:10 to 1:1) to afford the title compound 23 (0.025 g, 75%). 1H NMR (500 MHz, CD3OD) δ 3.62–3.50 (m, 1H), 3.02 (dd, J = 1H), 2.79 (d, J = 12.3 Hz, 1H), 2.41 (ddd, J = 15.3, 10.1, 5.5 Hz, 1H), 2.30–2.10 (m, 3H), 1.99–1.86 (m, 5H), 1.86–1.75 (m, 2H), 1.72–1.62 (m, 2H), 1.56–1.48 (m, 1H), 1.39–1.09 (overlapping signals: m, 8H; 1.31, d, J = 7.5 Hz, 3H; 1.21, s, 3H), 0.90 (d, J = 6.6 Hz, 3H), 0.81 (d, J = 4.9 Hz, 1H), 0.22 (d, J = 4.6 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 214.3, 177.9, 71.5, 53.6, 51.7, 46.8, 46.1, 41.0, 40.5, 38.6, 35.8, 35.5, 35.0, 35.0, 34.1, 32.5, 32.3, 30.8, 28.7, 25.4, 24.1, 23.0, 21.0, 19.0; HRMS(ESI) m/z calcd. for C25H37O4− [M − H]−: 401.2692, found 401.2713.
3α,7β-dihydroxy-12β-methyl-13α,14α-methylene-18-nor-5β-cholan-24-oic Acid (24). To a solution of 23 (0.022 g, 0.056 mmol) in isopropanol (5 mL) was added an excess of Na(s) (added until complete dissolution) and the reaction was heated to 100 °C (5 h). Once cooled, HCl (2M) was added until the pH = 1 and the product was extracted with ethyl acetate (3 × 15 mL). The organic layers were combined, washed with brine (10 mL), dried (MgSO4) and purified by flash chromatography on silica gel (acetone/dichloromethane + 1% acetic acid, 0:10 to 1:4) to afford the title compound 24 (5.0 mg, 21%). 1H NMR (500 MHz, CD3OD) δ 3.63 (td, J = 11.6, 4.8 Hz, 1H), 3.52 (tt, J = 10.5, 4.8 Hz, 1H), 2.39 (ddd, J = 15.1, 10.2, 5.4 Hz, 1H), 2.29–2.22 (m, 1H), 2.18–2.10 (m, 3H), 1.98–1.76 (m, 5H), 1.70–1.42 (m, 9H), 1.33–1.16 (m, 9H), 1.13–1.04 (m, 2H), 0.97 (s, 3H), 0.91 (d, J = 6.7 Hz, 3H), 0.63 (d, J = 4.4 Hz, 1H), 0.22 (dd, J = 4.5, 1.6 Hz, 1H). 13C NMR (126 MHz, CD3OD) δ 177.9, 72.4, 72.2, 51.3, 47.8, 43.8, 41.7, 38.6, 38.4, 37.3, 36.49, 36.46, 35.6, 35.5, 35.2, 34.3, 34.0, 31.1, 29.6, 28.8, 24.3, 24.3, 23.6, 21.1, 19.1; HRMS(ESI) m/z calcd. for C25H39O4 [M + H]+: 403.2848, found 403.2837.
