
Accelerating AutoDock Vina with GPUs

 , , , and
, , , and
Abstract
:
1. Introduction
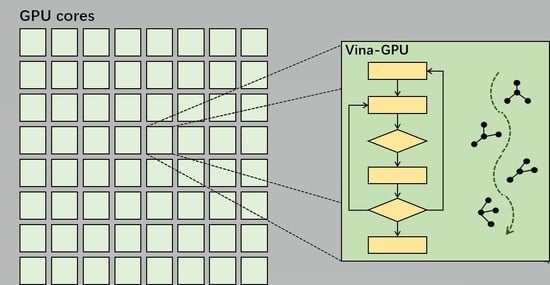
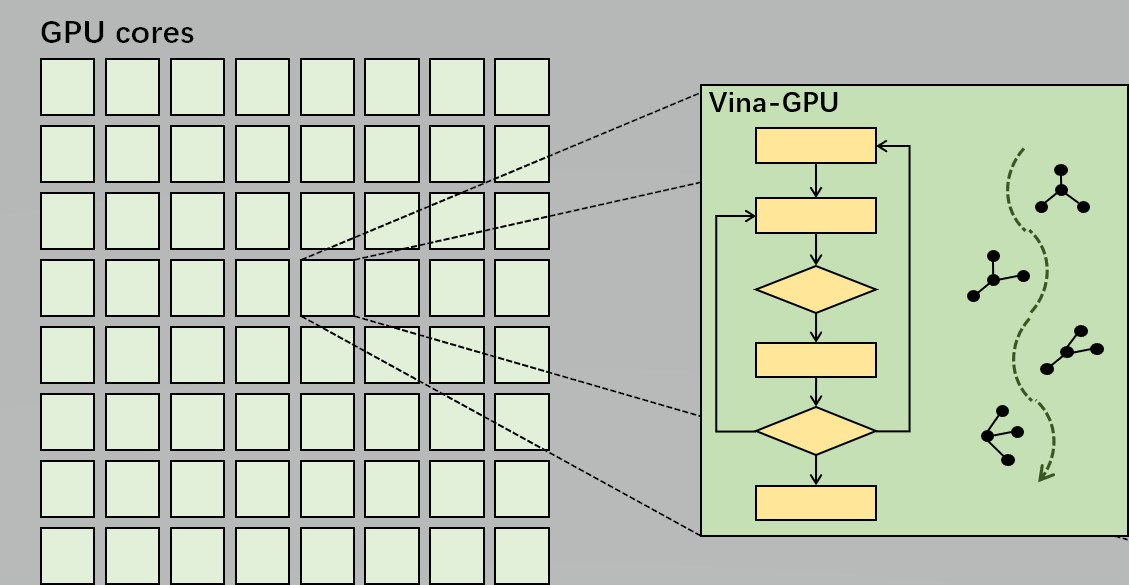
2. Methodology
2.1. Host Part
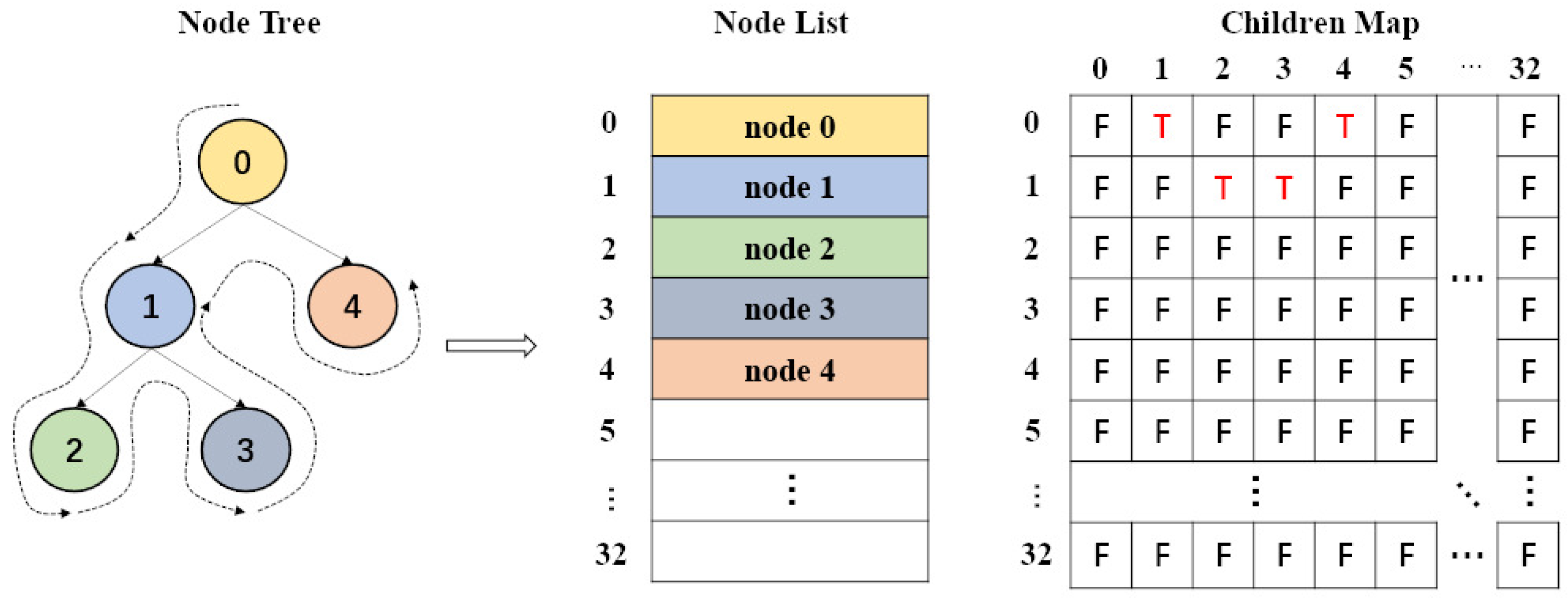
2.2. Device Part
| Algorithm 1 Vina-GPU method |
Input: random ligand conformations: |
Output: top k ligand conformations |
|
3. Results and Discussion
3.1. Experimental Settings
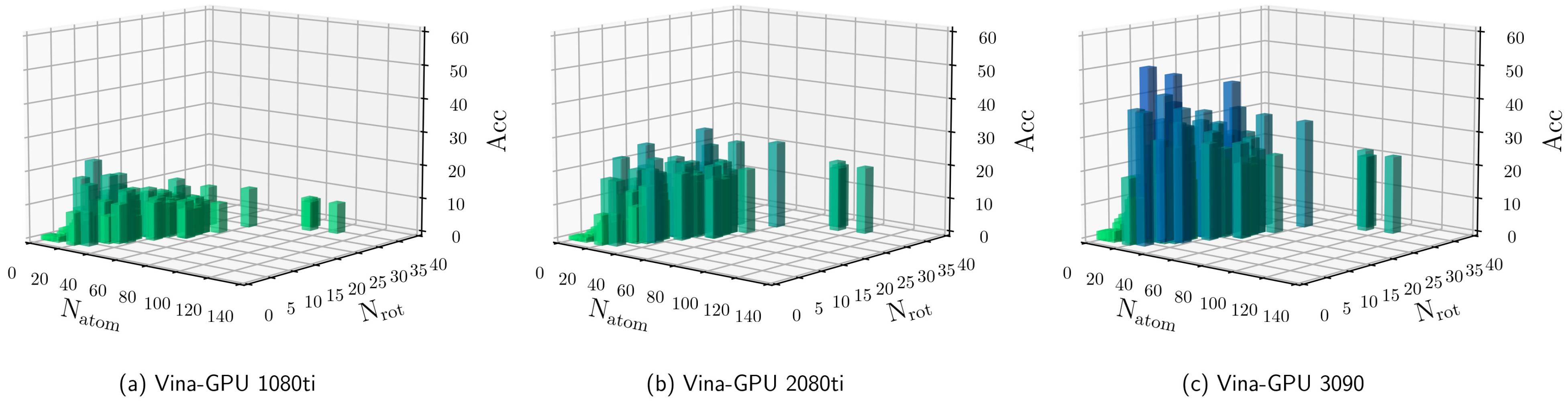
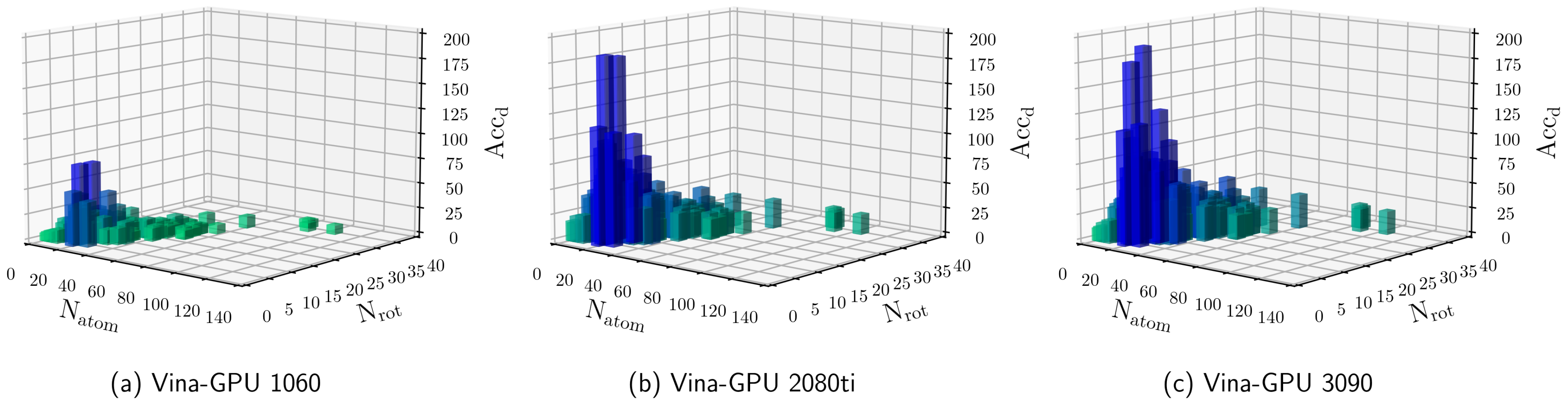
3.2. Influence of Hyperparameters
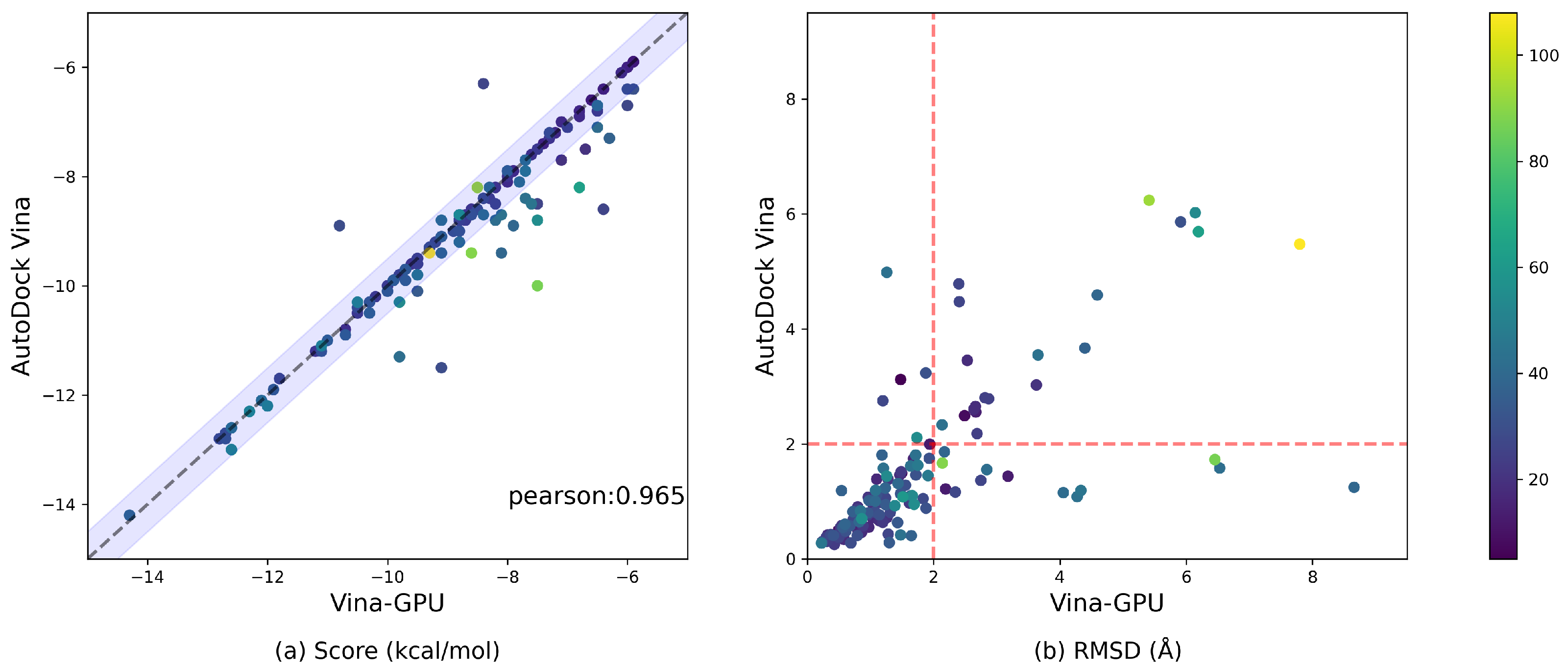
3.3. Docking Accuracy
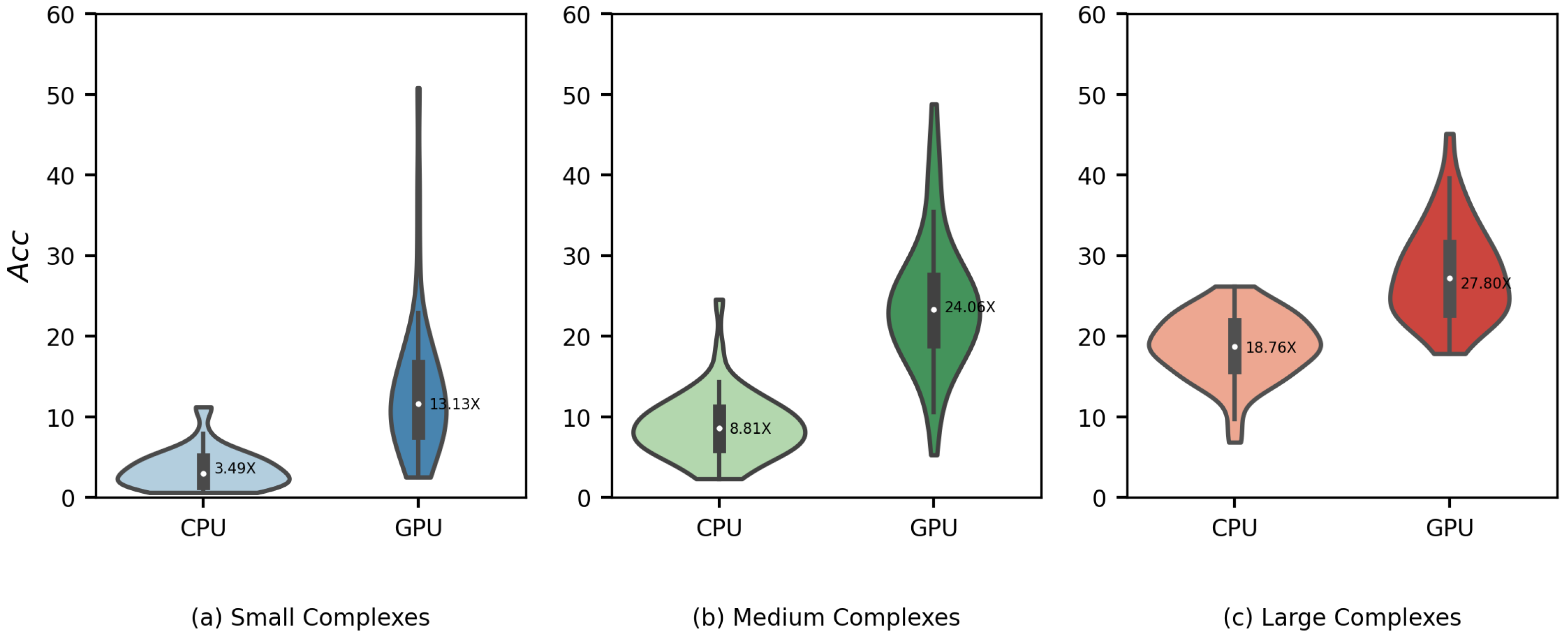
3.4. Runtime Comparison
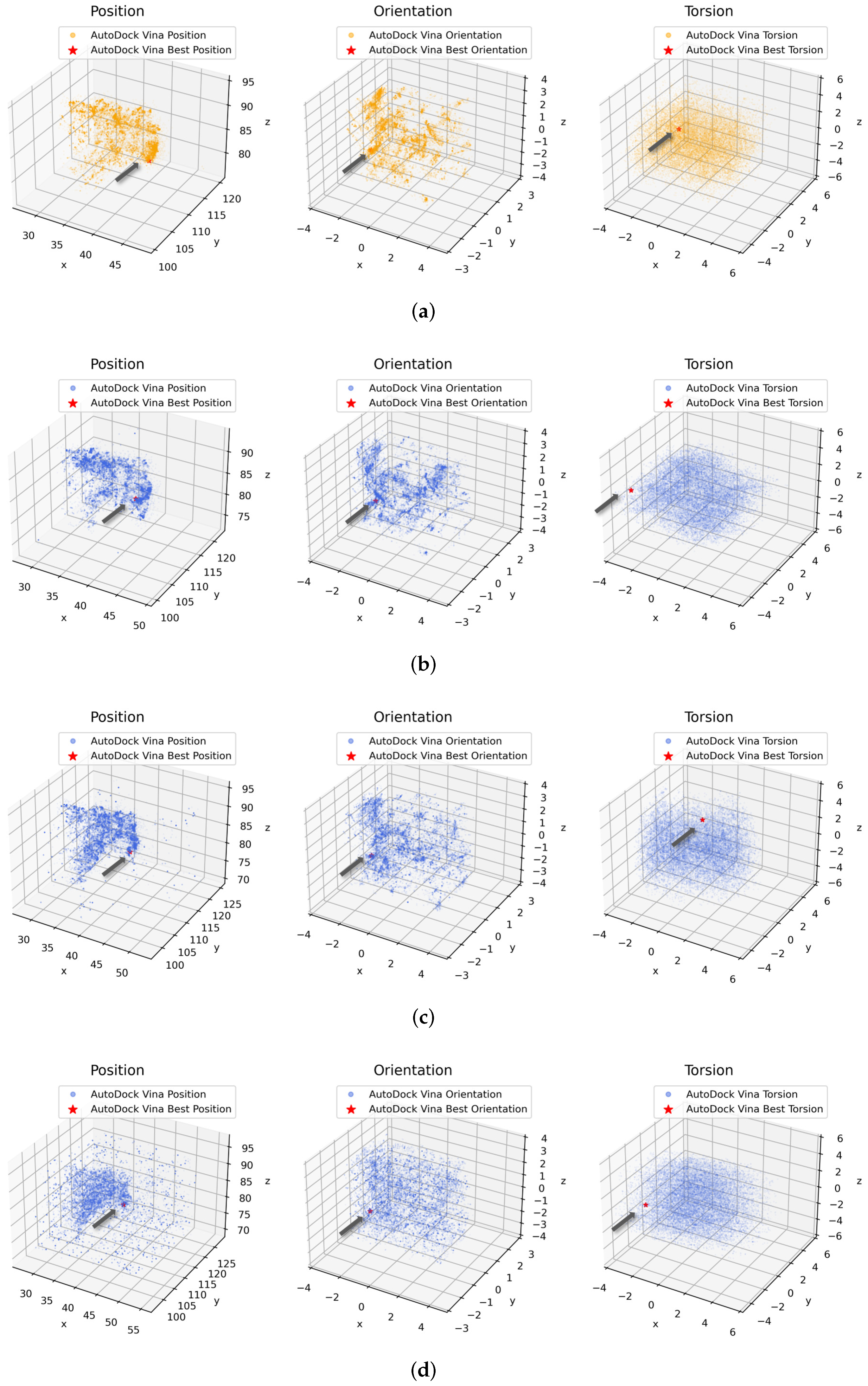
3.5. Conformation Spaces Analysis
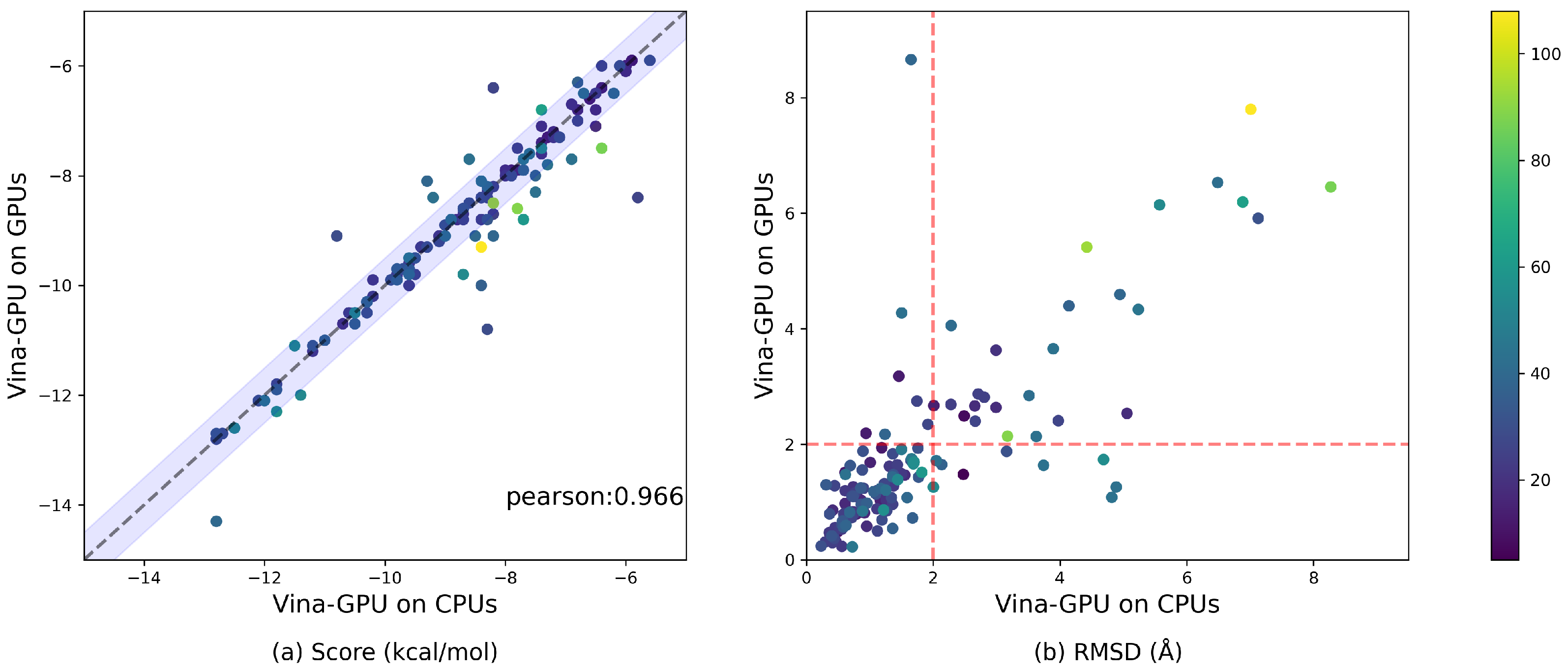
3.6. Comparison with the Implementation of Vina-GPU on CPUs
