
Synthesis of Functionalized Six-Membered-Ring Azahelicenes


Abstract
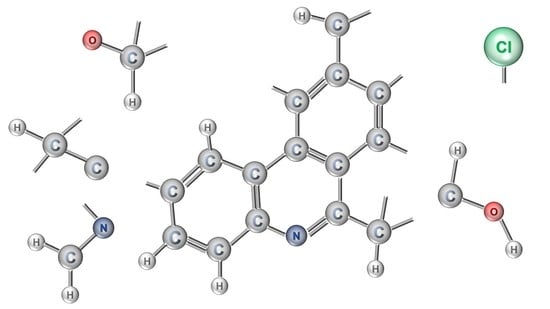
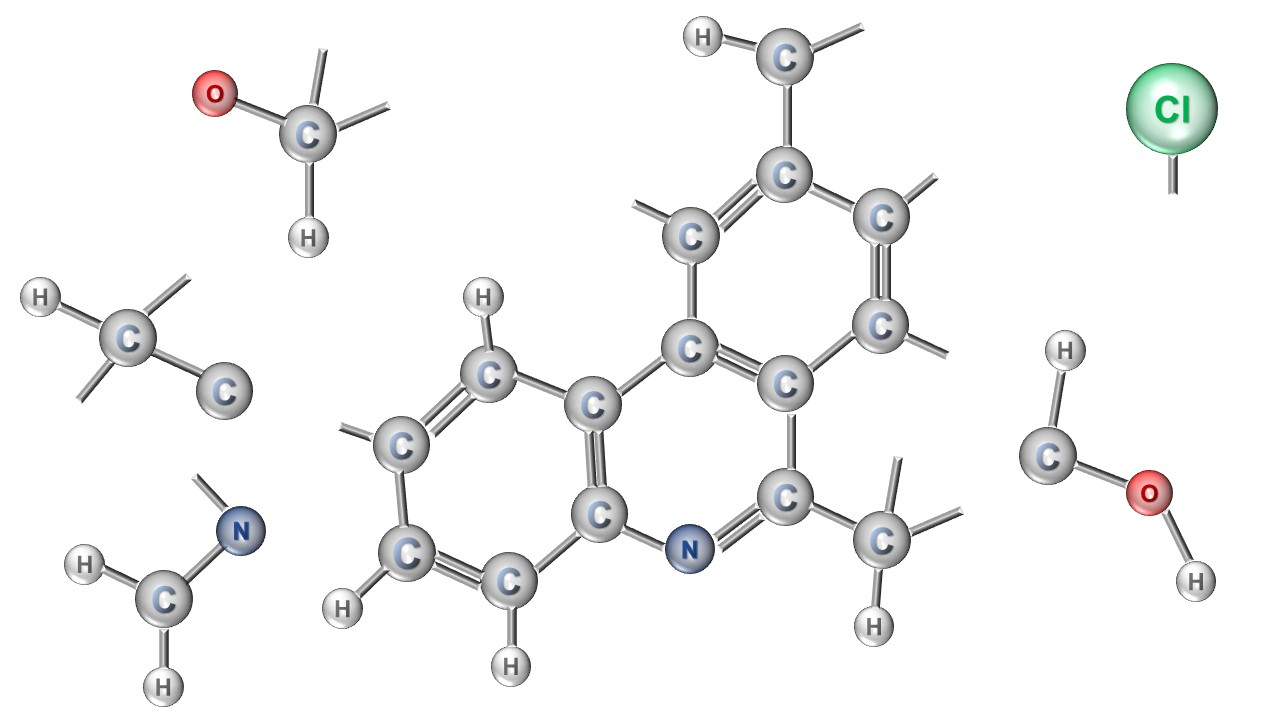
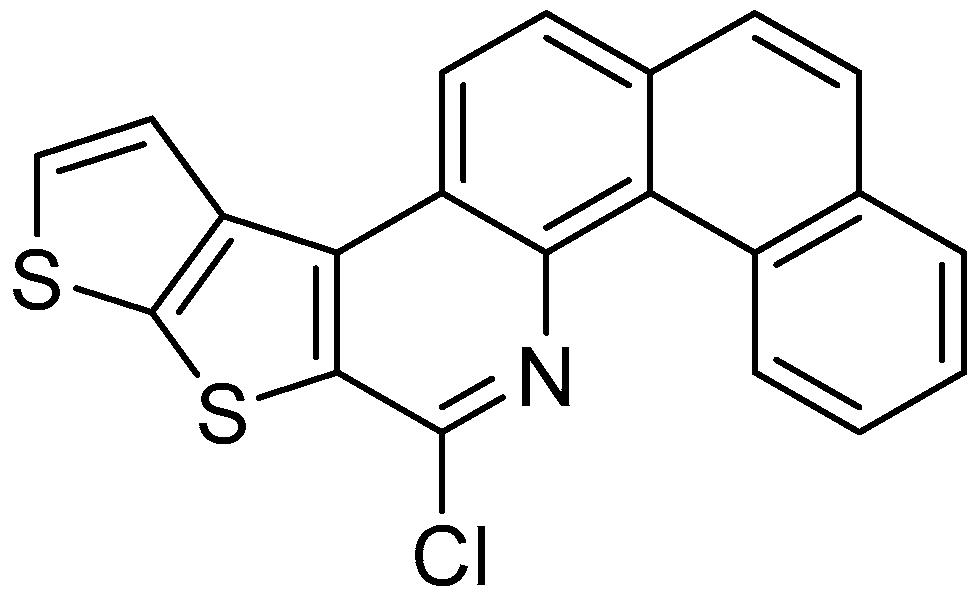
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
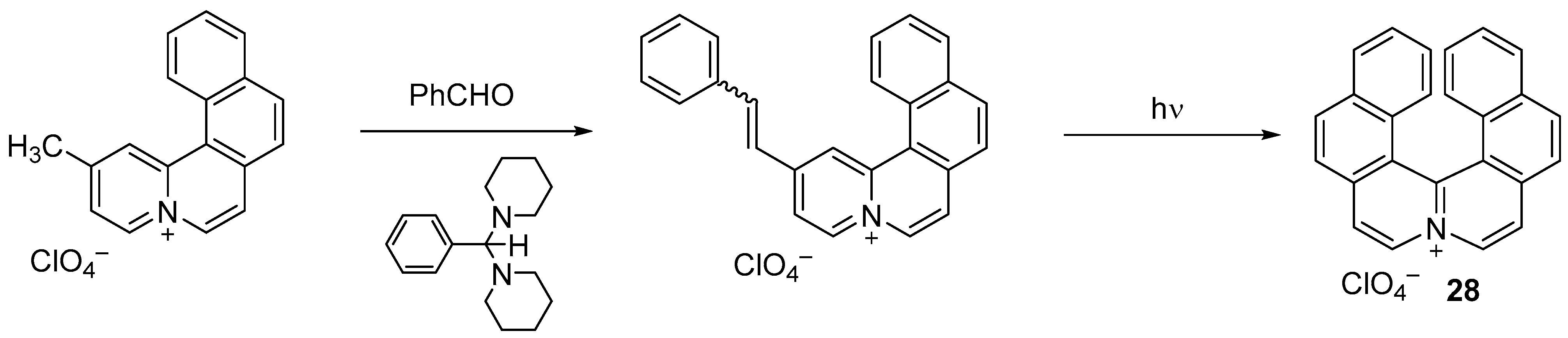{kind=link}
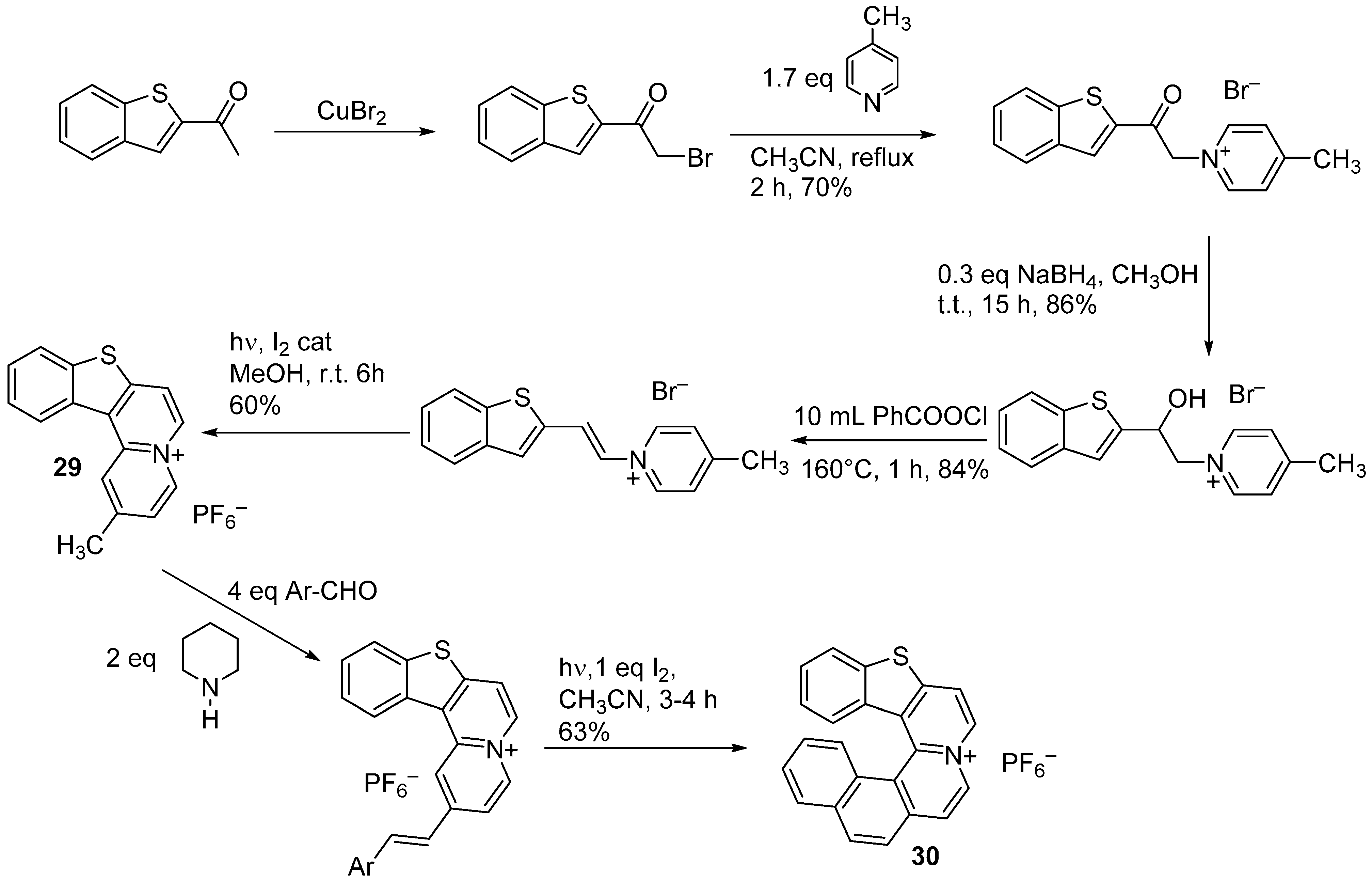{kind=link}
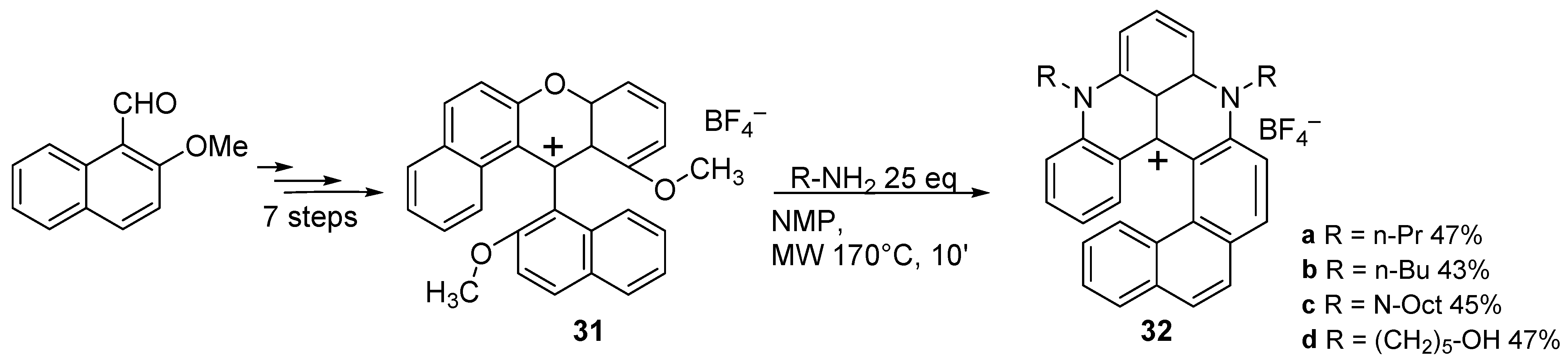{kind=link}
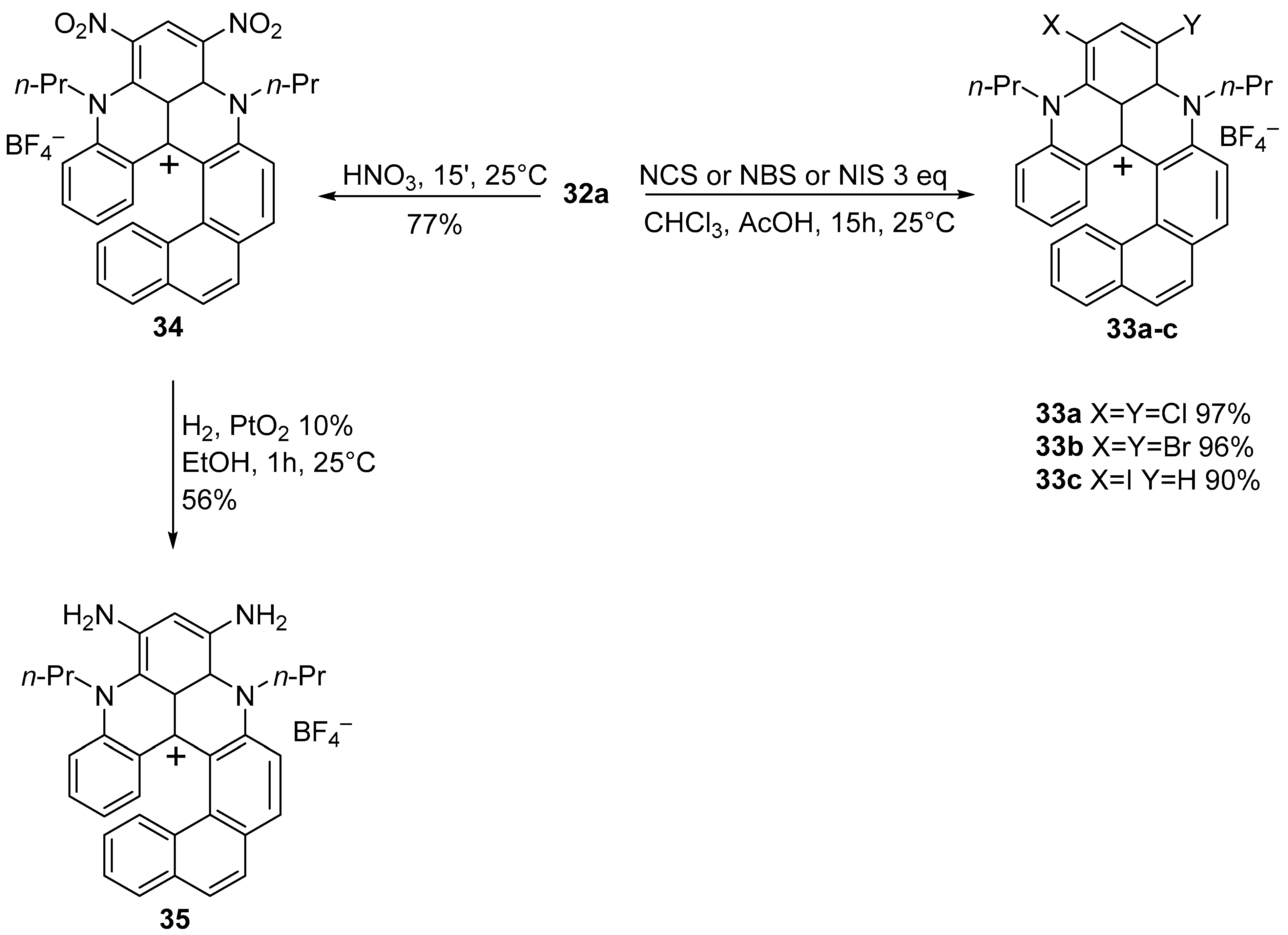{kind=link}
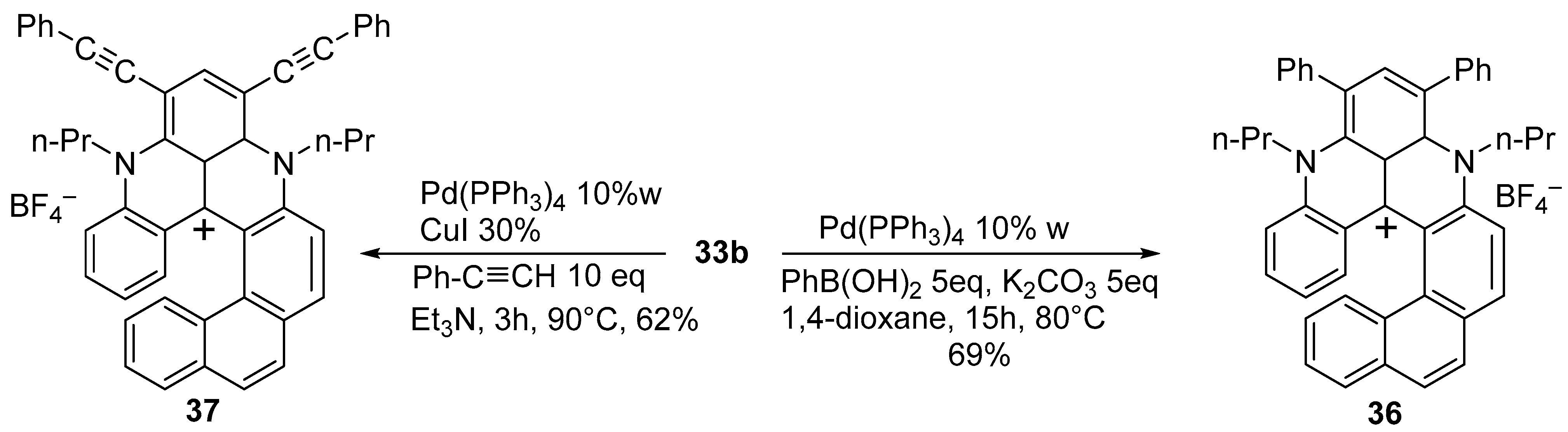{kind=link}
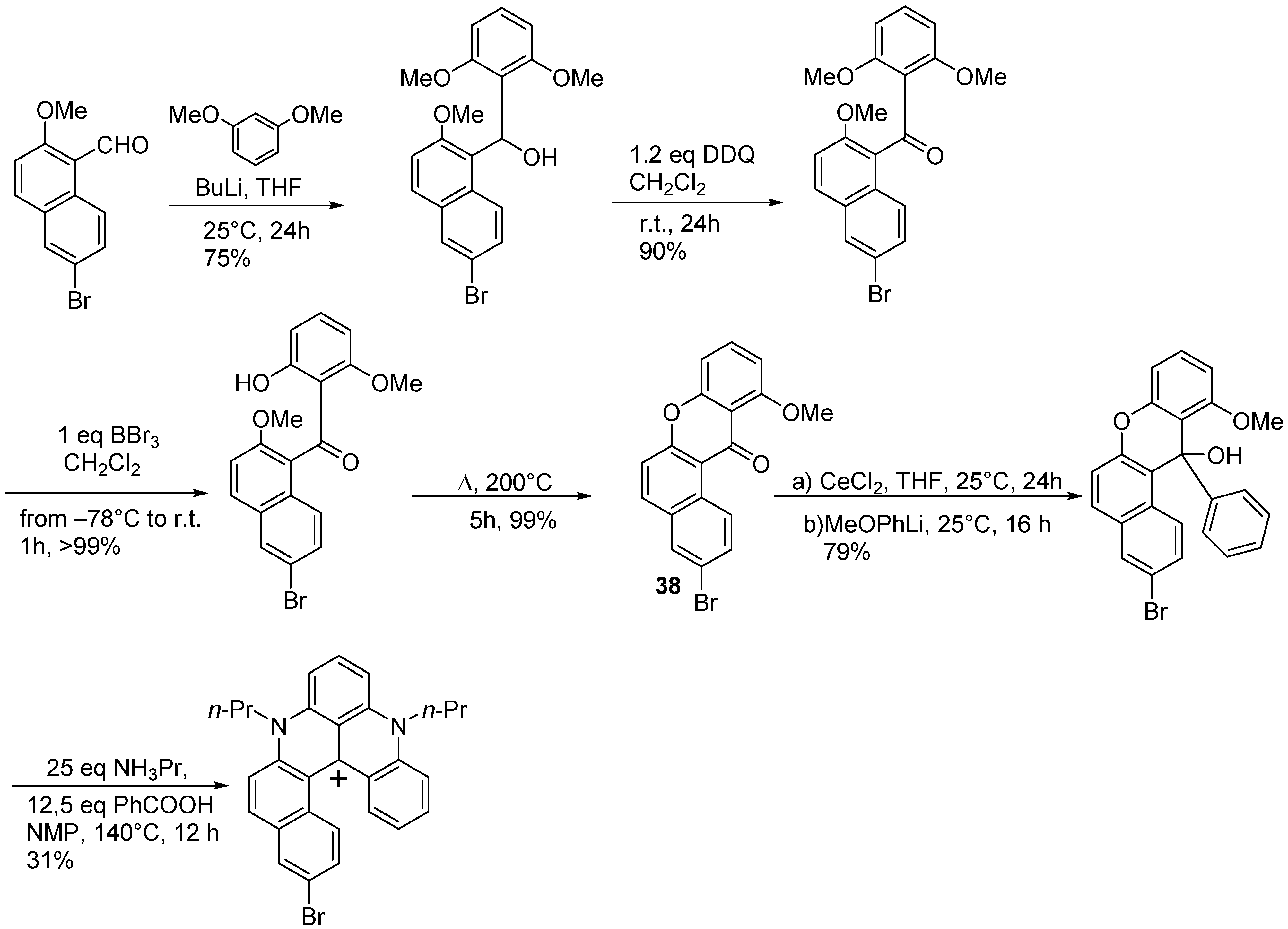{kind=link}
1. Introduction
2. Direct Synthesis of Functionalized Azahelicenes
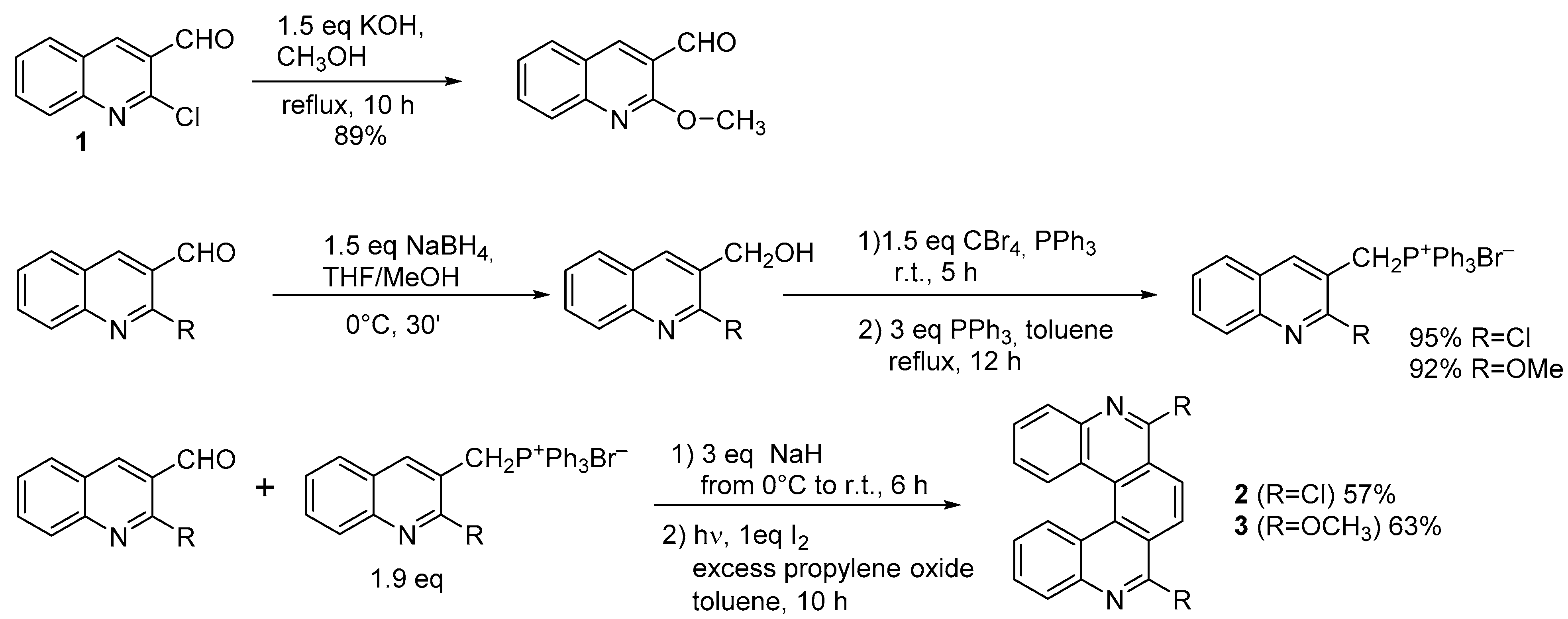
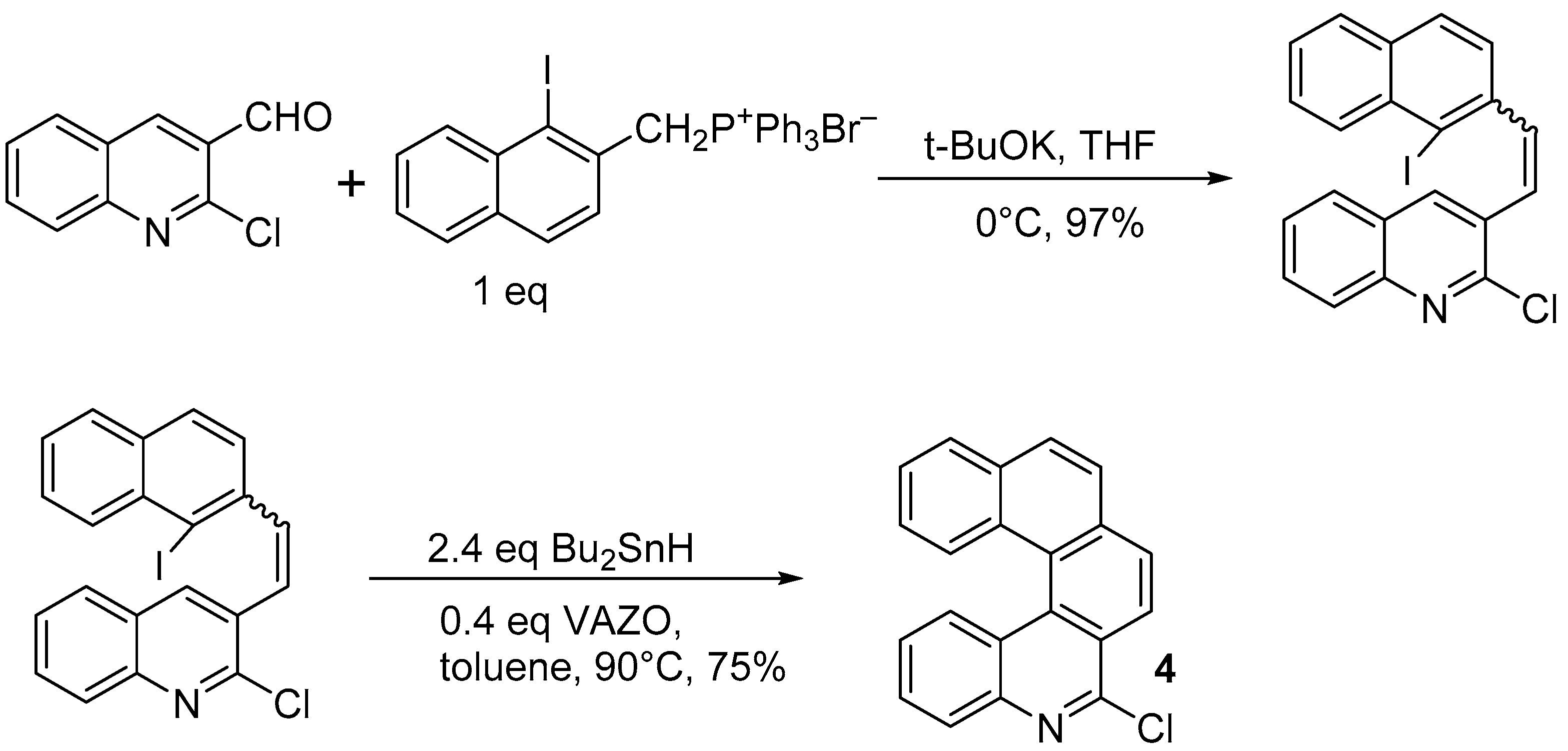
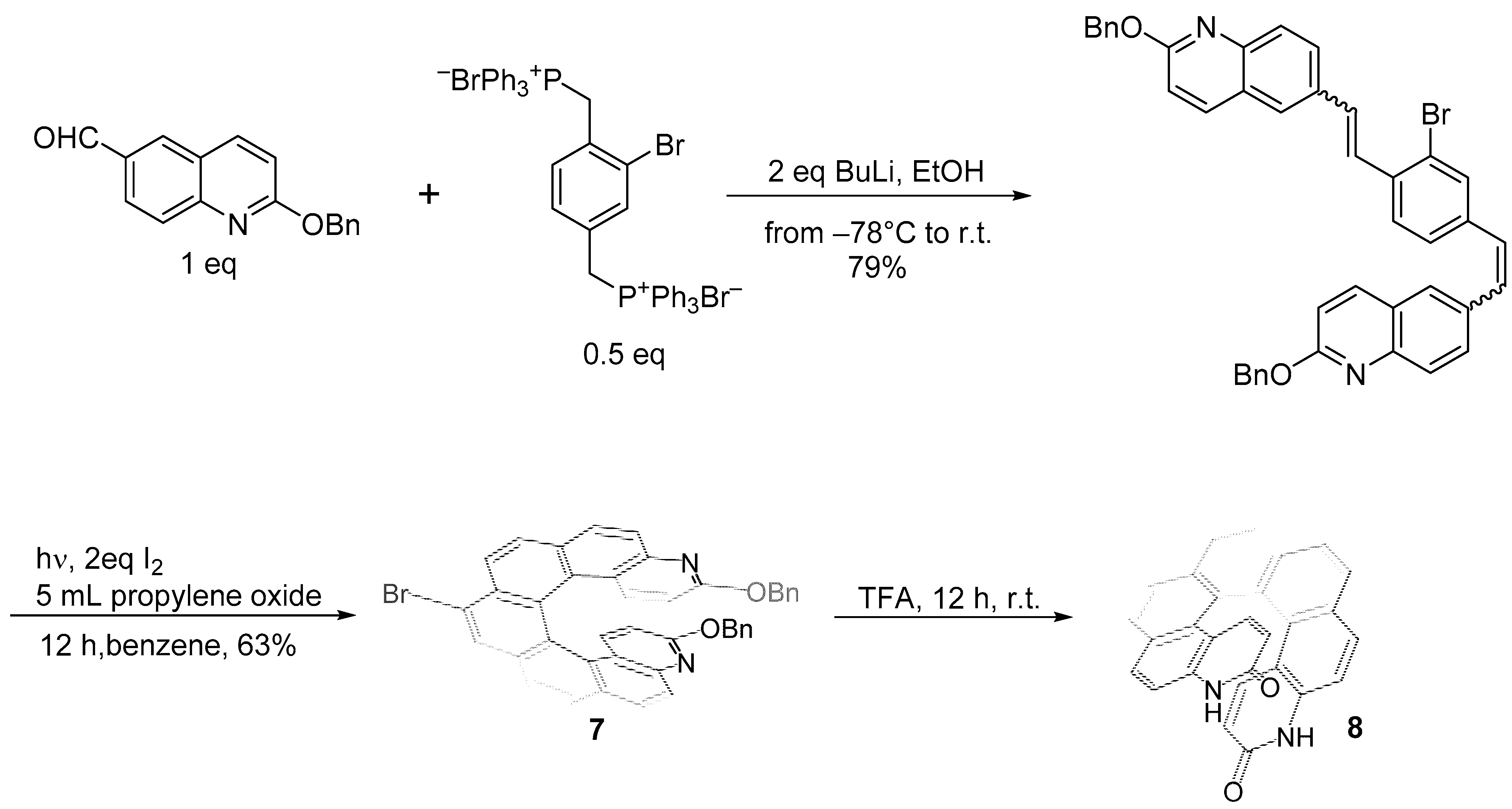
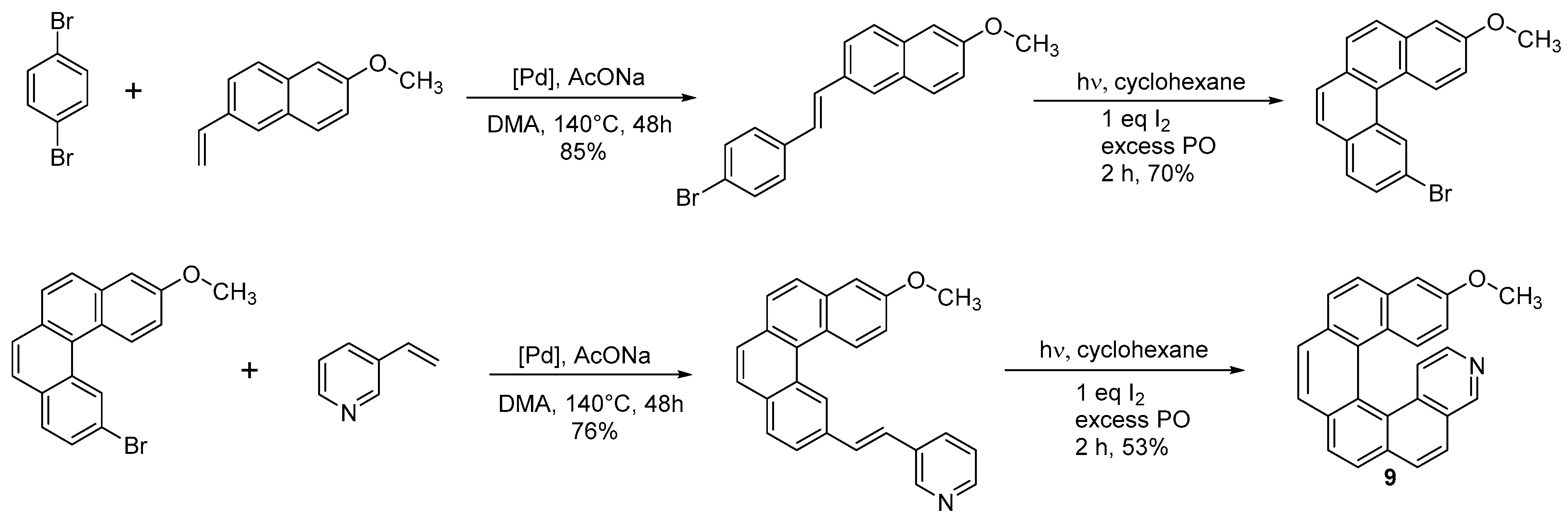
2.1. Diarylethene Cyclization
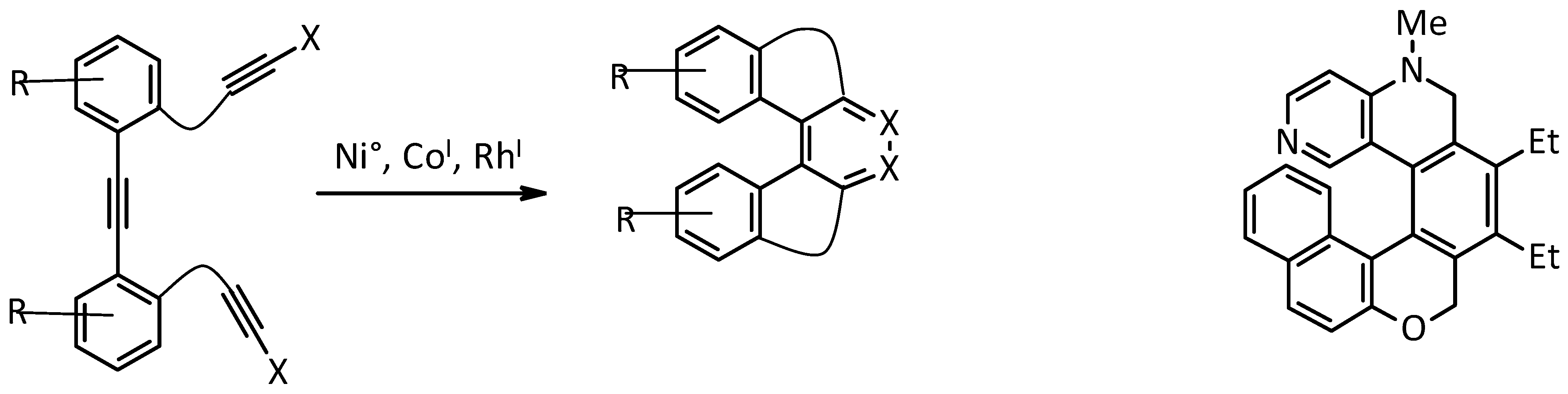
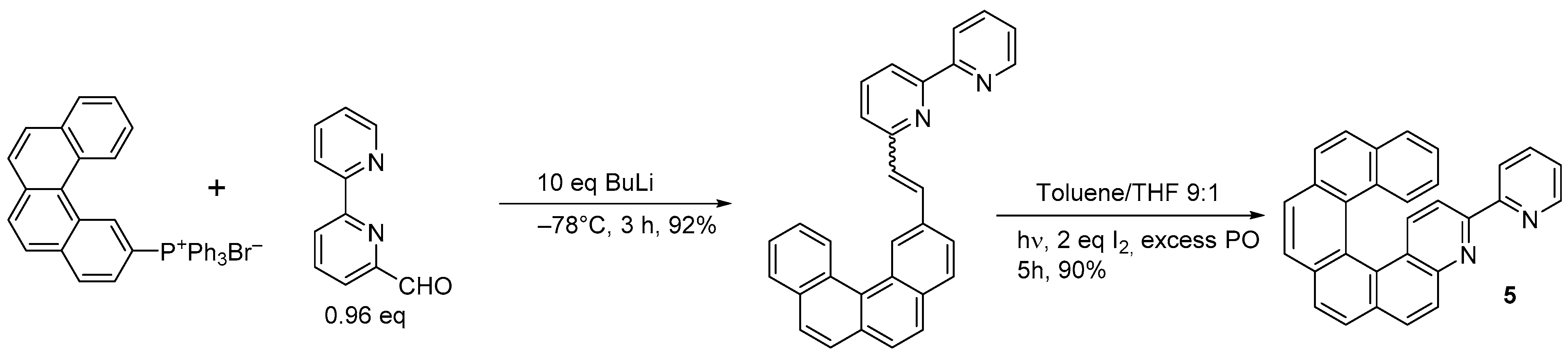
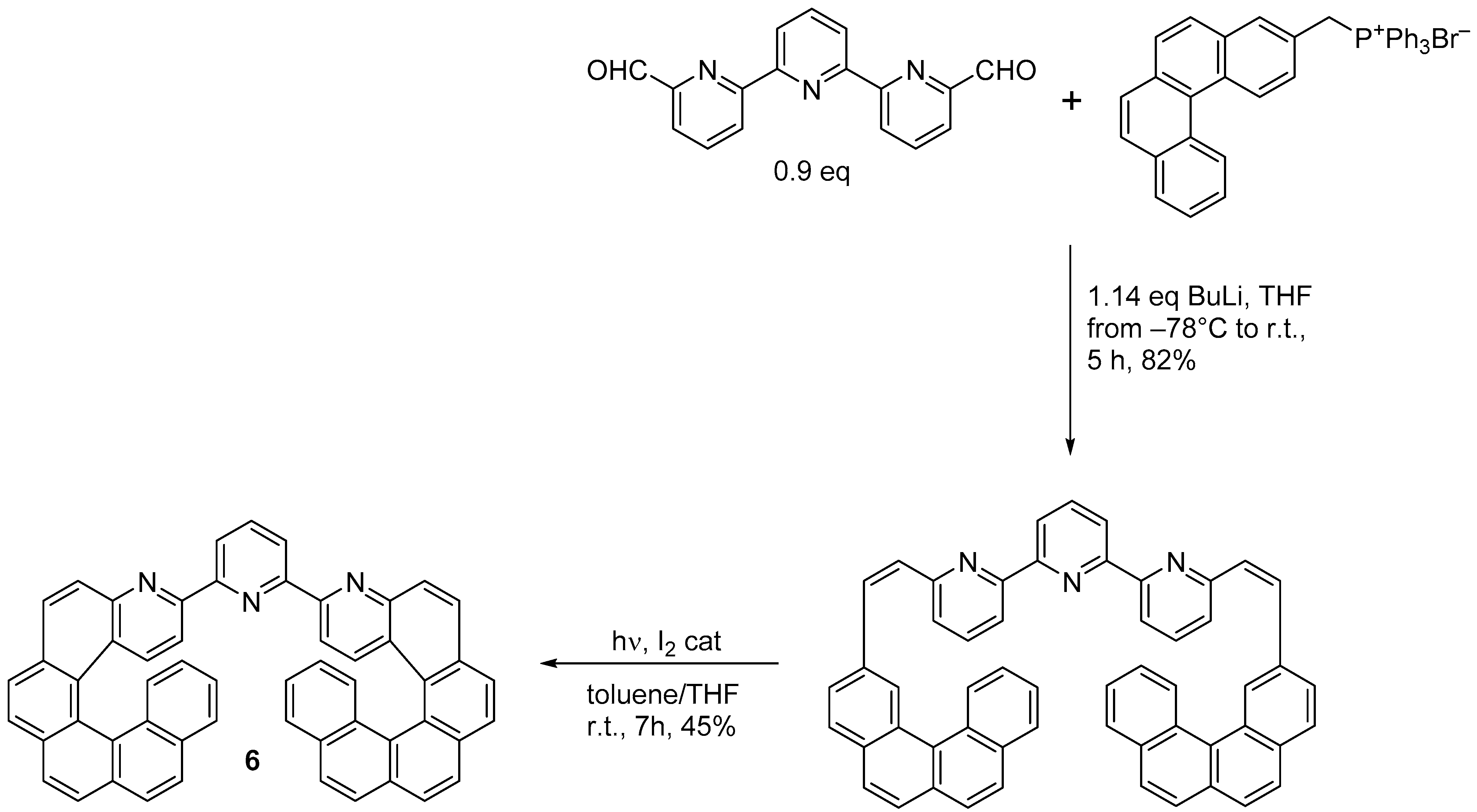
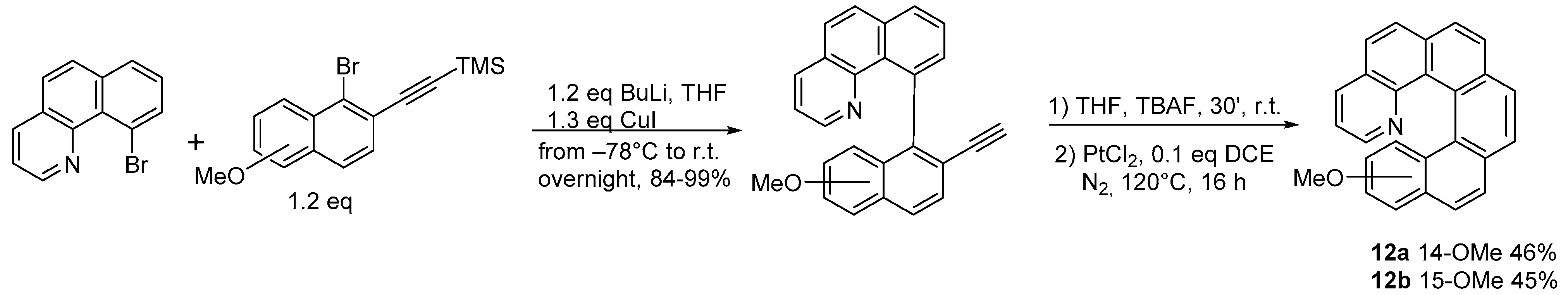
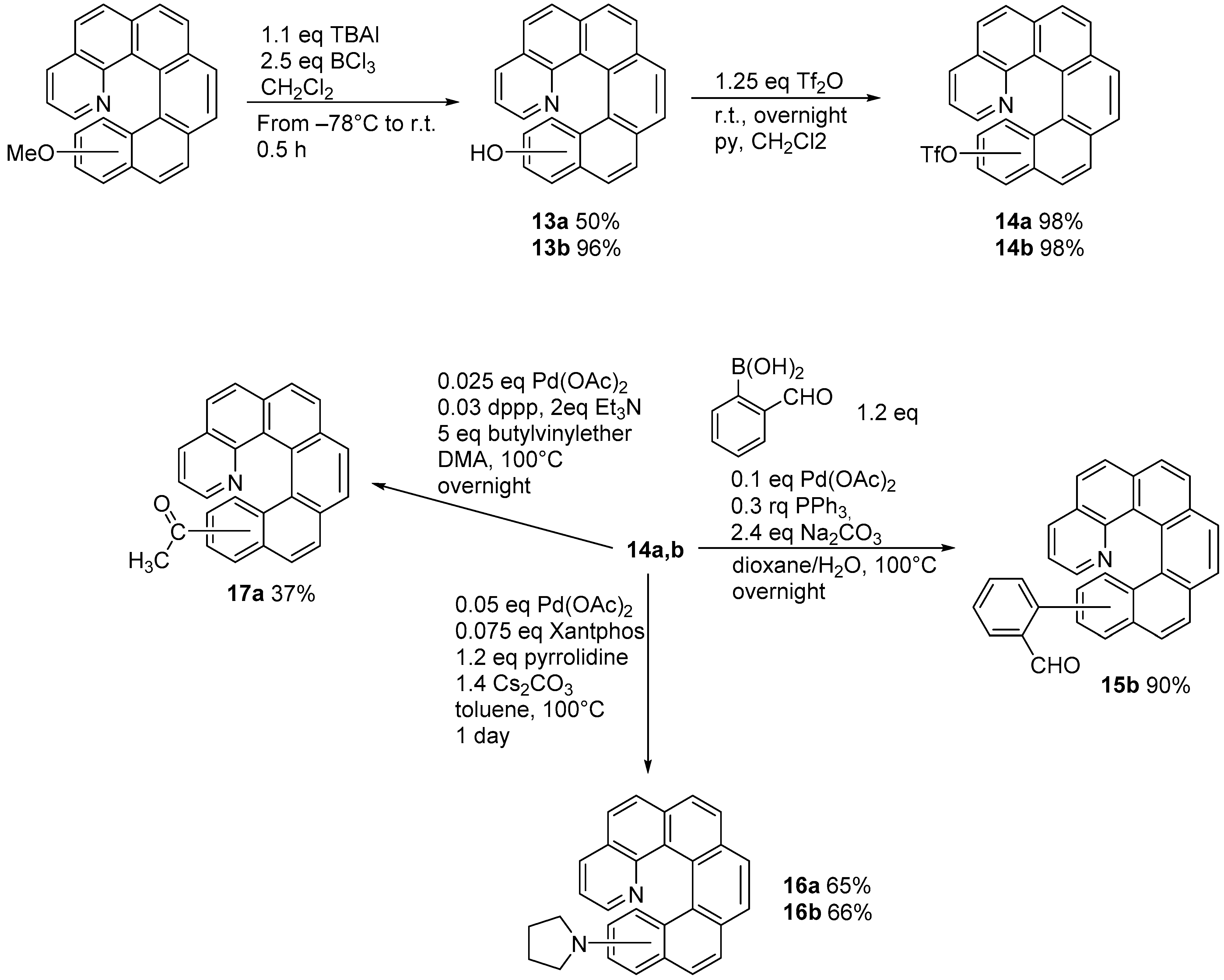
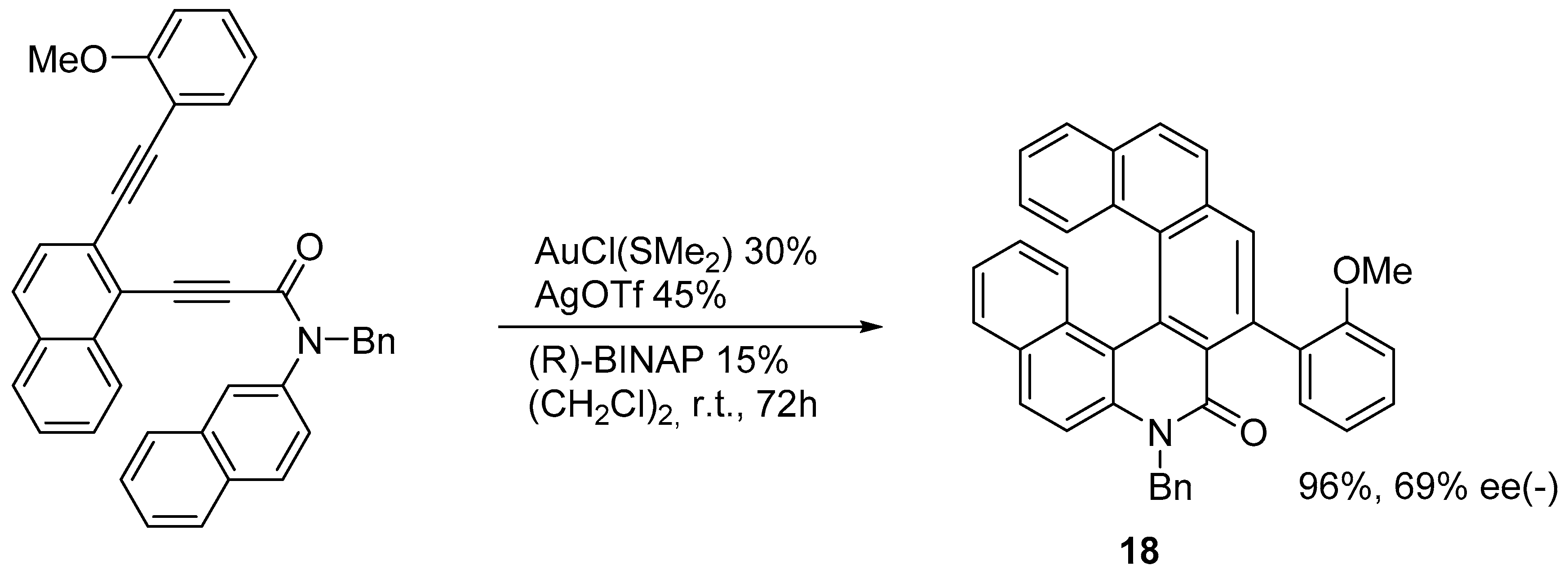
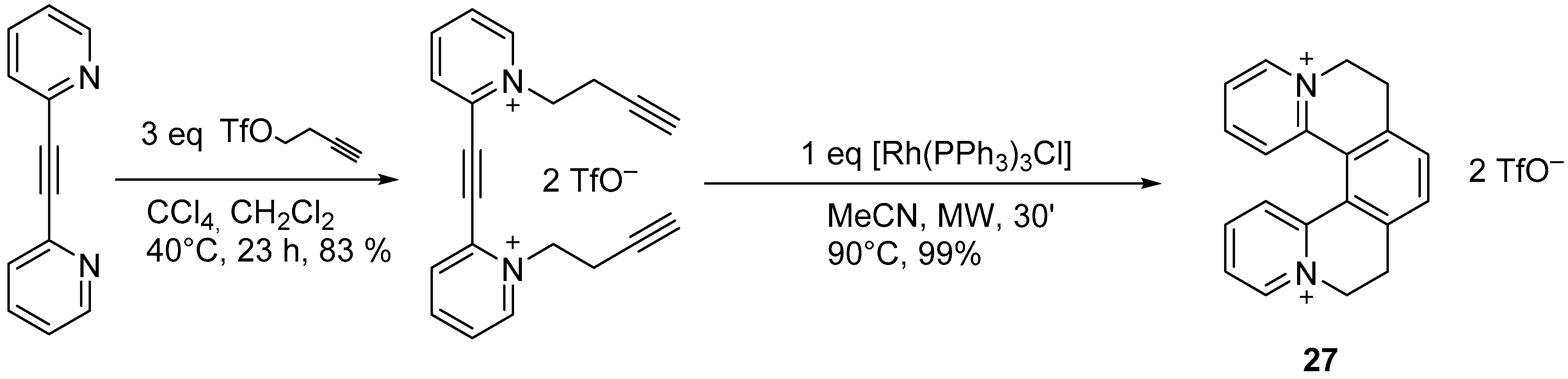
2.2. [2 + 2 + 2] Cycloisomerization
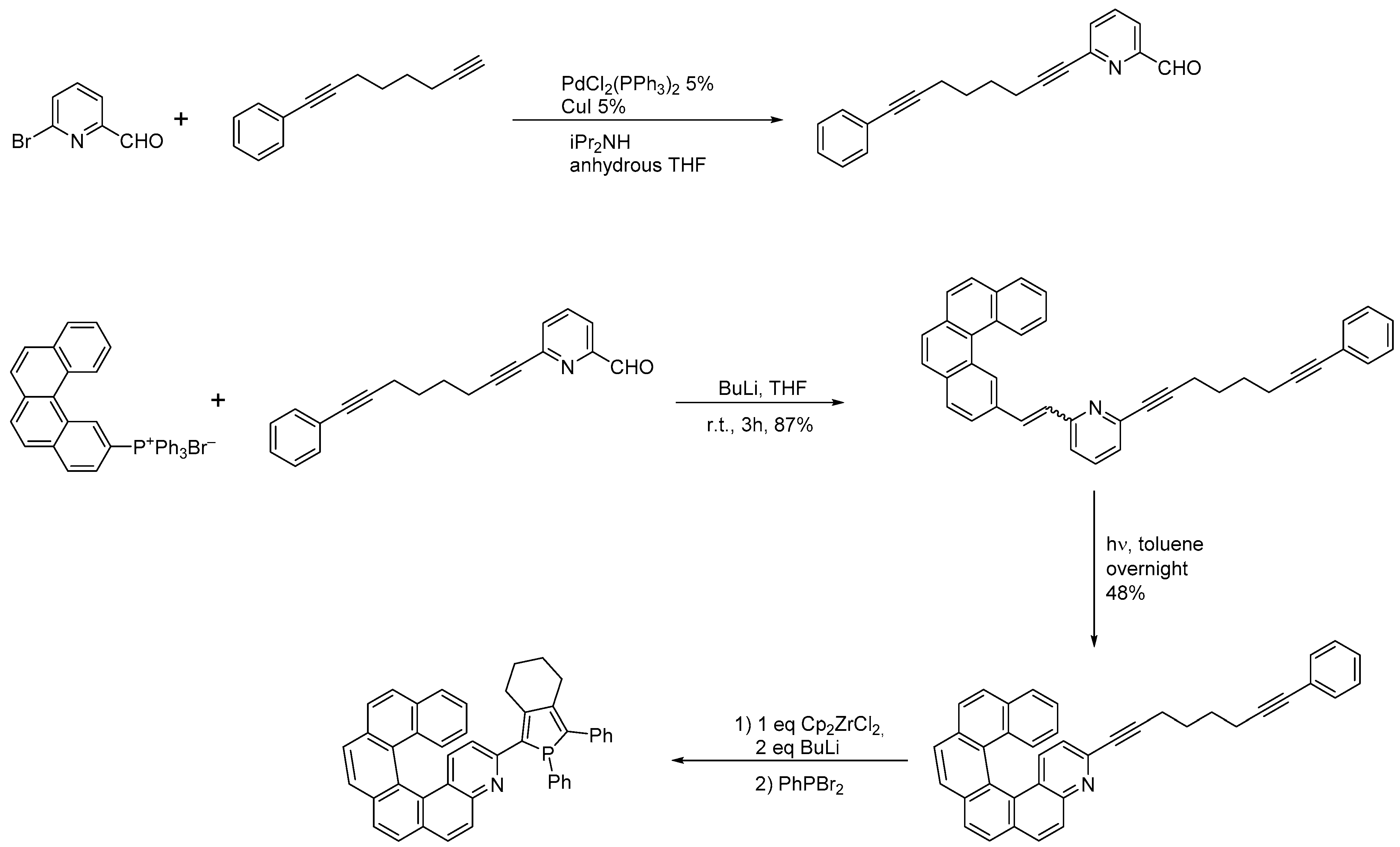
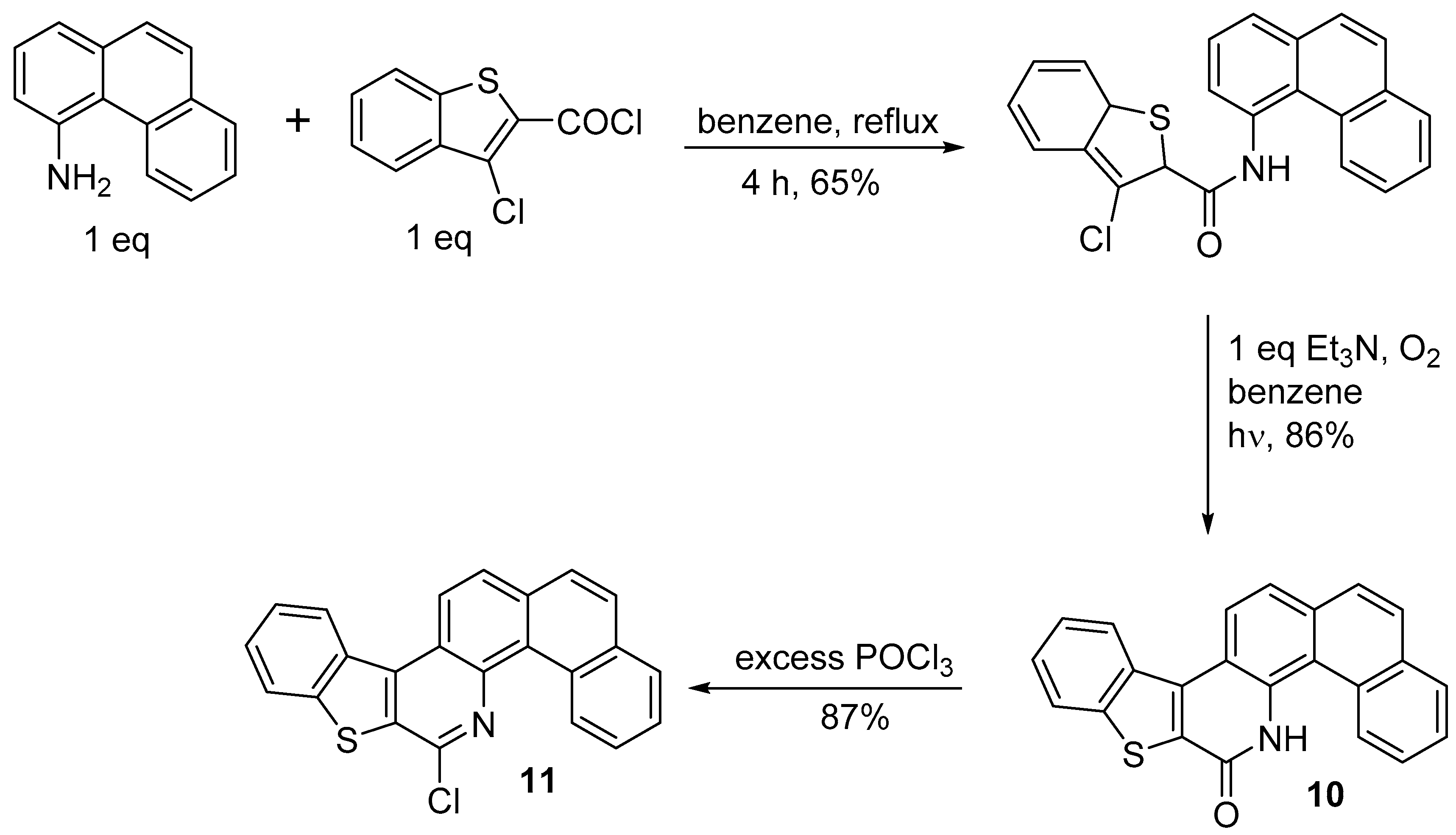
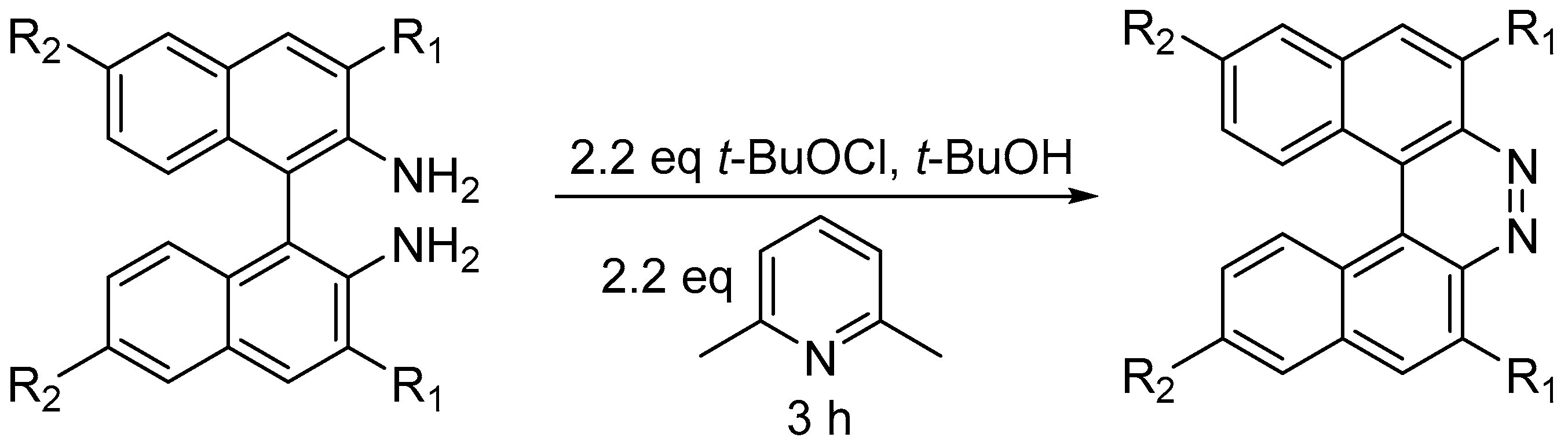
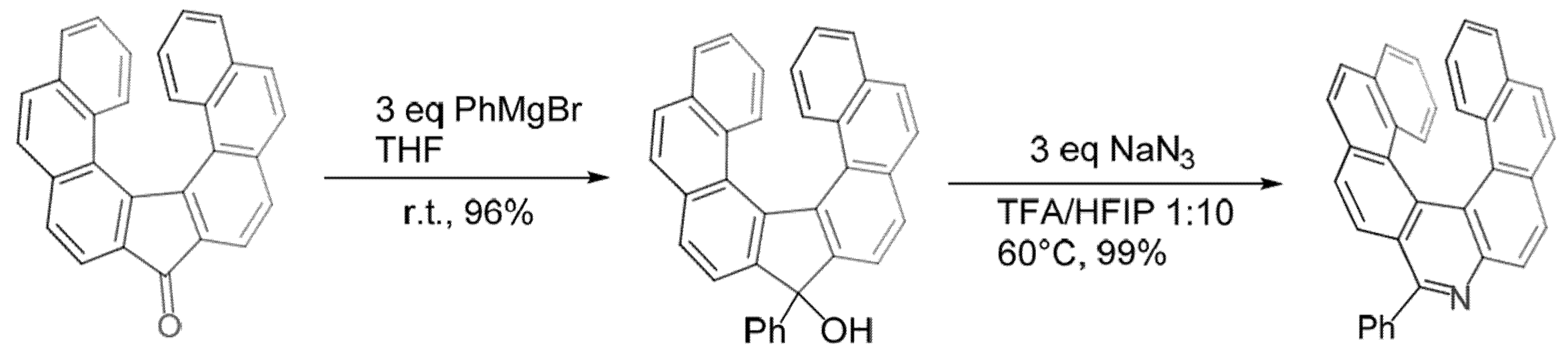
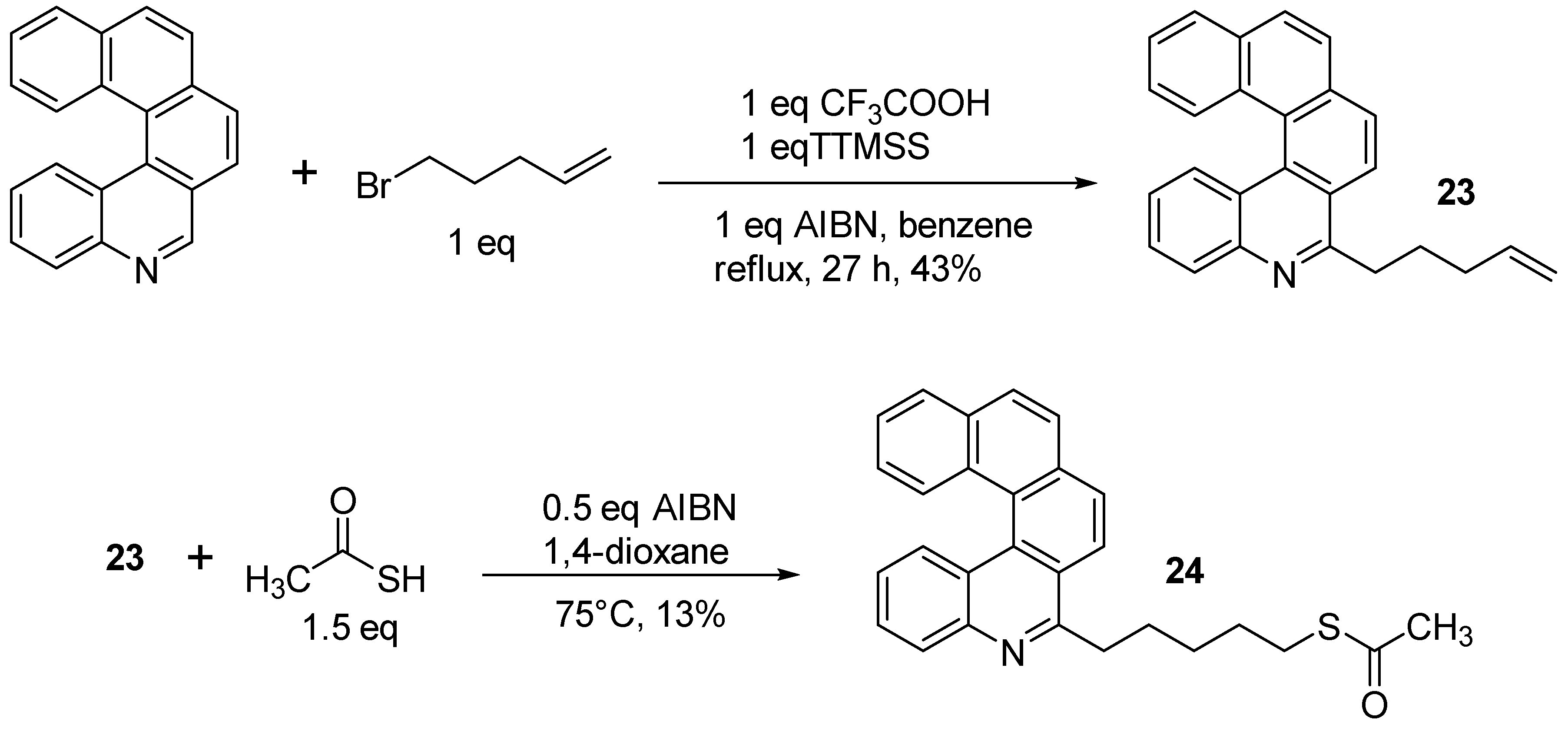
2.3. Other Synthetic Methods
3. Attaching Substituents to Azahelicenes
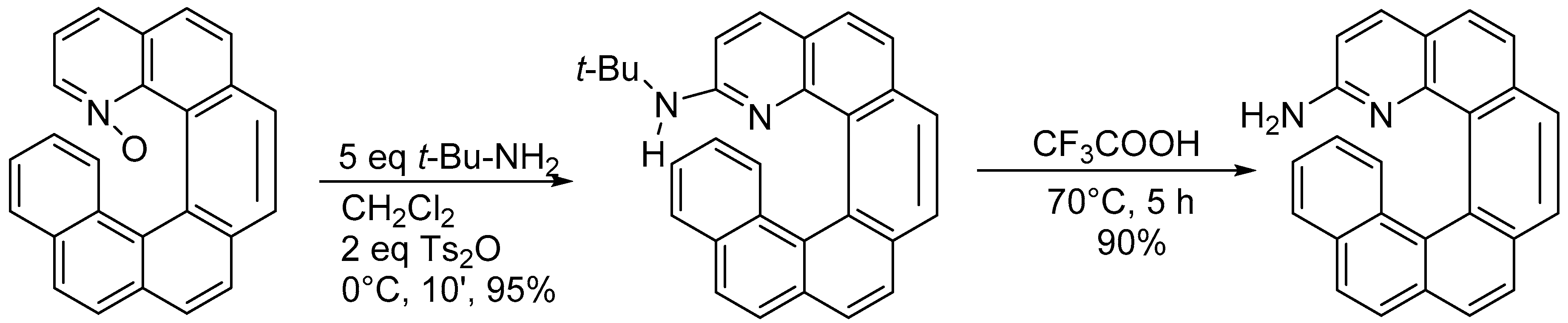
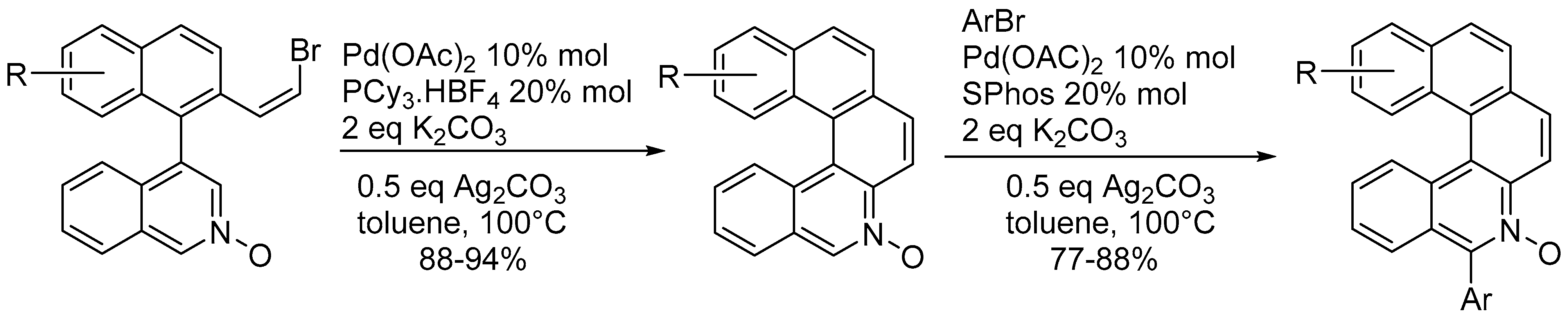
3.1. Reactions of N-Oxides
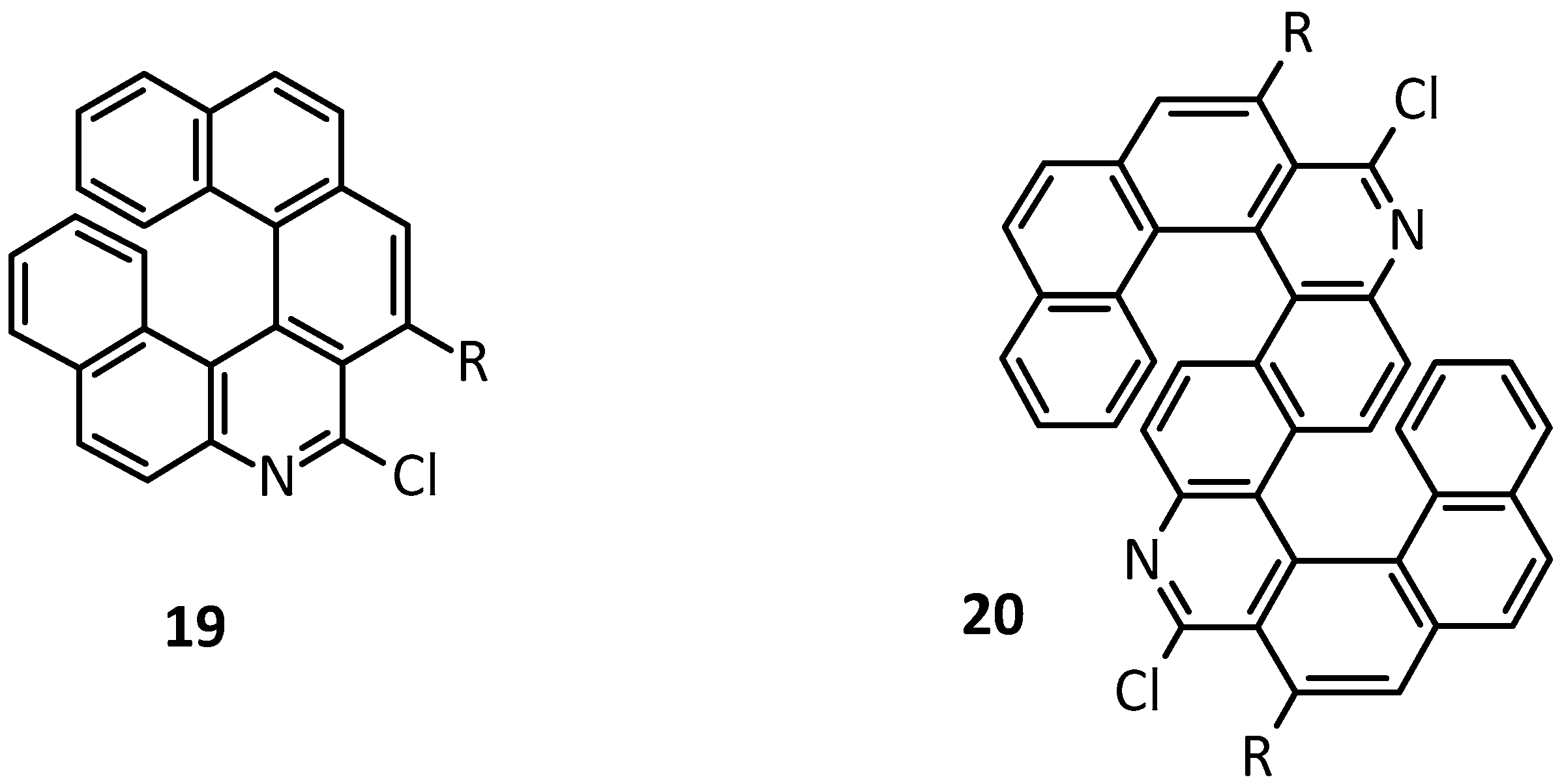
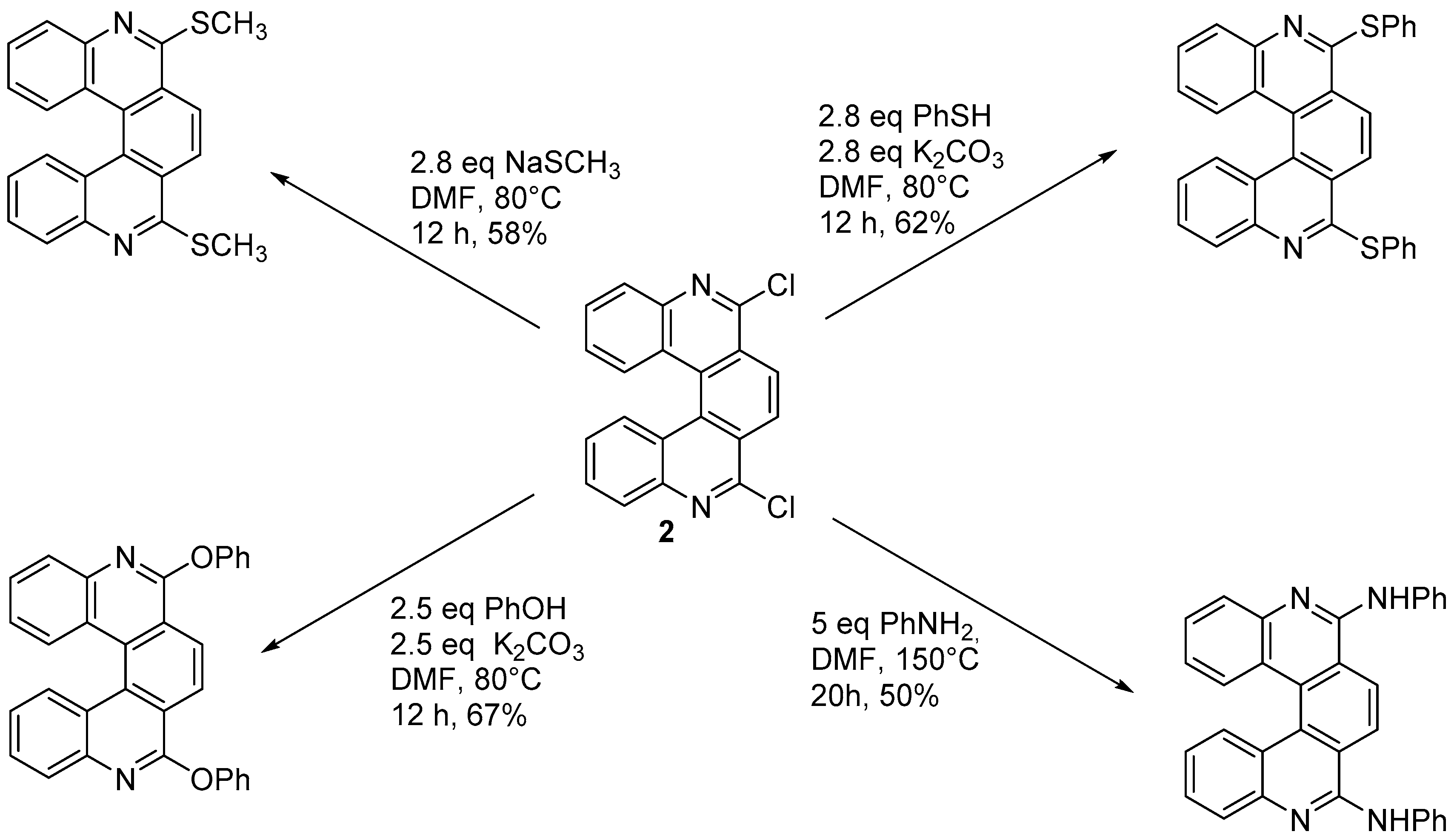
3.2. Substitution Reactions
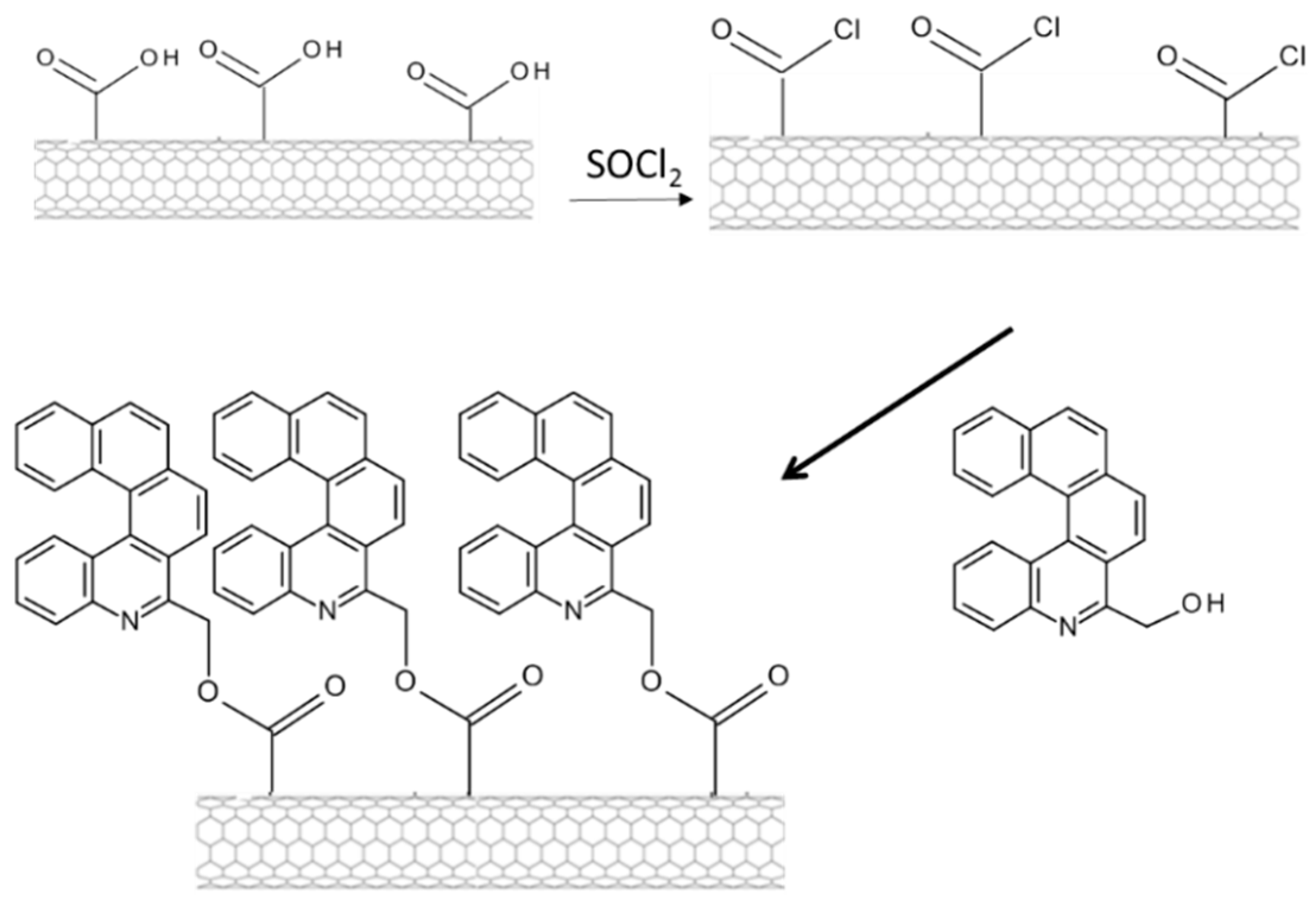
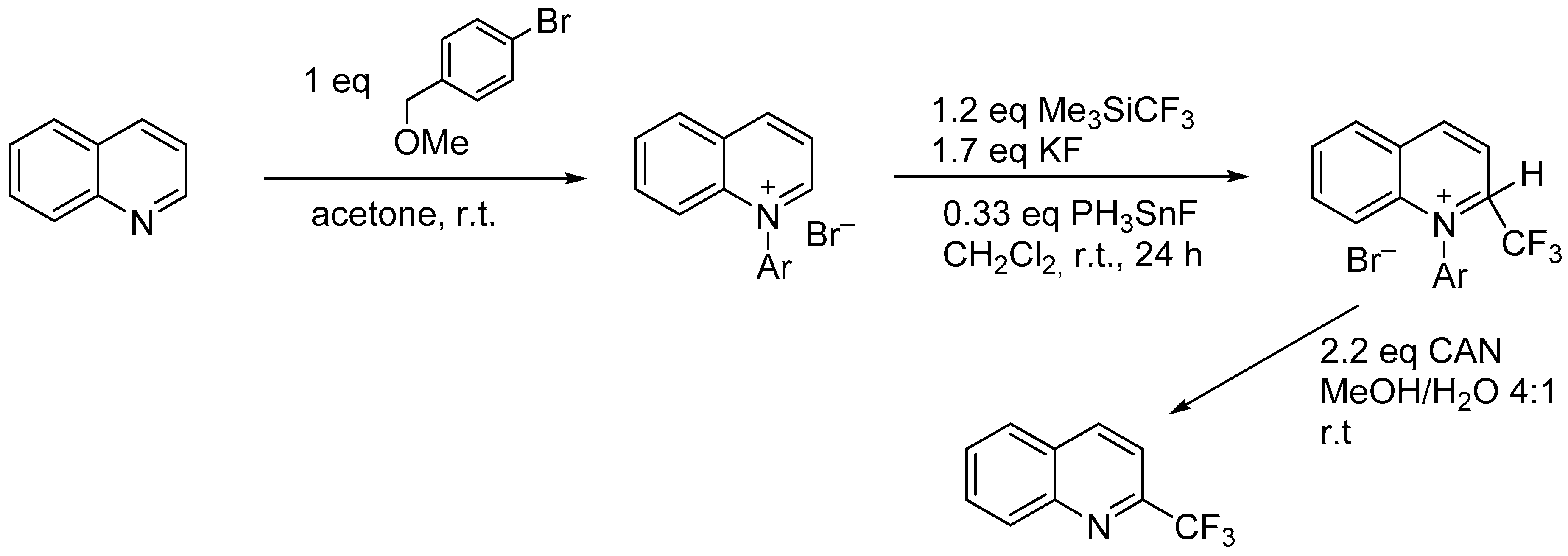
3.3. The Minisci Reaction
4. Positively Charged Azahelicenes
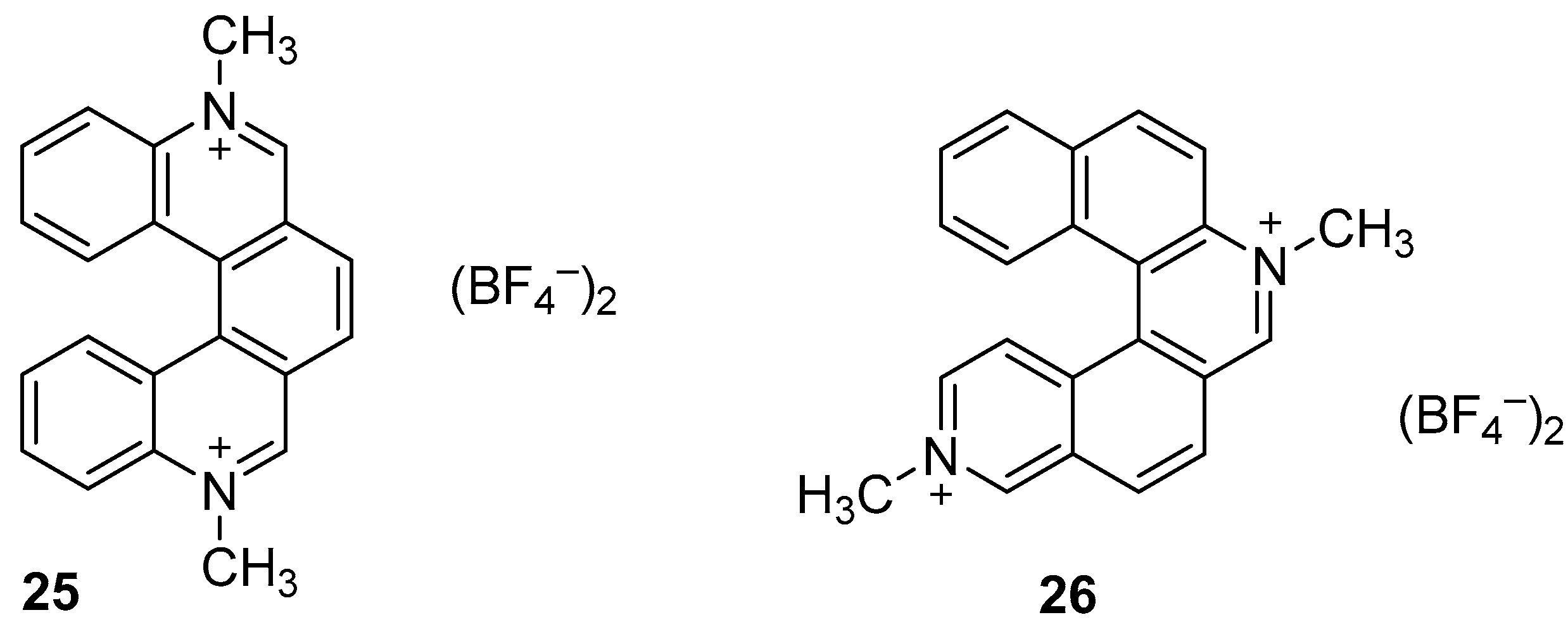
4.1. Quaternary Azahelicenium Salts
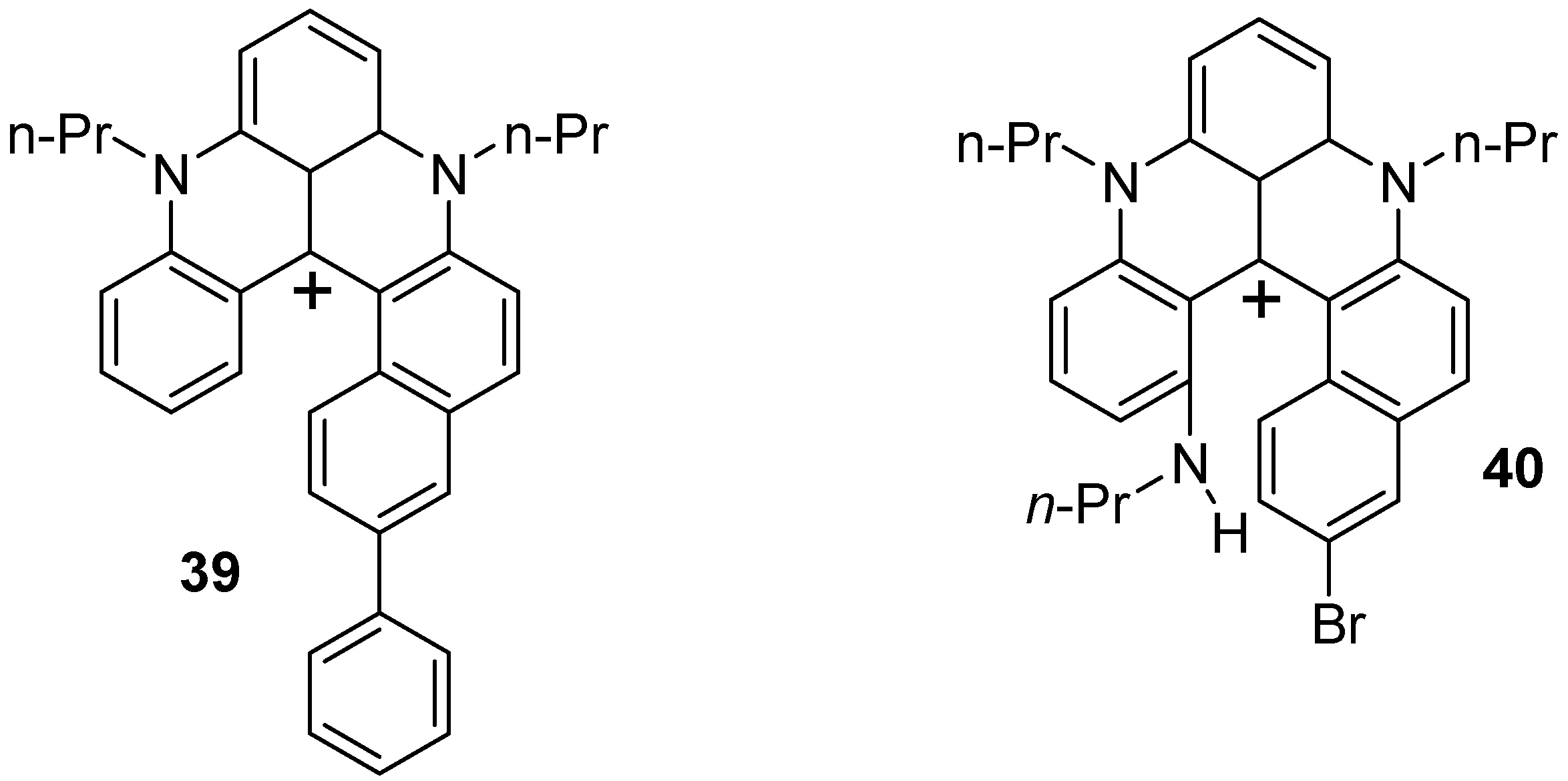
4.2. Helquats and Azoniahelicenes
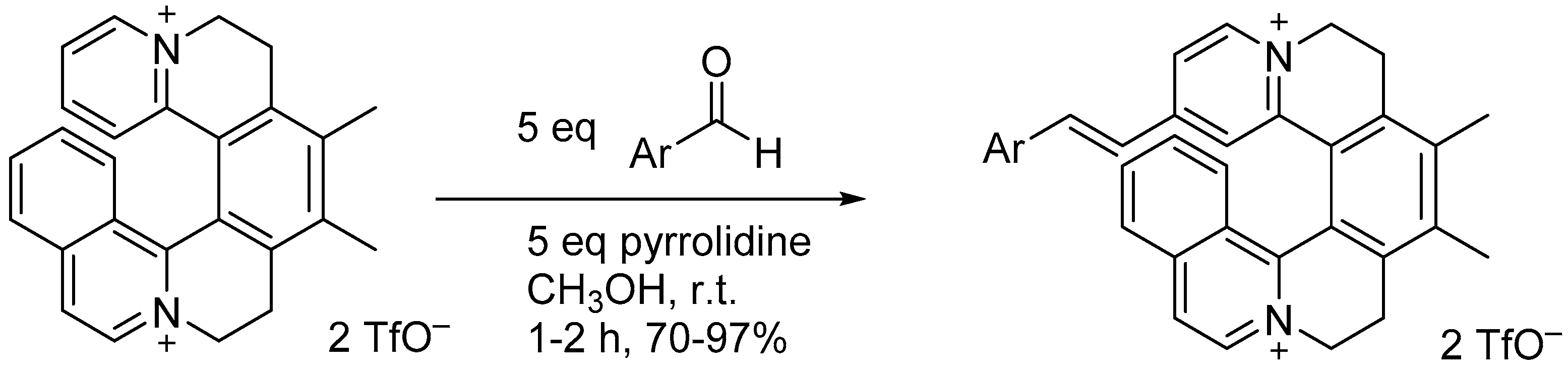
4.3. Azahelicenes with Cationic Framework