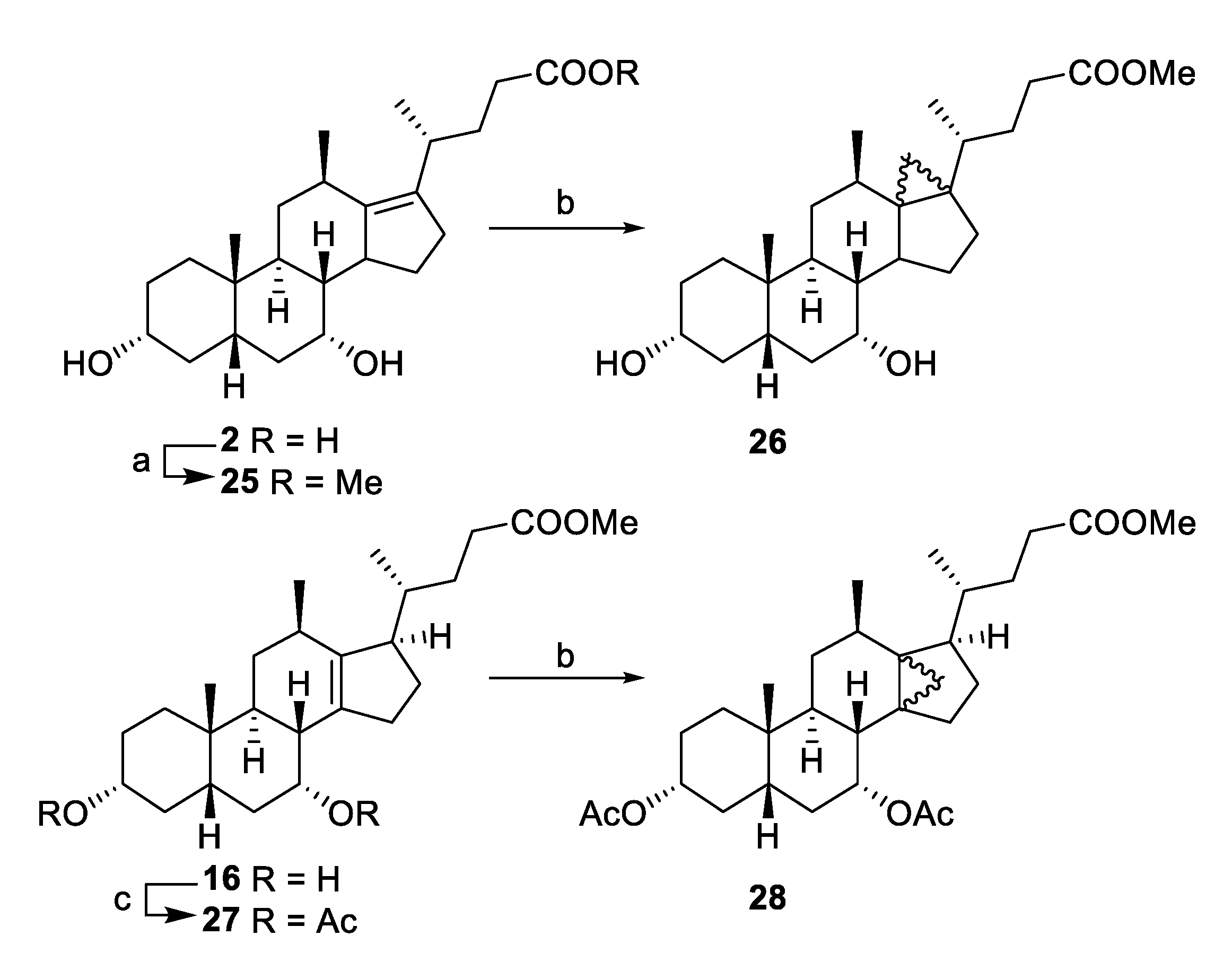
Methyl 3α,7α-dihydroxy-12β-methyl-18-nor-5β-chol-13(17)-en-24-oate (25). An ethereal solution of diazomethane was prepared by distilling a solution of Diazald (1.00 g, 2.56 mmol) in a mixture of Et2O (15 mL) and 1.8M KOH (aq) (2:8 mL water/EtOH, 17.8 mmol). The freshly distilled diazomethane was slowly added to a suspension of 2 (0.459 g, 1.18 mmol) in ethyl acetate (100 mL) at r.t. until completion. Excess diazomethane was quenched with acetic acid and the solution was concentrated from toluene (50 mL) followed by methanol (50 mL) before concentrating to dryness. The product was purified by flash chromatography on silica gel (ethyl acetate/petroleum ether, 3:7 to 7:3) to afford the title compound 25 (0.446 g, 93%). 1H NMR (500 MHz, CDCl3) δ 3.92–3.85 (b.s., 1H), 3.60 (s, 3H), 3.40 (tt, J = 11.1, 4.4 Hz, 1H), 2.95 (h, J = 6.9 Hz, 1H), 2.66–2.23 (m, 3H), 2.23–2.08 (m, 6H), 2.00–1.93 (m, 1H), 1.93–1.85 (m, 2H), 1.84–1.77 (m, 1H), 1.70–1.61 (m, 2H), 1.61–1.54 (m, 3H), 1.54–1.48 (m, 1H), 1.39–1.27 (m 2H), 1.27–1.20 (m, 1H), 1.19 (d, J = 7.0 Hz, 3H), 1.05 (td, J = 11.2 Hz, 3.1 Hz, 1H), 0.97–0.89 (overlapping signals: m, 1H and 0.93, d, J = 6.7 Hz, 3H), 0.82–0.73 (overlapping signals: m, 1H and 0.76, s, 3H); 13C NMR (126 MHz, CDCl3) δ 174.3, 139.9, 134.2, 71.8, 67.0, 51.3, 49.3, 48.8, 41.2, 39.5, 36.3, 35.6, 35.1, 34.8, 34.4, 32.9, 32.5, 31.0, 30.6, 30.3, 30.0, 25.9, 22.8, 20.7, 19.6; HRMS(ESI) m/z calcd. for C25H40O4Na [M + Na]+: 427.2824, found 427.2824.
