
Development of a Novel Electrochemical Sensor Based on Gold Nanoparticle-Modified Carbon-Paste Electrode for the Detection of Congo Red Dye


Abstract
:1. Introduction
2. Results and Discussion
2.1. Characterization
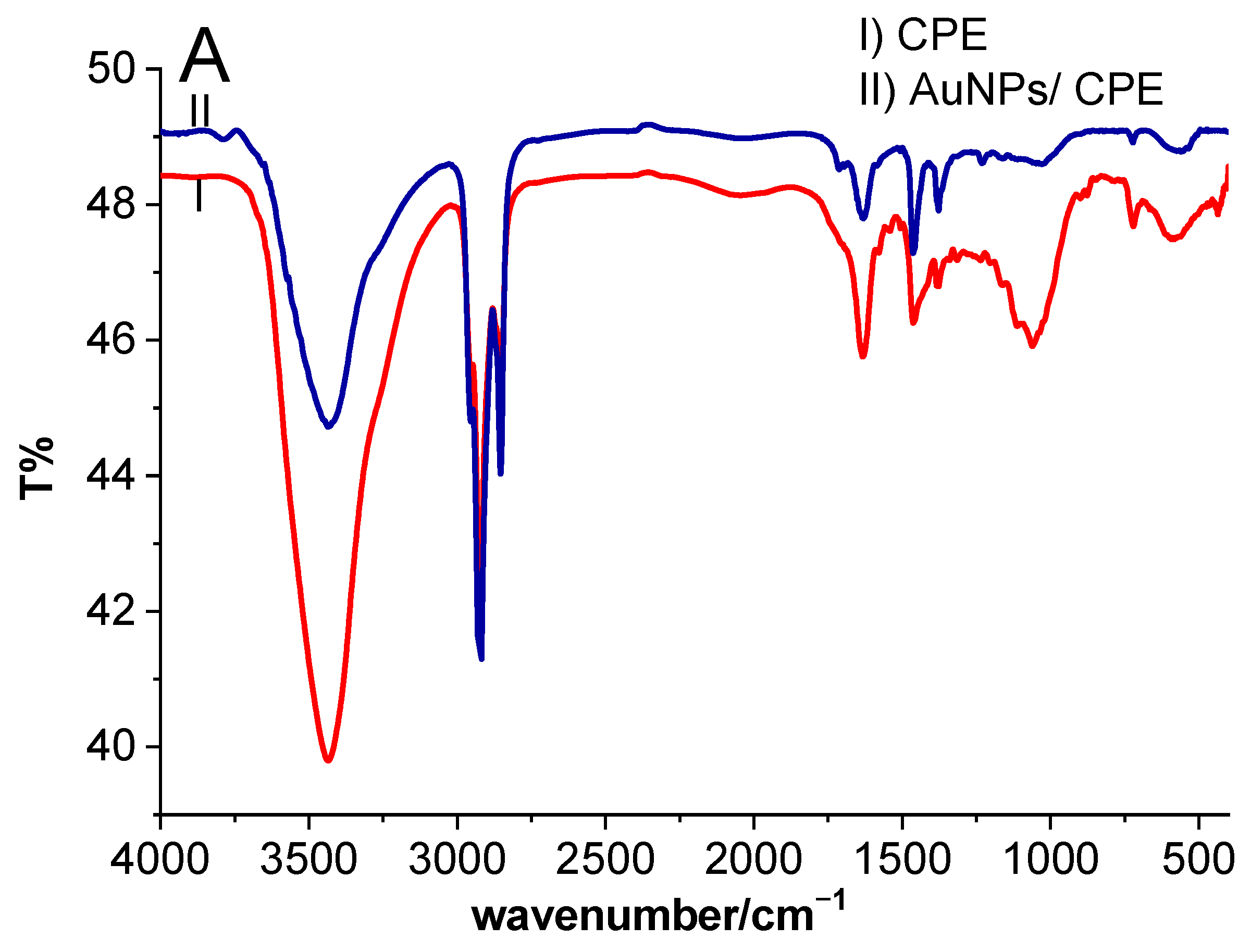
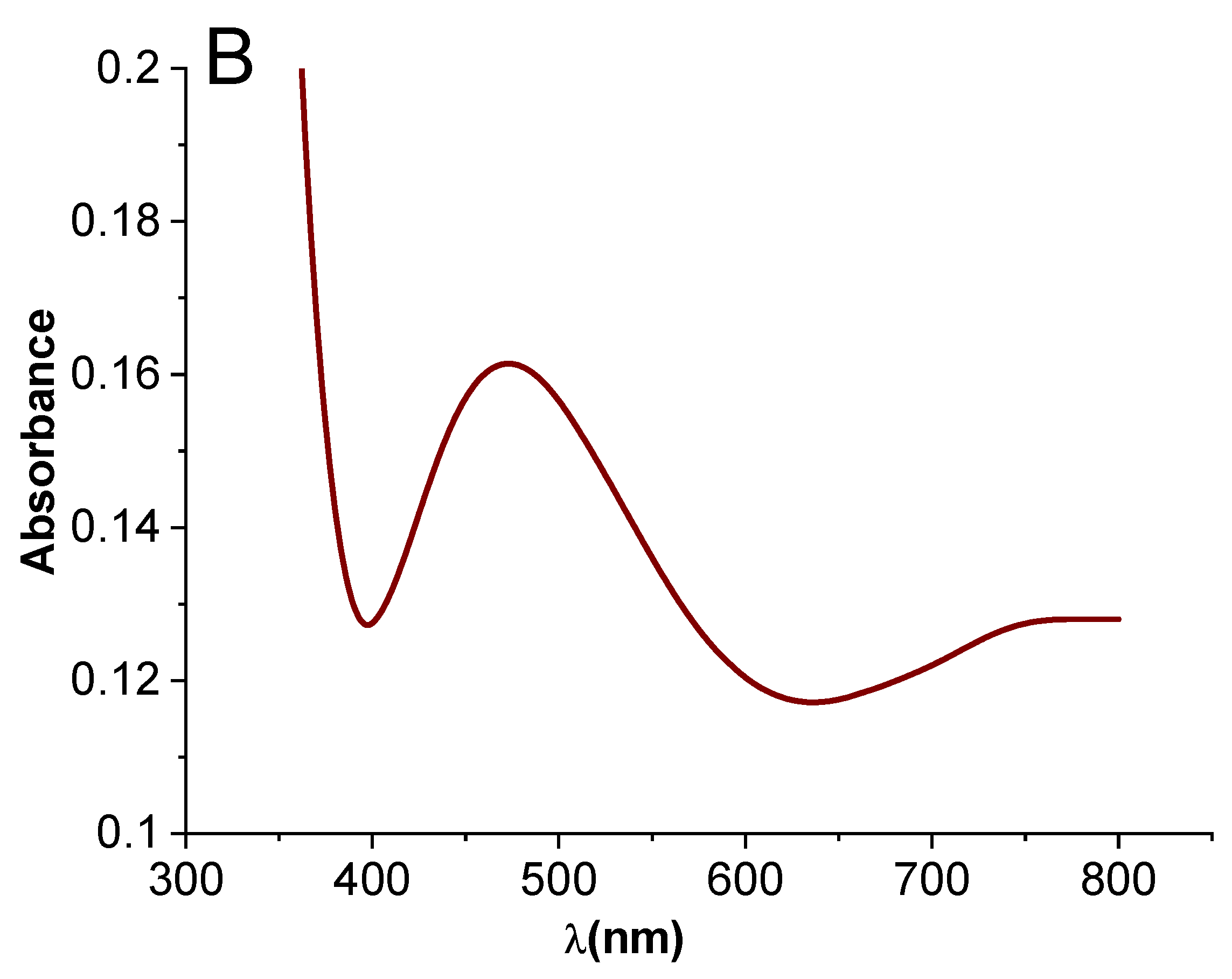