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dumitrascu, F.; Dumitrescu, D.G.; Aron, I. Azahelicenes and other similar tri and tetracyclic helical molecules. Arkivoc 2010, 1, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Starý, I.; Stará, I.G. The synthesis of azahelicenes. Targets Heterocycl. Syst. 2017, 21, 23–53. [Google Scholar]
- Verbiest, T.; Van Elshocht, S.; Kauranen, M.; Hellemans, L.; Snauwaert, J.; Nuckolls, C.; Katz, T.J.; Persoons, A. Strong enhancement of nonlinear optical properties through supramolecular chirality. Science 1998, 282, 913–915. [Google Scholar] [CrossRef] [PubMed]
- Šámal, M.; Míšek, J.; Stará, I.G.; Starý, I. Organocatalysis with azahelicenes: The first use of helically chiral pyridine-based catalysts in the asymmetric acyl transfer reaction. Collect. Czechoslov. Chem. Commun. 2009, 74, 1151–1159. [Google Scholar] [CrossRef]
- Fontana, F.; Melone, F.; Iannazzo, D.; Leonardi, S.G.; Neri, G. Synthesis, characterization and electrochemical properties of 5-aza[5]helicene-CH2O-CO-MWCNTs nanocomposite. Nanotechnology 2017, 28, 135501. [Google Scholar] [CrossRef] [PubMed]
- Caronna, T.; Mele, A.; Famulari, A.; Mendola, D.; Fontana, F.; Juza, M.; Kamuf, M.; Zawatzky, K.; Trapp, O. A Combined Experimental and Theoretical Study on the Stereodynamics of Monoaza[5]Helicenes: Solvent-Induced Increase of the Enantiomerization Barrier in 1-Aza-[5] Helicene. Chem.-Eur. J. 2015, 21, 13919–13924. [Google Scholar] [CrossRef]
- Bazzini, C.; Brovelli, S.; Caronna, T.; Gambarotti, C.; Giannone, M.; Macchi, P.; Meinardi, F.; Mele, A.; Panzeri, W.; Recupero, F.; et al. Tubino Synthesis and Characterization of Some Aza[5]Helicenes. Eur. J. Org. Chem. 2005, 1247–1257. [Google Scholar] [CrossRef]
- Caronna, T.; Castiglione, F.; Fontana, F.; Famulari, A.; Malpezzi, L.; Mele, A.; Mendola, D.; Sora, I.N. Quantum Mechanics Calculations, Basicity and Crystal Structure: The Route to Transition Metal Complexes of Azahelicenes. Molecules 2012, 17, 463–479. [Google Scholar] [CrossRef] [Green Version]
- Napagoda, M.; Rulíšek, L.; Jančařík, A.; Klívar, J.; Šámal, M.; Stará, I.G.; Starý, I.; Šolínová, V.; Kašička, V.; Svatoš, A. Azahelicene Superbases as MAILD Matrices for Acidic Analytes. ChemPlusChem 2013, 78, 937–942. [Google Scholar] [CrossRef]