Methyl 3α,7α-diacetyloxy-12β-methyl-18-nor-5β-chol-13(14)-en-24-oate (27). To a solution of 16 (83 mg, 0.21 mmol) in toluene (4.5 mL) was added pyridine (1 mL) followed by 4-(dimethylamino)pyridine (8 mg, 0.065 mmol) then acetic anhydride (0.2 mL) at r.t. After 3 h, the reaction mixture was concentrated, dissolved in ethyl acetate (20 mL) then washed with 2M HCl (10 mL). The organic layer was washed with water, sat. NaHCO3 (20 mL), brine (20 mL), dried (MgSO4) and concentrated. The crude residue was purified by flash chromatography on silica gel (ethyl acetate/petroleum ether, 0:10 to 3:7) to afford the title compound 27 (68 mg, 68% yield). 1H NMR (500 MHz, CDCl3) δ 5.06 (q, J = 3.1 Hz, 1H), 4.63–4.54 (m, 1H), 3.63 (s, 3H), 2.55 (d, J = 9.1 Hz, 1H), 2.40 (d, J = 11.7 Hz, 1H), 2.32 (ddd, J = 15.0, 9.6, 5.3 Hz, 2H), 2.22–2.09 (m, 2H), 2.08–1.93 (overlapping signals: m, 4H and 1.99, s, 3H and 1.99, s, 3H), 1.92–1.81 (m, 2H), 1.78–1.60 (m, 6H), 1.60–1.54 (m, 1H), 1.53–1.40 (m, 2H), 1.26–1.17 (m, 1H), 1.11 (td, J = 14.4, 3.5 Hz, 1H), 1.02 (d, J = 6.8 Hz, 3H), 1.01–0.84 (overlapping signals: m, 1H and 0.91, d, J = 6.8 Hz, 3H and 0.90, s, 3H); 13C NMR (126 MHz, CDCl3) δ 174.3, 170.42, 170.39, 140.8, 137.6, 74.0, 70.8, 53.3, 51.3, 40.4, 39.3, 35.5, 34.6, 34.5, 33.4, 33.2, 32.53, 32.48, 31.5, 31.0, 26.8, 25.2, 25.0, 22.2, 21.4, 21.3, 21.0, 18.7; HRMS(ESI) m/z calcd. for C29H44O6Na [M + Na]+: 511.3036, found 511.3039.