2.1.1. Fourier Transform Infrared (FTIR) and Ultraviolet-Visible (UV–Vis) Spectroscopy
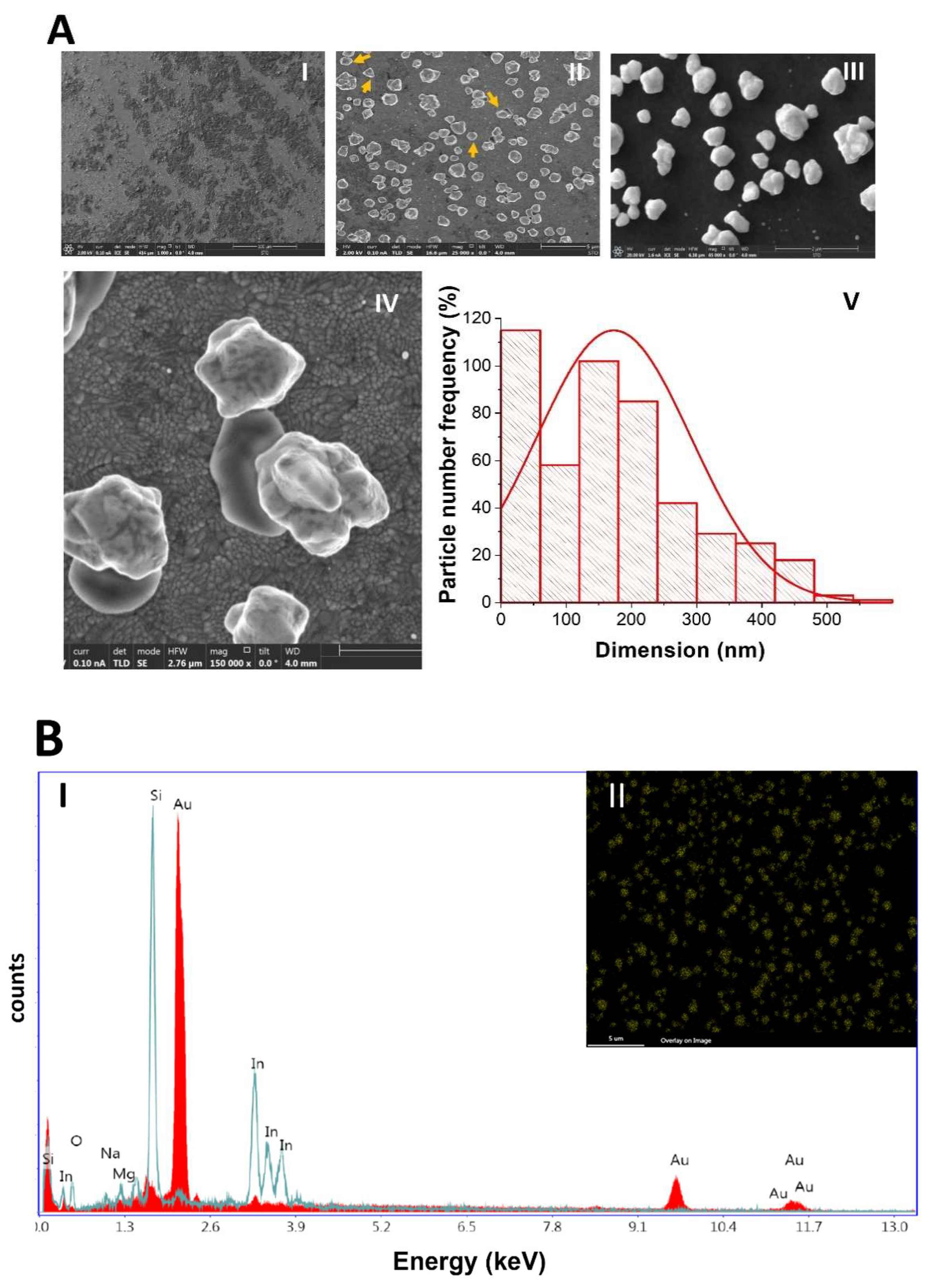