- Lightner, D.A.; Hefelfinger, D.T.; Powers, T.W.; Frank, G.W.; Trueblood, K.N. Hexahelicenes. The absolute configuration. J. Am. Chem. Soc. 1972, 94, 3492–3497. [Google Scholar] [CrossRef]
- Waghray, D.; Zhang, J.; Jacobs, J.; Nulens, W.; Basarić, N.; Van Meervelt, L.; Dehaen, W. Synthesis and Structural Elucidation of Diversely Functionalized 5,10-Diaza[5]Helicenes. J. Org. Chem. 2012, 77, 10176–10183. [Google Scholar] [CrossRef]
- Baruah, B.; Bhuyan, P.J. Synthesis of some complex pyrano[2,3-b]-and pyrido[2,3-b]quinolines from simple acetanilides via intramolecular domino hetero Diels–Alder reactions of 1-oxa-1,3-butadienes in aqueous medium. Tetrahedron 2009, 65, 7099–7104. [Google Scholar] [CrossRef]
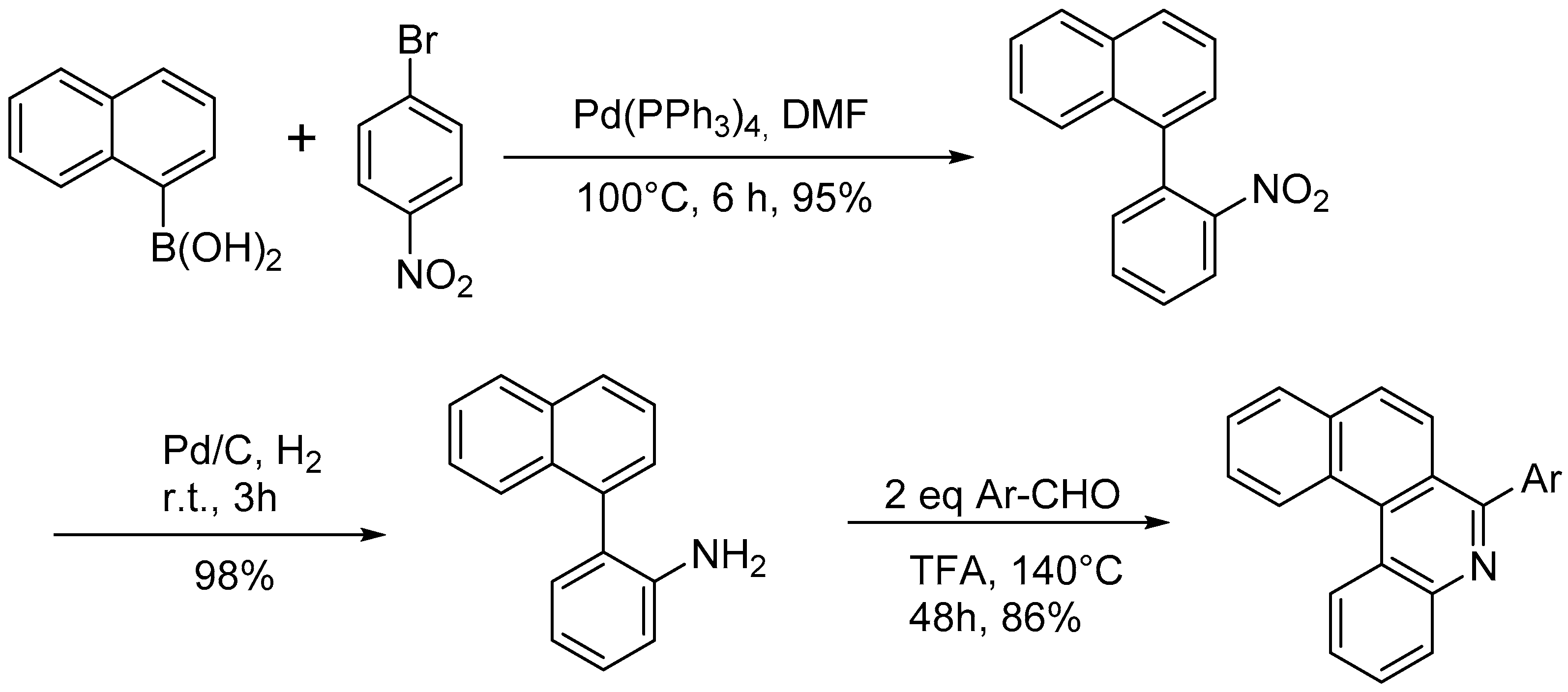
- Harrowven, D.C.; Guy, I.L.; Nanson, L. Efficient Phenanthrene, Helicene, and Azahelicene Syntheses. Angew. Chem. Int. Ed. 2006, 45, 2242–2245. [Google Scholar] [CrossRef]
- Isla, H.; Saleh, N.; Ou-Yang, J.-K.; Dhbaibi, K.; Jean, M.; Dziurka, M.; Favereau, L.; Vanthuyne, N.; Toupet, L.; Jamoussi, B.; et al. Bis-4-Aza[6]Helicene: A Bis-Helicenic 2,2′-Bipyridine with Chemically Triggered Chiroptical Switching Activity. J. Org. Chem. 2019, 84, 5383–5393. [Google Scholar] [CrossRef] [PubMed]
- Saleh, N.; Moore, B., II; Srebro, M.; Vanthuyne, N.; Toupet, L.; Williams, J.A.G.; Roussel, C.; Deol, K.K.; Muller, G.; Autschbach, J.; et al. Acid/Base-Triggered Switching of Circularly Polarized Luminescence and Electronic Circular Dichroism in Organic and Organometallic Helicenes. Chem. Eur. J. 2015, 21, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Isla, H.; Srebro-Hooper, M.; Jean, M.; Vanthuyne, N.; Roisnel, T.; Lunkley, L.; Muller, G.; Williams, J.A.G.; Autschbach, J.; Crassous, J. Conformational changes and chiroptical switching of enantiopure bis-helicenic terpyridine upon Zn2+ binding. Chem. Commun. 2016, 52, 5932–5935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graule, S.; Rudolph, M.; Shen, W.; Gareth Williams, J.A.; Lescop, C.; Autschbach, J.; Crassous, J.; Réau, R. Assembly of π-Conjugated Phosphole Azahelicene Derivatives into Chiral Coordination Complexes: An Experimental and Theoretical Study. Chem. Eur. J. 2010, 16, 5976–6005. [Google Scholar] [CrossRef] [PubMed]