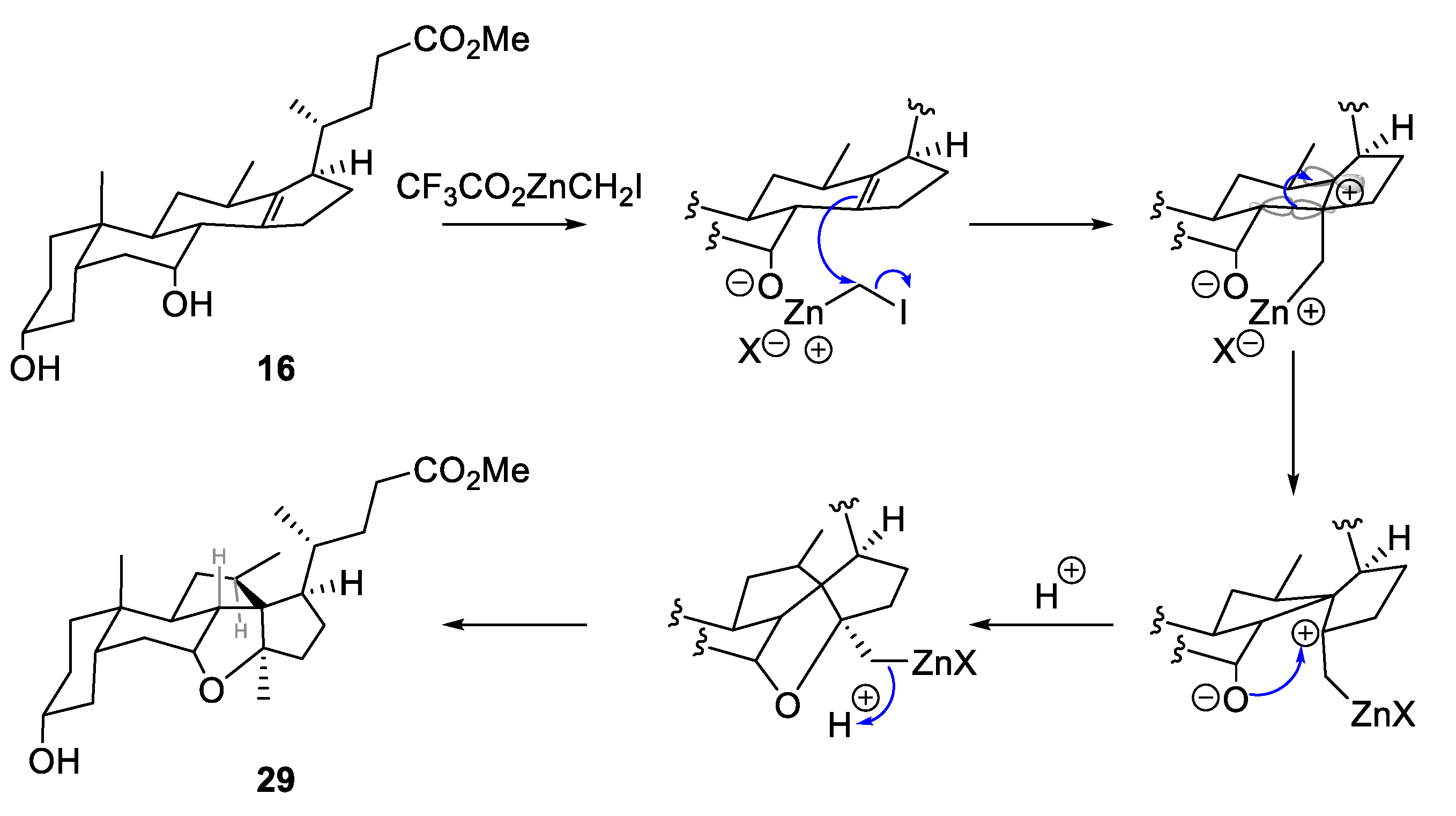
Methyl (R)-4-((1S,3aR,4aR,4a1R,5aS,7R,9aS,9bS,11R,11aR)-7-hydroxy-3a,9a,11-trimethylhexadecahydrobenzo[4,5]indeno[7,1-bc]cyclopenta[d]furan-1-yl)pentanoate (29) and methyl (R)-4-((1S,3aR,4aR,4a1R,5aS,7R,9aS,9bS,11R,11aR)-7-methoxy-3a,9a,11-trimethylhexadecahydrobenzo[4,5]indeno[7,1-bc]cyclopenta[d]furan-1-yl)pentanoate (30). To a three-neck flask under argon and cooled to 0 °C was added dichloromethane (5 mL) then diethylzinc (1 M in hexanes, 10 mL) followed trifluoroacetic acid (0.4 mL in 10 mL of dichloromethane) and the mixture was left to stir for 10 min. A solution of diiodomethane (0.8 mL, 10 mmol) in dichloromethane (5 mL) was then added dropwise at 0 °C and the mixture stirred for 20 min before the addition of 16 (0.431 g, 1.07 mmol) in dichloromethane (5 mL) and the reaction was allowed to warm to r.t. After 4 h, the reaction was quenched with saturated NH4Cl (40 mL) and the aqueous layer was washed with ethyl acetate (2 × 30 mL). The combined organic layers were washed with NaHCO3 (40 mL), water (40 mL), brine (40 mL), dried (MgSO4) and concentrated. The crude residue was purified by flash chromatography on silica gel (ethyl acetate/petroleum ether, 1:4 to 4:1) to afford 29 (0.031 g, 7%) and 30 (0.103 g, 30% yield, based on a purity of 90%) as separate compounds. Compound 29: 1H NMR (500 MHz, CDCl3) δ 4.21–4.13 (m, 1H), 3.65 (s, 3H), 3.55 (tt, J = 9.6, 4.9 Hz, 1H), 2.41 (ddd, J = 14.9, 9.4, 5,2 Hz, 1H), 2.32–2.19 (m, 3H), 2.01–1.97 (m, 1H), 1,91–1.85 (m, 1H), 1.85–1.81 (m, 1H), 1.81–1.76 (m, 3H), 1.76–1.70 (m, 1H), 1.70–1.64 (m, 4H), 1.59–1.50 (m, 2H), 