3.7. A Case for Virtual Screening
3.8. Usage of Vina-GPU
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, T.; Rarey, M. Computational methods for biomolecular docking. Curr. Opin. Struct. Biol. 1996, 6, 402–406. [Google Scholar] [CrossRef]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golbraikh, A.; Shen, M.; Xiao, Z.; Xiao, Y.D.; Lee, K.H.; Tropsha, A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput.-Aided Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.; Forli, S. AutoDock Vina 1.2. 0: New docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sanner, M.F. AutoDock CrankPep: Combining folding and docking to predict protein—Peptide complexes. Bioinformatics 2019, 35, 5121–5127. [Google Scholar] [CrossRef]
- Zhang, Y.; Sanner, M.F. Docking flexible cyclic peptides with AutoDock CrankPep. J. Chem. Theory Comput. 2019, 15, 5161–5168. [Google Scholar] [CrossRef]
- Santos-Martins, D.; Eberhardt, J.; Bianco, G.; Solis-Vasquez, L.; Ambrosio, F.A.; Koch, A.; Forli, S. D3R Grand Challenge 4: Prospective pose prediction of BACE1 ligands with AutoDock-GPU. J. Comput.-Aided Mol. Des. 2019, 33, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Santos-Martins, D.; Solis-Vasquez, L.; Tillack, A.F.; Sanner, M.F.; Koch, A.; Forli, S. Accelerating AutoDock4 with GPUs and gradient-based local search. J. Chem. Theory Comput. 2021, 17, 1060–1073. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S.; Sanner, M.F.; Olson, A.J.; Forli, S. The AutoDock suite at 30. Protein Sci. 2021, 30, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Yang, Q.; Du, Y.; Feng, G.; Liu, Z.; Li, Y.; Wang, R. Comparative assessment of scoring functions: The CASF-2016 update. J. Chem. Inf. Model. 