- Murguly, E.; McDonald, R.; Branda, N.R. Chiral Discrimination in Hydrogen-Bonded [7]Helicenes. Org. Lett. 2000, 2, 3169–3172. [Google Scholar] [CrossRef]
- Aloui, F.; El Abed, R.; Marinetti, A.; Hassine, B.B. Synthesis and resolution of a new helically chiral azahelicene. Tetrahedron Lett. 2008, 49, 4092–4095. [Google Scholar] [CrossRef]
- Sasaki, K.; Castle, R.N. The synthesis of novel polycyclic heterocyclic ring systems via photocyclization. 7. [1]benzothieno[2,3-c]naphtho[2,1-h]quinoline and [1]benzothieno[2,3-c]naphtho[2,1-h][1,2,4]triazolo[4,3-a]quinoline. J. Heterocycl. Chem. 1992, 29, 963. [Google Scholar] [CrossRef]
- Stará, I.G.; Starý, I. Helically Chiral Aromatics: The Synthesis of Helicenes by [2 + 2 + 2] Cycloisomerization of π-Electron Systems. Acc. Chem. Res. 2020, 53, 144–158. [Google Scholar] [CrossRef]
- Songis, O.; Míšek, J.; Schmid, M.B.; Kollárovič, A.; Stará, I.G.; Šaman, D.; Císařová, I.; Starý, I. A versatile synthesis of functionalized pentahelicenes. J. Org. Chem. 2010, 75, 6889–6899. [Google Scholar] [CrossRef]
- Storch, J.; Čermák, J.; Karban, J.; Císařová, I.; Sýkora, J. Synthesis of 2-Aza[6]Helicene and Attempts to Synthesize 2,14-Diaza[6]Helicene Utilizing Metal-Catalyzed Cycloisomerization. J. Org. Chem. 2010, 75, 3137–3140. [Google Scholar] [CrossRef]
- Weimar, M.; Correa da Costa, R.; Lee, F.-H.; Fuchter, M.J. A Scalable and Expedient Route to 1-Aza[6]Helicene Derivatives and its Subsequent Application to a Chiral-Relay Asymmetric Strategy. Org. Lett. 2013, 15, 1706–1709. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Furumi, S.; Takeuchi, M.; Shibuya, T.; Tanaka, K. Enantioselective Synthesis and Enhanced Circularly Polarized Luminescence of S-Shaped Double Azahelicenes. J. Am. Chem. Soc. 2014, 136, 5555–5558. [Google Scholar] [CrossRef]
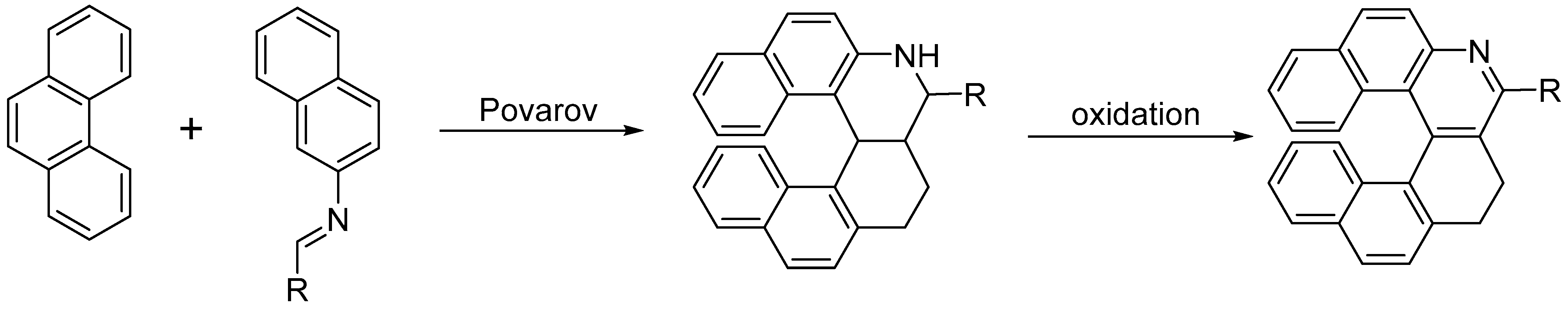
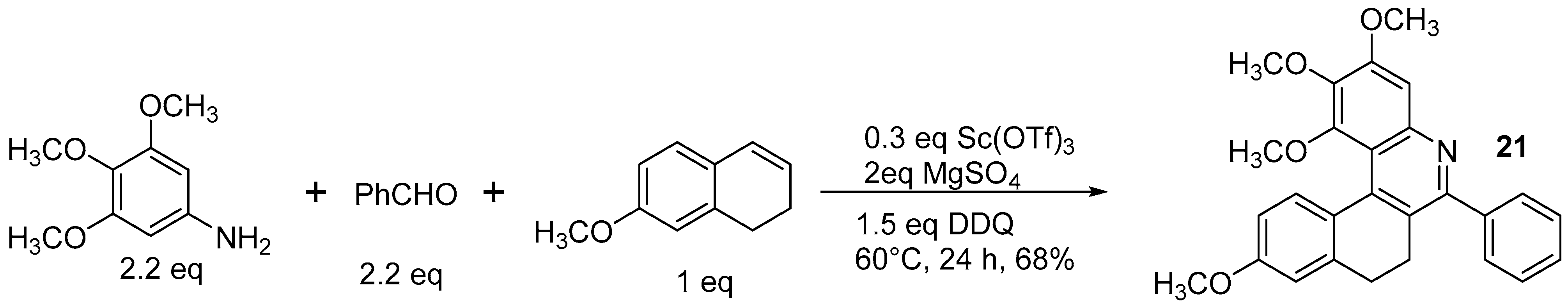