1.49–1.43 (m, 1H), 1.43–1.39 (m, 1H), 1.39–1.33 (m, 2H), 1.33–1.26 (m, 1H), 1.25 (s, 3H), 1.22–1.15 (m, 1H), 1.14–1.04 (m, 1H), 1.00 (d, J = 7.3 Hz, 3H), 0.97 (d, J = 6.9 Hz, 3H), 0.96–0.89 (m, 1H), 0.89–0.78 (overlapping signals: m, 1H and 0.84, s, 3H); 13C NMR (126 MHz, CDCl3) δ 174.1, 94.4, 77.1, 70.4, 64.3, 51.49, 51.45, 50.7, 42.1, 39.7, 39.0, 38.6, 38.1, 35.8, 35.2, 33.6, 33.3, 32.7, 32.3, 30.2, 29.8, 27.5, 25.6, 20.4, 20.2, 16.9; HRMS(ESI) m/z calcd. for C26H43O4 [M+H]+: 419.3161, found 419.3155. Compound 30: 1H NMR (500 MHz, CDCl3) δ 4.15 (dt, J = 5.2, 3.6 Hz, 1H), 3.64 (s, 3H), 3.26 (s, 3H), 3.13–3.05 (m, 1H), 2.40 (ddd, J = 14.9, 9.4, 5.2 Hz, 1H), 2.29–2.18 (m, 3H), 1.98–1.93 (m, 1H), 1.87–1.79 (m, 2H), 1.77–1.72 (m, 4H), 1.70–1.63 (m, 3H), 1.56–1.48 (m, 2H), 1.43–1.35 (m, 2H), 1.32–1.23 (m, 3H), 1.21 (s, 3H), 1.19–1.07 (m, 1H), 0.97 (ap. t, J = 7.0, 6H), 0.93–0.76 (overlapping signals: m, 2H and 0.83 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 174.0, 94.3, 78.5, 77.2, 64.0, 55.4, 51.4, 51.3, 50.7, 41.9, 39.8, 39.4, 38.3, 35.8, 34.7, 34.0, 33.5, 33.4, 32.8, 32.7, 29.8, 27.4, 26.8, 25.5, 20.4, 20.2, 16.8; HRMS(ESI) m/z calcd. for C27H44O4Na [M + Na]+: 455.3137, found 455.3134.
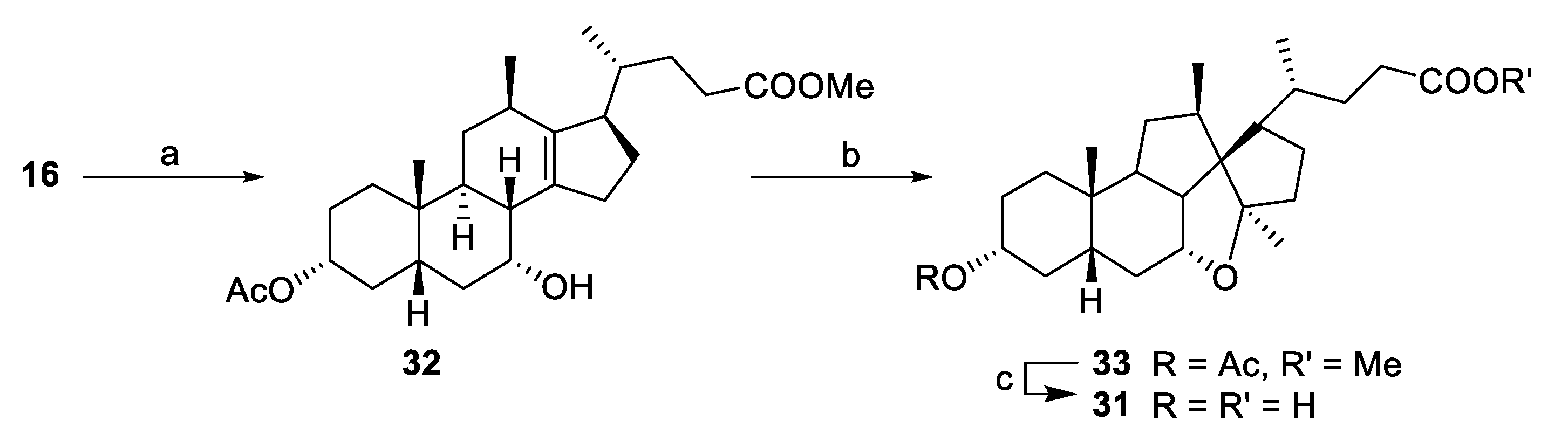
(R)-4-((1S,3aR,4aR,4a1R,5aS,7R,9aS,9bS,11R,11aR)-7-Hydroxy-3a,9a,11-trimethylhexadecahydrobenzo[4,5]indeno[7,1-bc]cyclopenta[d]furan-1-yl)pentanoic acid (31). From 29: To a solution of 29 (0.019 g, 0.045 mmol) in MeOH (2 mL) was added NaOH (aq) (2M, 0.5 mL, 1 mmol). The reaction was stirred at 70 °C (2 h) before being quenched with a solution of HCl (aq) (2M, 1 mL, 2 mmol). The