2018, 59, 895–913. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive evaluation of ten docking programs on a diverse set of protein–ligand complexes: The prediction accuracy of sampling power and scoring power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef]
- Fletcher, R. Practical Methods of Optimization; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef]
- Gorgulla, C.; Boeszoermenyi, A.; Wang, Z.F.; Fischer, P.D.; Coote, P.W.; Das, K.M.P.; Malets, Y.S.; Radchenko, D.S.; Moroz, Y.S.; Scott, D.A.; et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 580, 663–668. [Google Scholar] [CrossRef]
- Li, H.; Leung, K.S.; Wong, M.H. idock: A multithreaded virtual screening tool for flexible ligand docking. In Proceedings of the 2012 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), San Diego, CA, USA, 9–12 May 2012; pp. 77–84. [Google Scholar]
- Jaghoori, M.M.; Bleijlevens, B.; Olabarriaga, S.D. 1001 Ways to run AutoDock Vina for virtual screening. J. Comput.-Aided Mol. Des. 2016, 30, 237–249. [Google Scholar] [CrossRef] [Green Version]
- Mermelstein, D.J.; Lin, C.; Nelson, G.; Kretsch, R.; McCammon, J.A.; Walker, R.C. Fast and flexible gpu accelerated binding free energy calculations within the amber molecular dynamics package. J. Comput. Chem. 2018, 39, 1354–1358. [Google Scholar] [CrossRef] [PubMed]
- Hwu, W.M.W. GPU Computing Gems Emerald Edition; Morgan Kaufmann Publishers Inc.: Burlington, MA, USA, 2011. [Google Scholar]