- Viglianisi, C.; Biagioli, C.; Lippi, M.; Pedicini, M.; Villani, C.; Franzini, R.; Menichetti, S. Synthesis of Heterohelicenes by a Catalytic Multi-Component Povarov Reaction. Eur. J. Org. Chem. 2019, 1, 164–167. [Google Scholar] [CrossRef]
- Zheng, Y.-H.; Lu, H.-Y.; Li, M.; Chen, C.-F. Synthesis, structure and optical properties of aza[4]helicenes. Eur. J. Org. Chem. 2013, 15, 3059–3066. [Google Scholar] [CrossRef]
- Wallabregue, A.; Sherin, P.; Guin, J.; Besnard, C.; Vauthey, E.; Lacour, J. Modular Synthesis of pH-Sensitive Fluorescent Diaza[4]Helicenes. Eur. J. Org. Chem. 2014, 29, 6431–6438. [Google Scholar] [CrossRef]
- Takeda, Y.; Okazaki, M.; Maruoka, Y.; Minakata, S. A facile synthesis of functionalized 7,8-diaza[5]helicenes through an oxidative ring-closure of 1,1’-binaphthalene-2,2’-diamines (BINAMs). Beilstein J. Org. Chem. 2015, 11, 9–15. [Google Scholar] [CrossRef] [Green Version]
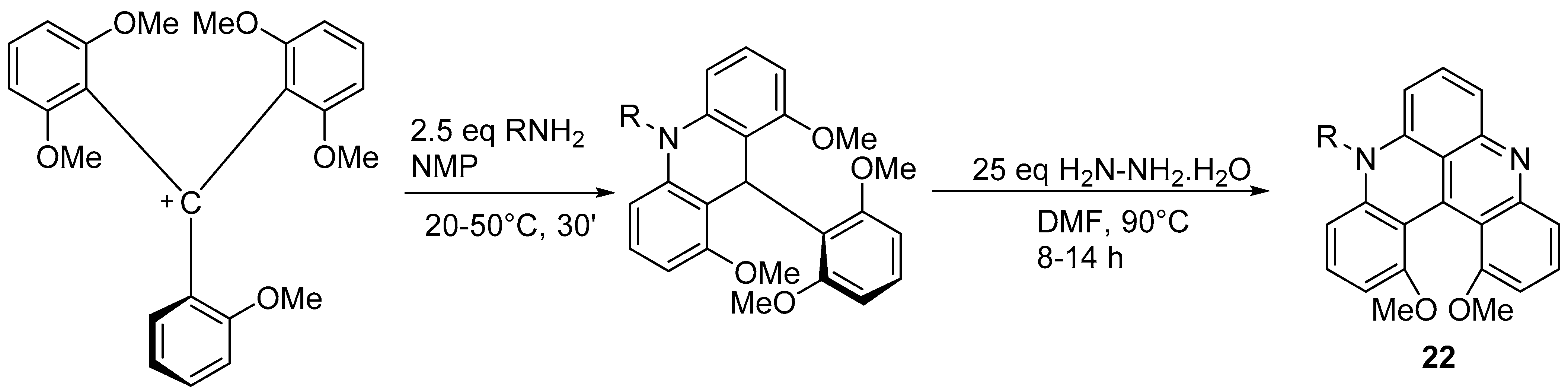
- Feng, J.; Wang, L.; Xue, X.; Chao, Z.; Hong, B.; Gu, Z. Ring-Expansion Strategy for α-Aryl Azahelicene Construction: Building Blocks for Optoelectronic Materials. Org. Lett. 2021, 23, 8056–8061. [Google Scholar] [CrossRef]
- Laarhoven, W.H.; Prinsen, W.J.C. Carbohelicenes and heterohelicenes. Top. Curr. Chem. 1984, 125, 63–130. [Google Scholar]
- Takenaka, N.; Chen, J.S.; Captain, B.; Sarangthem, R.S.; Chandrakumar, A. Helical chiral 2-aminopyridinium ions: A new class of hydrogen bond donor catalysts. J. Am. Chem. Soc. 2010, 132, 4536–4537. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Takenaka, N. Applications of Helical-Chiral Pyridines as Organocatalysts in Asymmetric Synthesis. Chem. Rec. 2013, 13, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, E.; Matsumoto, Y.; Kamikawa, K. Synthesis of azahelicene N-oxide by palladium-catalyzed direct C-H annulation of a pendant (Z)-bromovinyl side chain. Eur. J. Org. Chem. 2011, 19, 11837–11841. [Google Scholar] [CrossRef]
- Loska, R.; Majcher, M.; Makosza, M. Synthesis of trifluoromethylated azines via nucleophilic oxidative substitution of hydrogen by trifluoromethyl carbanion. J. Org. Chem. 2007, 72, 5574–5580. [Google Scholar] [CrossRef]