2.1.2. Scanning Electron Microscopy (SEM) and Energy-Dispersive X-ray Spectroscopy (EDX)
2.1.3. Transmission Electron Microscopy (TEM)
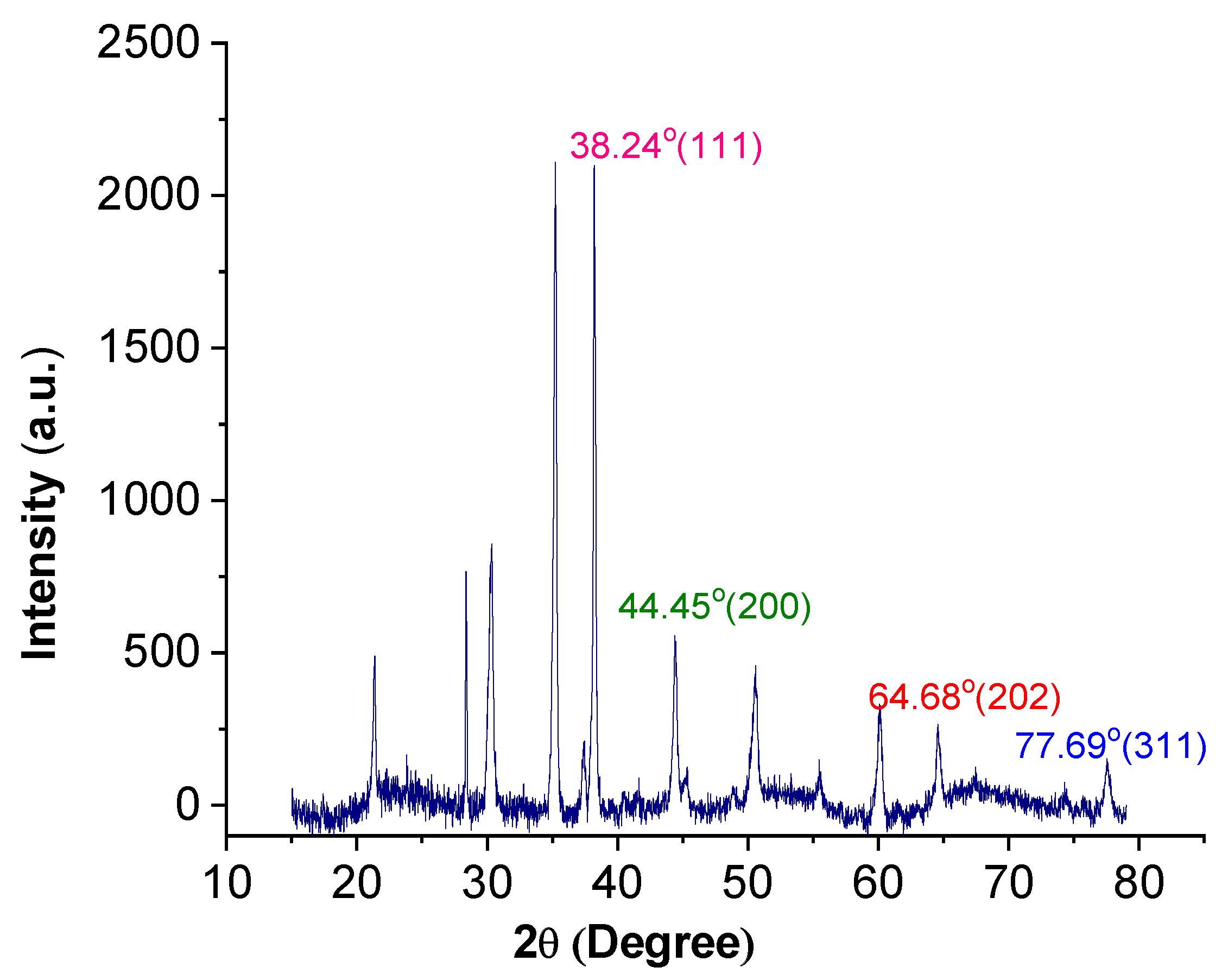
2.1.4. X-ray Diffraction Spectroscopy (XRD)
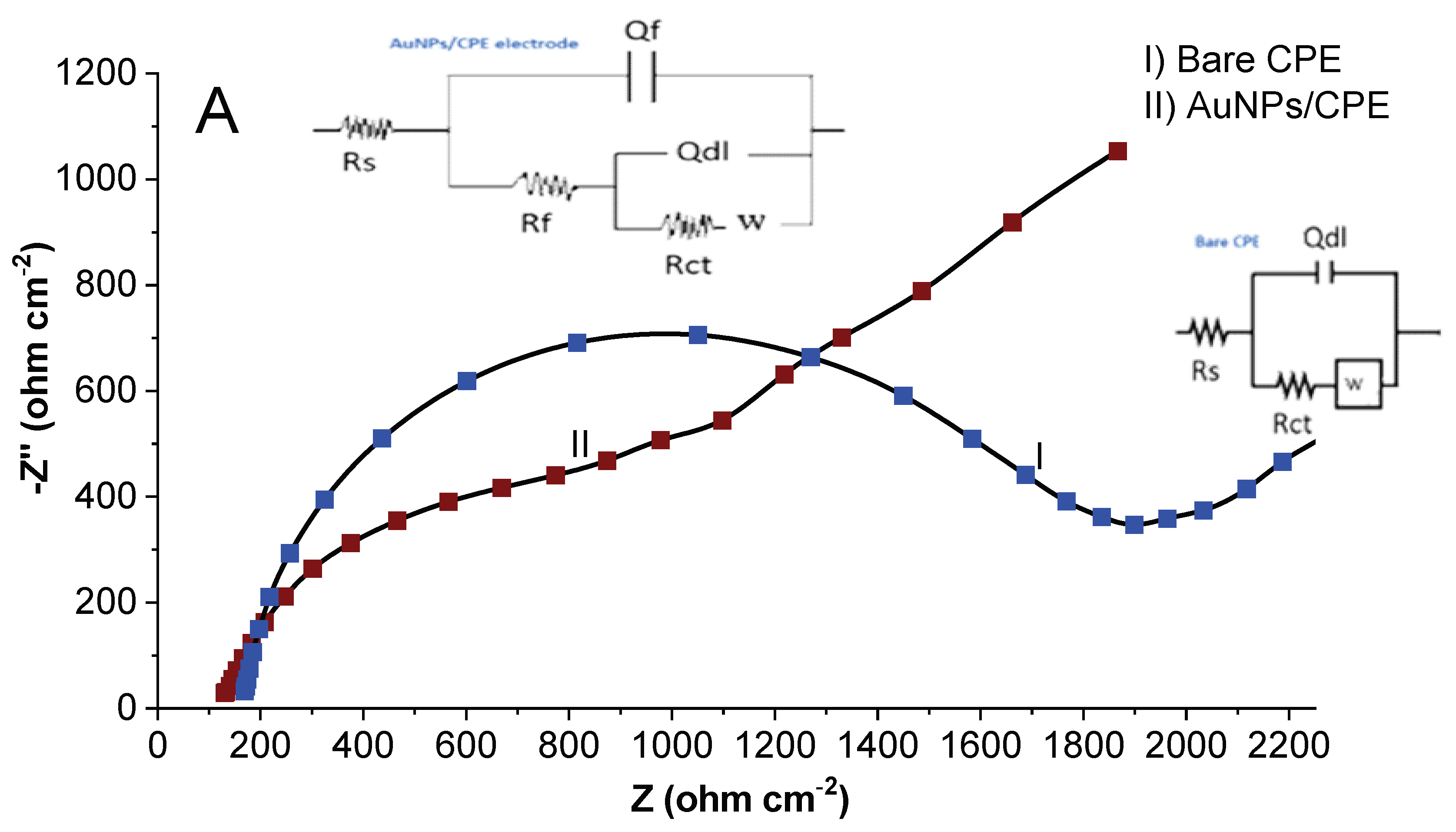