product was extracted with ethyl acetate (10 mL), washed with water (10 mL) and brine (10 mL), dried (MgSO4) filtered and concentrated to afford compound 31 (0.017 g, 92%). From 33: To a solution of 33 (0.030 g, 0.059 mmol based on purity of 90%,) in MeOH (2 mL) was added NaOH (aq) (2M, 1.5 mL, 3 mmol). The reaction was stirred at 70 °C (2 h) before being quenched with a solution of HCl (aq) (2M, 1 mL, 2 mmol). The product was extracted with ethyl acetate (10 mL), washed with water (10 mL) and brine (10 mL), dried (MgSO4) and concentrated to dryness. The residue was recrystallized from ethyl acetate/petroleum ether to afford compound 31 (0.022 g, 91%). 1H NMR (500 MHz, CD3OD) δ 4.27–4.23 (m, 1H), 3.50 (tt, J = 10.0, 4.2 Hz, 1H), 2.49–2.36 (m, 2H), 2.36–2.22 (m, 2H), 1.99–1.86 (m, 7H), 1.75–1.60 (m, 6H), 1.55–1.47 (m, 2H), 1.45–1.33 (m, 3H), 1.32–1.17 (overlapping signals: m, 3H and 1.31, s, 3H), 1.08 (d, J = 7.4 Hz, 3H), 1.06 (d, J = 6.9 Hz, 3H), 1.01–0.95 (m, 1H), 0.91 (s, 3H); 13C NMR (126 MHz, MeOD) δ 177.5, 96.2, 79.0, 71.4, 65.8, 52.8, 52.3, 43.2, 41.3, 40.0, 39.5, 36.84, 36.79, 34.7, 34.5, 33.7, 33.2, 31.0, 27.8, 26.4, 21.2, 20.5, 17.4; HRMS(ESI) m/z calcd. for C25H40O4Na [M + Na]+: 427.2824, found 427.2824. Crystal data for C25H40O4 (M = 404.57 g/mol): monoclinic, space group P21, a = 10.0027(5) Å, b = 9.9958(5) Å, c = 11.4173(4) Å, V = 1112.61(9) Å3, Z = 2, T = 120(2) K, μ(CuKα) = 0.627 mm−1, Dcalc = 1.208 g/cm3, 11,986 reflections measured (5.951° ≤ Θ ≤ 71.714°), 3745 unique (Rint = 0.0358), which were used in all calculations. The final R1 was 0.0548 (I > 2σ(I)) and wR2 was 0.1472 (all data).