- Minisci, F.; Bernardi, R.; Bertini, F.; Galli, R.; Perchinunno, M. Nucleophilic character of alkyl radicals VI: A new convenient selective alkylation of heteroaromatic bases. Tetrahedron 1971, 27, 3575–3579. [Google Scholar] [CrossRef]
- Minisci, F.; Vismara, E.; Fontana, F. Recent Developments of Free-Radical Substitutions of Heteroaromatic Bases. Heterocycles 1989, 28, 489–519. [Google Scholar] [CrossRef] [Green Version]
- Giordano, C.; Minisci, F.; Vismara, E.; Levi, S. A General, Selective, and Convenient Procedure of Homolytic Formylation of Heteroaromatic Bases. J. Org. Chem. 1986, 51, 536. [Google Scholar] [CrossRef]
- Minisci, F.; Vismara, E.; Romano, U. Silver-mediated oxidative decarboxylation of carboxylic acids by peroxocompounds: New sources of carbon-centered radicals for heteroaromatic substitution. Tetrahedron Lett. 1985, 26, 4803–4806. [Google Scholar] [CrossRef]
- Minisci, F.; Fontana, F.; Vismara, E. Substitutions by nucleophilic free radicals: A new general reaction of heteroaromatic bases. J. Heterocycl. Chem. 1990, 27, 79–96. [Google Scholar] [CrossRef]
- Minisci, F.; Fontana, F.; Pianese, G.; Yan, Y.-M. Polar Effects in Free-Radical Reactions. Homolytic Heteroaromatic Substitutions by Alkyl Bromides. J. Org. Chem. 1993, 58, 4207–4211. [Google Scholar] [CrossRef]
- Minisci, F.; Fontana, F.; Caronna, T.; Zhao, L. A novel substitution reaction by photoinduced electron-transfer between pyridine derivatives and alkyltin compounds. Tetrahedron Lett. 1992, 33, 3201–3204. [Google Scholar] [CrossRef]
- Chatgilialoglu, C. Organosilanes as radical-based reducing agents in synthesis. Acc. Chem. Res. 1992, 25, 188–194. [Google Scholar] [CrossRef]
- Zanchi, C.; Lucotti, A.; Cancogni, D.; Fontana, F.; Trusso, S.; Ossi, P.M.; Tommasini, M. Functionalization of nanostructured gold substrates with chiral chromophores for SERS applications: The case of 5-aza[5]helicene. Chirality 2018, 30, 875–882. [Google Scholar] [CrossRef]