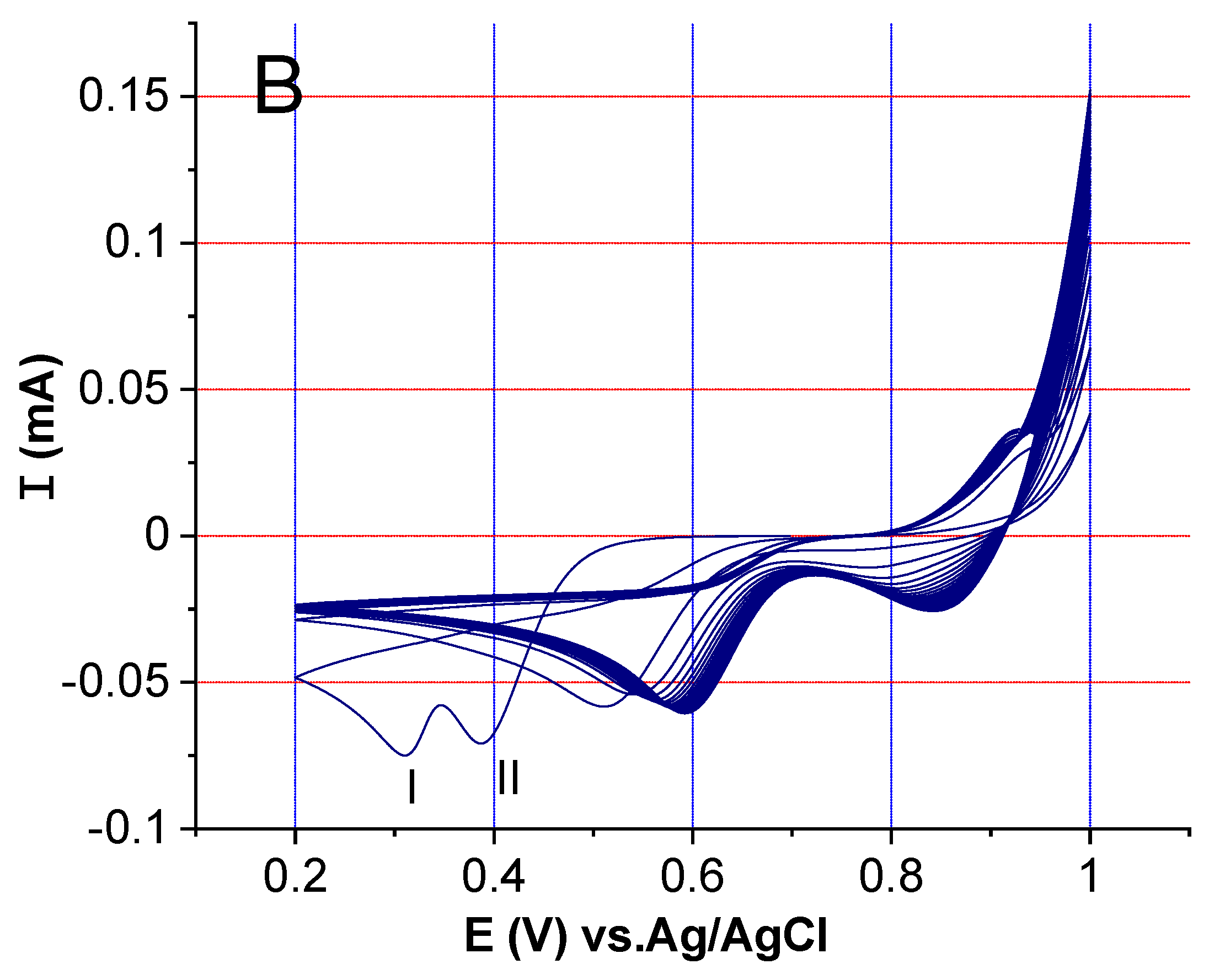
2.2. Electrochemical Activity Investigation
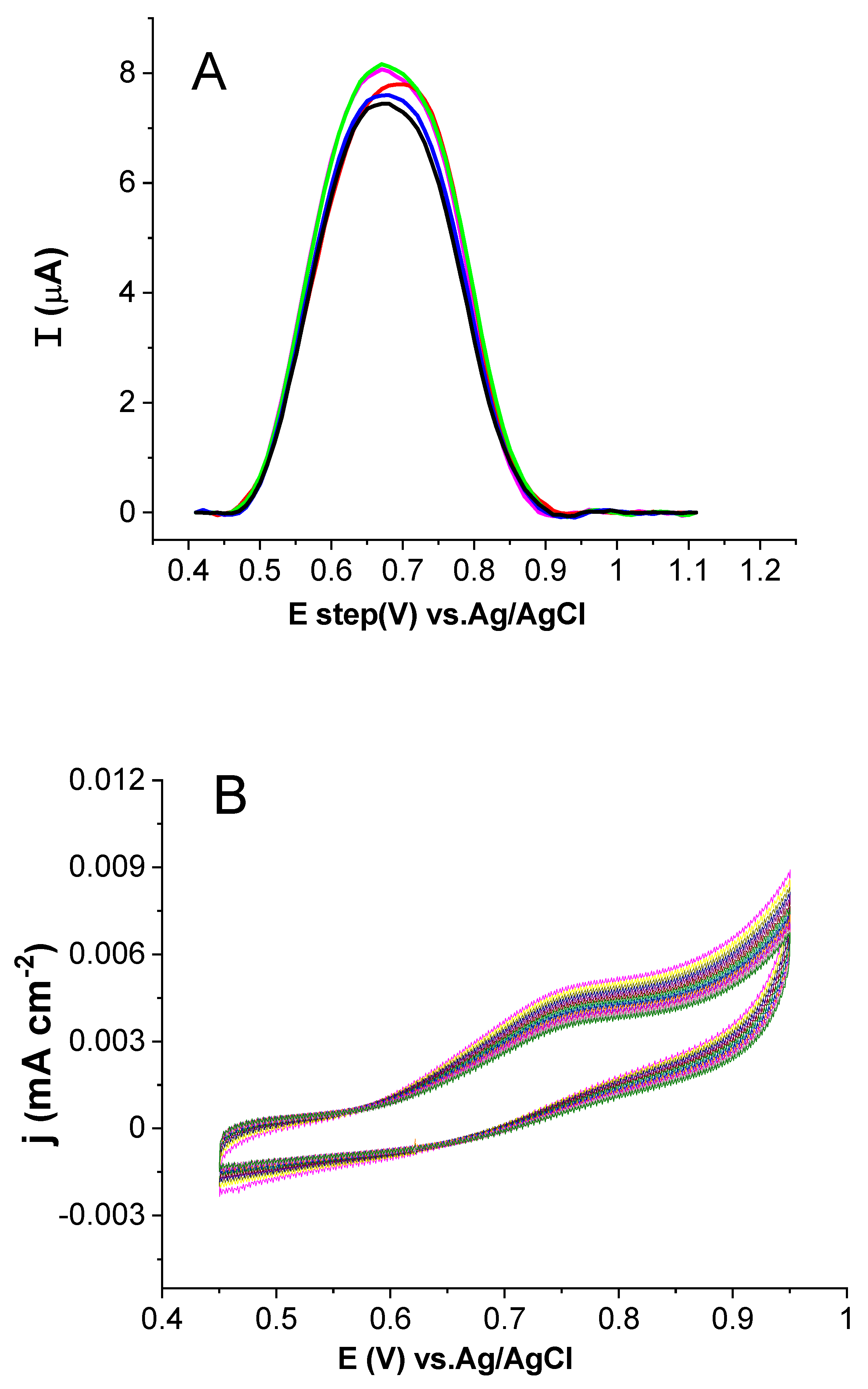
2.3. Optimization of Investigational Parameters