Methyl 3α-acetoxy-7α-hydroxyl-12β-methyl-18-nor-5β-chol-13(14)-en-24-oate (32). To a stirred solution of 16 (0.097 g, 0.24 mmol) in THF (15 mL) was added NaHCO3 (0.440 g, 5.2 mmol) and acetic anhydride (0.5 mL, 5.0 mmol) and the mixture was heated at 40 °C (18 h). The reaction was quenched with water (30 mL) and extracted with ethyl acetate (30 mL). The organic layer was washed with brine, dried (MgSO4), concentrated and the residue was purified by flash chromatography on silica gel (ethyl acetate/petroleum ether, 0:10 to 1:1) to afford the title compound 32 (0.035 g, 33%). 1H NMR (500 MHz, CDCl3) δ 4.58 (tt, J = 11.4, 4.6 Hz, 1H), 4.11 (q, J = 2.9 Hz, 1H), 3.64 (s, 3H), 2.65 (d, J = 8.6 Hz, 1H), 2.42–2.30 (m, 4H), 2.29–2.10 (m, 4H), 2.01–1.88 (overlapping signals: m, 3H; 1.98, s, 3H), 1.87–1.63 (m, 6H), 1.57–1.40 (m, 3H), 1.29–1.18 (m, 2H), 1.14–0.96 (overlapping signals: m, 2H; 1.04, d, J = 7.08 Hz, 3H), 0.93 (d, J = 6.95 Hz, 3H), 0.90 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 174.5, 170.7, 143.1, 137.7, 74.2, 68.2, 53.5, 51.4, 41.6, 40.6, 35.7, 35.3, 35.0, 34.8, 33.9, 33.6, 33.0, 32.7, 31.9, 31.4, 26.7, 25.3, 24.9, 22.3, 21.4, 21.1, 18.9; HRMS(ESI) m/z calcd. for C27H42O5Na [M + Na]+: 469.2930, found 469.2931.
Methyl (4R)-4-((1S,3aR,4aR,5aS,7R,9aS,11R,11aR)-7-acetoxy-3a,9a,11-trimethylhexadecahydrobenzo[4,5]indeno[7,1-bc]cyclopenta[d]furan-1-yl)pentanoate (33). Diethylzinc (1 M in hexanes, 1.3 mL, 1.3 mmol) was added to dichloromethane (2.5 mL) and cooled to 0 °C before trifluoroacetic acid (0.1 mL, 1.4 mmol) and diiodomethane (0.4 mL, 5 mmol) were added to the mixture. After 10 min, a solution of 32 (0.057 g, 0.13 mmol) in dichloromethane (3 mL) was added and the reaction mixture was stirred at r.t. (2 h) before being quenched with NH4Cl sat. (25 mL). The aqueous layer was washed with ethyl acetate (3 × 20 mL) and the combined organic layers were washed with NaHCO3 (20 mL), water (20 mL), brine (25 mL), dried (MgSO4) and concentrated. The crude product was purified by flash chromatography on silica gel (ethyl acetate/petroleum ether, 0:10 to 3:7) to afford the title compound 33 (0.030 g, 51% based on a purity of 90%). 1H NMR (500 MHz, CDCl3) δ 4.71 (tt, J = 8.3, 3.9 Hz, 1H), 4.16 (dt, J = 5.6, 4.0 Hz, 1H), 3.65 (s, 3H), 2.41 (ddd, J = 15.0, 9.4, 5.2 Hz, 1H), 2.31–2.20 (m, 3H), 2.02–1.94 (overlapping signals, m, 1H and 1.98, s, 3H), 1.87–1.74 (m, 6H), 1.70–1.63 (m, 4H), 1.58–1.46 (m, 3H), 1.46–1.34 (m, 3H), 1.29–1.22 (overlapping signals, m, 2H and 1.25, s, 3H), 0.99 (d, J = 7.4 Hz, 3H), 0.97 (d, J = 6.9 Hz, 3H), 0.94–0.88 (m, 1H), 0.86 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 174.1, 170.7, 94.4, 72.0, 63.9, 51.5, 51.2, 50.7, 41.9, 40.9, 38.9, 38.5, 35.9, 33.9, 33.8, 33.4, 33.1, 33.0, 32.7, 29.8, 27.3, 26.3, 25.4, 21.5, 20.2, 20.1, 16.8; HRMS(ESI) m/z calcd. for C28H44O5Na [M + Na]+: 483.3086, found 483.3071.


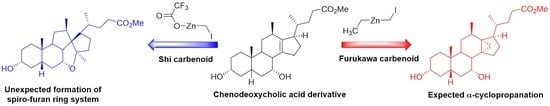
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