- Passeri, R.; Aloisi, G.G.; Latterini, L.; Elisei, F.; Caronna, T.; Fontana, F.; Natali Sora, I. Photophysical properties of N-alkylated aza-helicene derivatives as DNA intercalators: Counterion effects. Photochem. Photobiol. Sci. 2009, 8, 1574–1582. [Google Scholar] [CrossRef]
- Latterini, L.; Galletti, E.; Passeri, R.; Barbafina, A.; Urbanelli, L.; Emiliani, C.; Elisei, F.; Fontana, F.; Mele, A.; Caronna, T. Fluorescence properties of aza-helicenium derivatives for cell imaging. J. Photochem. Photobiol. A Chem. 2011, 222, 307–313. [Google Scholar] [CrossRef]
- Fontana, F.; Bertolotti, B.; Grecchi, S.; Mussini, P.R.; Micheli, L.; Cirilli, R.; Tommasini, M.; Rizzo, S. 2,12-diaza[6]helicene: An Efficient non-conventional stereogenic scaffold for enantioselective electrochemical interphases. Chemosensors 2021, 9, 216–230. [Google Scholar] [CrossRef]
- Zhang, X.; Clennan, E.L.; Arulsamy, N.; Weber, R.; Weber, J. Synthesis, Structure, and Photochemical Behavior of [5]Heli-Viologen Isomers. J. Org. Chem. 2016, 81, 5474–5486. [Google Scholar] [CrossRef]
- Huang, Q.; Jiang, L.; Liang, W.; Gui, J.; Xu, D.; Wu, W.; Nakai, Y.; Nishijima, M.; Fukuhara, G.; Mori, T.; et al. Inherently Chiral Azonia[6]Helicene-Modified β-Cyclodextrin: Synthesis, Characterization and Chirality Sensing of Underivatized Amino Acids in Water. J. Org. Chem. 2016, 81, 3430–3434. [Google Scholar] [CrossRef]
- Adriaenssens, L.; Severa, L.; Šálová, T.; Císařová, I.; Radek, P.; Šaman, D.; Rocha, S.V.; Finney, N.; Pospíšil, L.; Slavíček, P.; et al. Helquats: A Facile, Modular, Scalable Route to Novel Helical Dications. Chem. Eur. J. 2009, 15, 1072–1076. [Google Scholar] [CrossRef]
- Coe, B.J.; Rusanova, D.; Joshi, V.D.; Sanchez, S.; Vávra, J.; Khobragade, D.; Severa, L.; Císařová, I.; Šaman, D.; Radek, P.; et al. Helquat Dyes: Helicene-Like Push–Pull Systems with Large Second-Order Nonlinear Optical Responses. J. Org. Chem. 2016, 81, 1912–1920. [Google Scholar] [CrossRef] [PubMed]