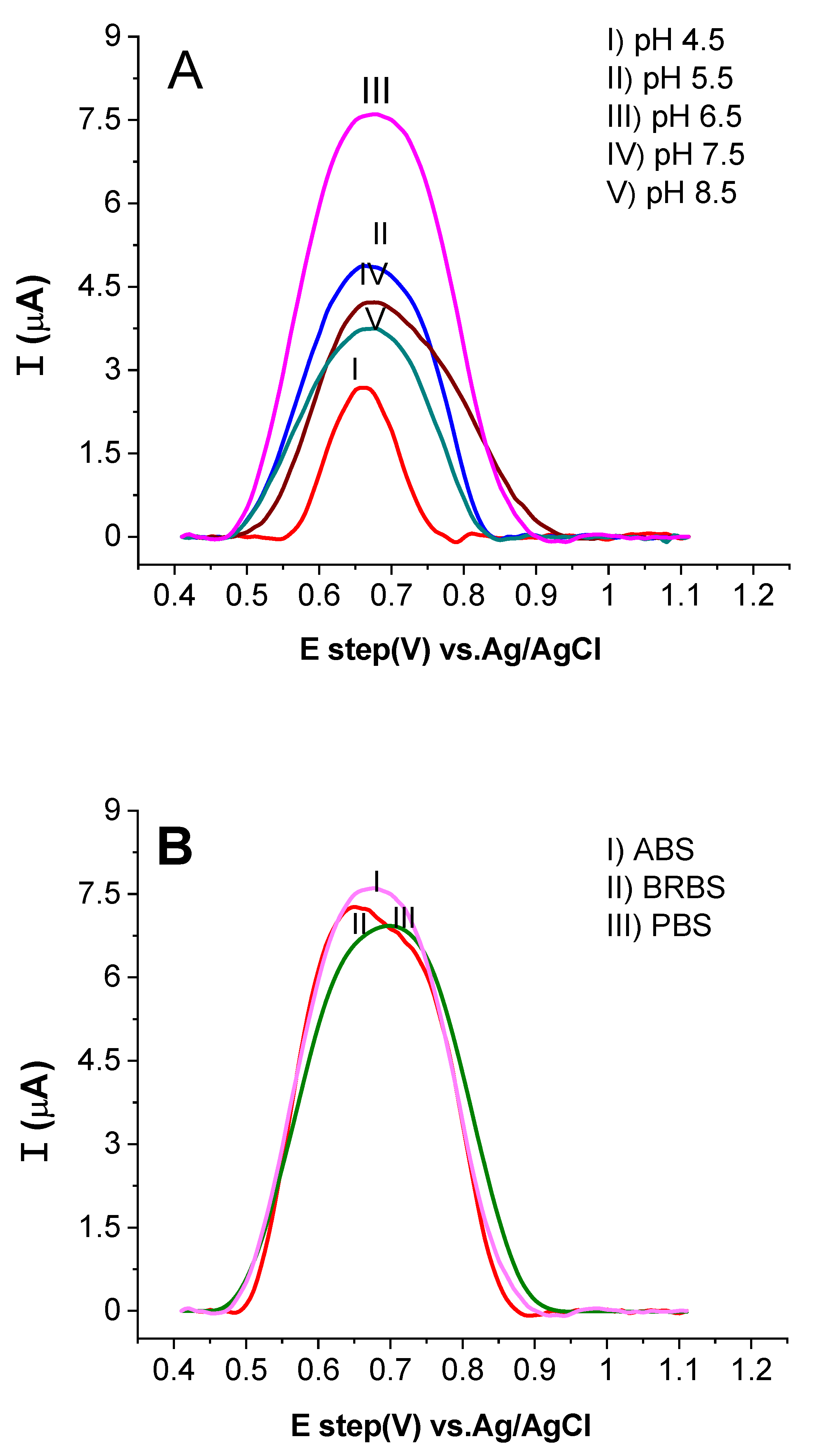
2.3.1. Influence of Changing pH and the Supporting Electrolyte