- Stone, J.E.; Hynninen, A.P.; Phillips, J.C.; Schulten, K. Early experiences porting the NAMD and VMD molecular simulation and analysis software to GPU-accelerated OpenPOWER platforms. In Proceedings of the International Conference on High Performance Computing, Frankfurt, Germany, 19–23 June 2016; pp. 188–206. [Google Scholar]
- LeGrand, S.; Scheinberg, A.; Tillack, A.F.; Thavappiragasam, M.; Vermaas, J.V.; Agarwal, R.; Larkin, J.; Poole, D.; Santos-Martins, D.; Solis-Vasquez, L.; et al. GPU-accelerated drug discovery with docking on the summit supercomputer: Porting, optimization, and application to COVID-19 research. In Proceedings of the 11th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Virtual Event, 21–24 September 2020; pp. 1–10. [Google Scholar]
- Fan, M.; Wang, J.; Jiang, H.; Feng, Y.; Mahdavi, M.; Madduri, K.; Kandemir, M.T.; Dokholyan, N.V. Gpu-accelerated flexible molecular docking. J. Phys. Chem. B 2021, 125, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Wu, Y.; Wang, Y.; Vilseck, J.Z.; Brooks III, C.L. Accelerated CDOCKER with GPUs, parallel simulated annealing, and fast Fourier transforms. J. Chem. Theory Comput. 2020, 16, 3910–3919. [Google Scholar] [CrossRef] [PubMed]
- Imbernón, B.; Serrano, A.; Bueno-Crespo, A.; Abellán, J.L.; Pérez-Sánchez, H.; Cecilia, J.M. METADOCK 2: A high-throughput parallel metaheuristic scheme for molecular docking. Bioinformatics 2021, 37, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Kim, J.; Chae, J.; Yun, S.J. GPU-Accelerated Autodock Vina: Viking. 2020. Available online: https://www.morressier.com/o/event/5e733c5acde2b641284a7e27/article/5e73656bcde2b641284aa4e5 (accessed on 27 March 2022).