- Buckley, L.E.R.; Coe, B.J.; Rusanova, D.; Joshi, V.D.; Sanchez, S.; Jirásek, M.; Vávra, J.; Khobragade, D.; Severa, L.; Císařová, I.; et al. Tunable Chiral Second-Order Nonlinear Optical Chromophores Based on Helquat Dications. J. Phys. Chem. A 2017, 121, 5842–5855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Gutiérrez, P.E.; Jirásek, M.; Severa, L.; Novotná, P.; Koval, D.; Sázelová, P.; Vávra, J.; Meyer, A.; Císařová, I.; Šaman, D.; et al. Functional helquats: Helical cationic dyes with marked switchable chiroptical properties in the visible region. Chem. Commun. 2015, 51, 1583–1586. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Yafune, T.; Ōkubo, M.; Hida, M. Synthesis of azonia derivative of hexahelicene. Tetrahedron Lett. 1989, 30, 7217–7218. [Google Scholar] [CrossRef]
- Sato, K.; Katayama, Y.; Yamagishi, T.; Arai, S. The Synthesis of New Azoniathiahelicenes. J. Heterocycl. Chem. 2006, 43, 177–181. [Google Scholar] [CrossRef]
- Torricelli, F.; Bosson, J.; Besnard, C.; Chekini, M.; Bürgi, T.; Lacour, J. Modular Synthesis, Orthogonal Post-Functionalization, Absorption, and Chiroptical Properties of Cationic [6]Helicenes. Angew. Chem. Int. Ed. Eng. 2013, 52, 1796–1800. [Google Scholar] [CrossRef] [PubMed]
- Marinova, M.; Pascal, S.; Guénée, L.; Besnard, C.; Shivachev, B.; Kostova, K.; Villani, C.; Franzini, R.; Dimitrov, V.; Lacour, J. Synthesis, Resolution, Configurational Stability, and Properties of Cationic Functionalized [5]Helicenes. J. Org. Chem. 2020, 85, 11908–11923. [Google Scholar] [CrossRef]




































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontana, F.; Bertolotti, B. Synthesis of Functionalized Six-Membered-Ring Azahelicenes. Molecules 2022, 27, 2522. https://doi.org/10.3390/molecules27082522
Fontana F, Bertolotti B. Synthesis of Functionalized Six-Membered-Ring Azahelicenes. Molecules. 2022; 27(8):2522. https://doi.org/10.3390/molecules27082522
Chicago/Turabian StyleFontana, Francesca, and Benedetta Bertolotti. 2022. "Synthesis of Functionalized Six-Membered-Ring Azahelicenes" Molecules 27, no. 8: 2522. https://doi.org/10.3390/molecules27082522
APA StyleFontana, F., & Bertolotti, B. (2022). Synthesis of Functionalized Six-Membered-Ring Azahelicenes. Molecules, 27(8), 2522. https://doi.org/10.3390/molecules27082522