- Solis-Vasquez, L.; Santos-Martins, D.; Tillack, A.F.; Koch, A.; Eberhardt, J.; Forli, S. Parallelizing Irregular Computations for Molecular Docking. In Proceedings of the 2020 IEEE/ACM 10th Workshop on Irregular Applications: Architectures and Algorithms (IA3), Atlanta, GA, USA, 11 November 2020; pp. 12–21. [Google Scholar]
- Kannan, S.; Ganji, R. Porting autodock to CUDA. In Proceedings of the IEEE Congress on Evolutionary Computation, Barcelona, Spain, 18–23 July 2010; pp. 1–8. [Google Scholar]
- Hartshorn, M.J.; Verdonk, M.L.; Chessari, G.; Brewerton, S.C.; Mooij, W.T.; Mortenson, P.N.; Murray, C.W. Diverse, high-quality test set for the validation of protein-ligand docking performance. J. Med. Chem. 2007, 50, 726–741. [Google Scholar] [CrossRef]
- Li, Y.; Han, L.; Liu, Z.; Wang, R. Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. J. Chem. Inf. Model. 2014, 54, 1717–1736. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Handoko, S.D.; Ouyang, X.; Su, C.T.T.; Kwoh, C.K.; Ong, Y.S. QuickVina: Accelerating AutoDock Vina using gradient-based heuristics for global optimization. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1266–1272. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Jaccard, P. The distribution of the flora in the alpine zone. New Phytol. 1912, 11, 37–50. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Top i | Jacard Index | ||
|---|---|---|---|
| 15 | 14 | 16 | 0.875 |
| 50 | 46 | 54 | 0.852 |
| 100 | 91 | 109 | 0.835 |
| 200 | 187 | 213 | 0.878 |
| 300 | 277 | 323 | 0.858 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, S.; Chen, R.; Lin, M.; Lin, Q.; Zhu, Y.; Ding, J.; Hu, H.; Ling, M.; Wu, J. Accelerating AutoDock Vina with GPUs. Molecules 2022, 27, 3041. https://doi.org/10.3390/molecules27093041
Tang S, Chen R, Lin M, Lin Q, Zhu Y, Ding J, Hu H, Ling M, Wu J. Accelerating AutoDock Vina with GPUs. Molecules. 2022; 27(9):3041. https://doi.org/10.3390/molecules27093041
Chicago/Turabian StyleTang, Shidi, Ruiqi Chen, Mengru Lin, Qingde Lin, Yanxiang Zhu, Ji Ding, Haifeng Hu, Ming Ling, and Jiansheng Wu. 2022. "Accelerating AutoDock Vina with GPUs" Molecules 27, no. 9: 3041. https://doi.org/10.3390/molecules27093041
APA StyleTang, S., Chen, R., Lin, M., Lin, Q., Zhu, Y., Ding, J., Hu, H., Ling, M., & Wu, J. (2022). Accelerating AutoDock Vina with GPUs. Molecules, 27(9), 3041. https://doi.org/10.3390/molecules27093041