2.3.2. Effect of AuNP Thickness Layer
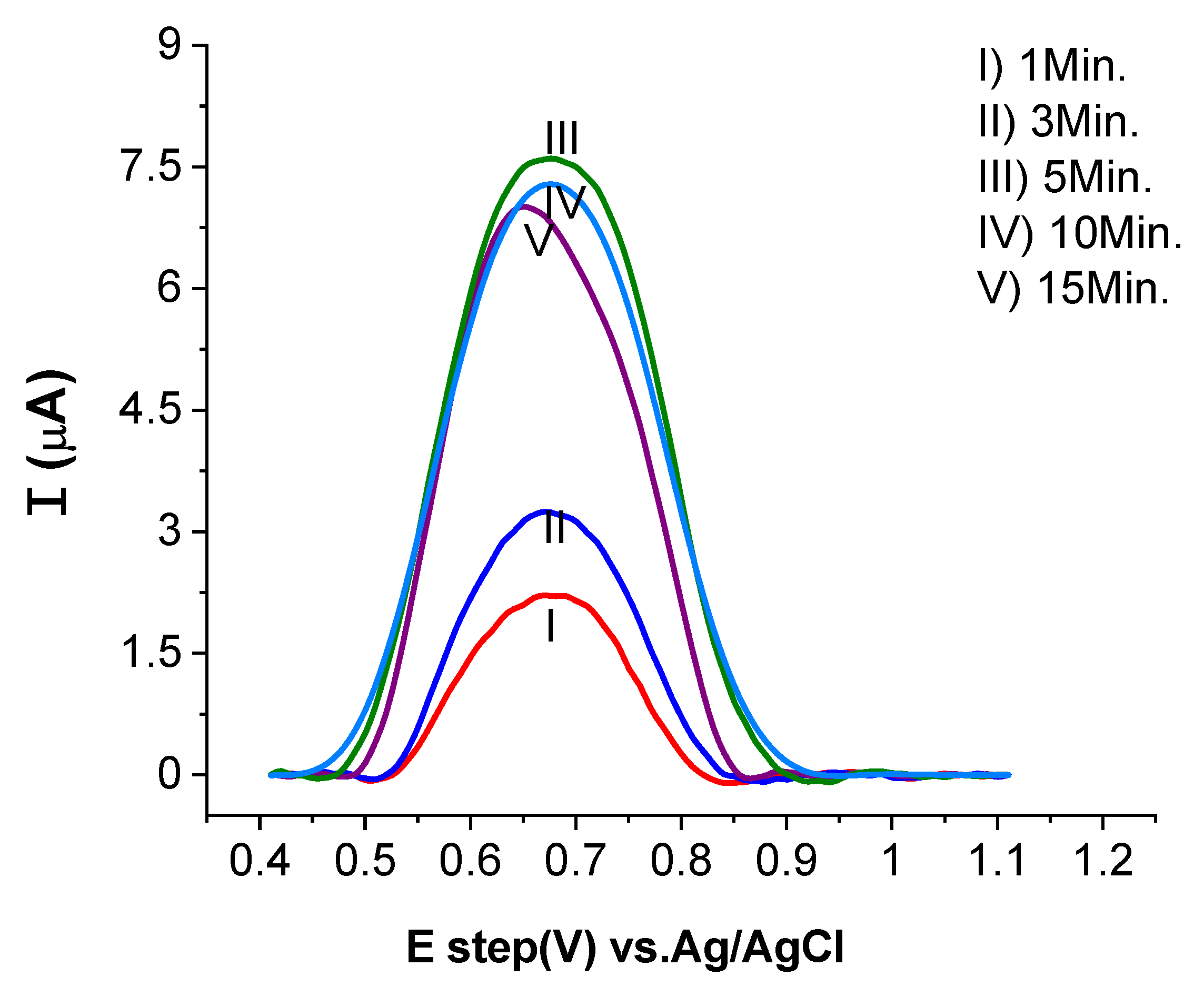
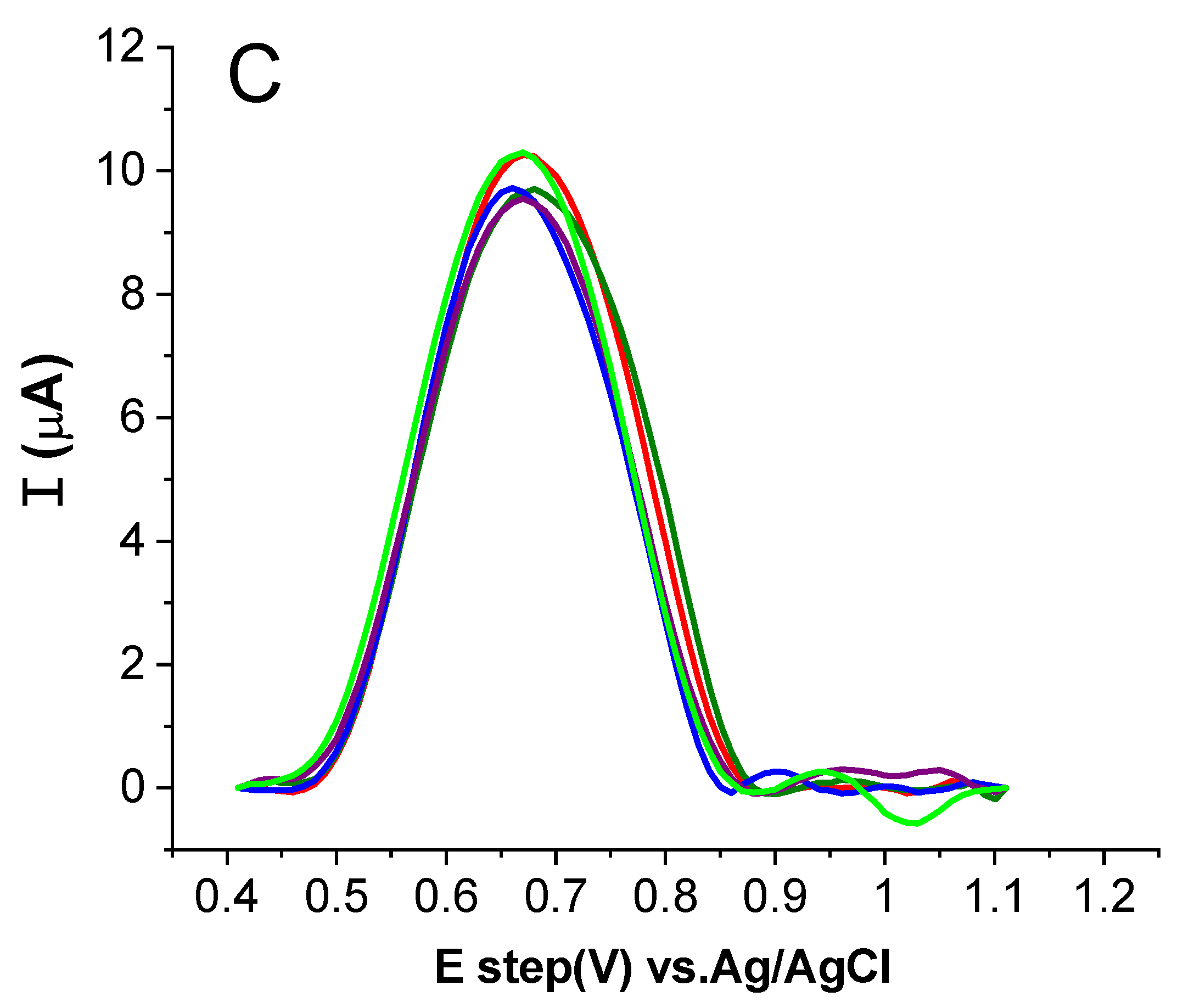
2.3.3. Influence of Accumulation Time
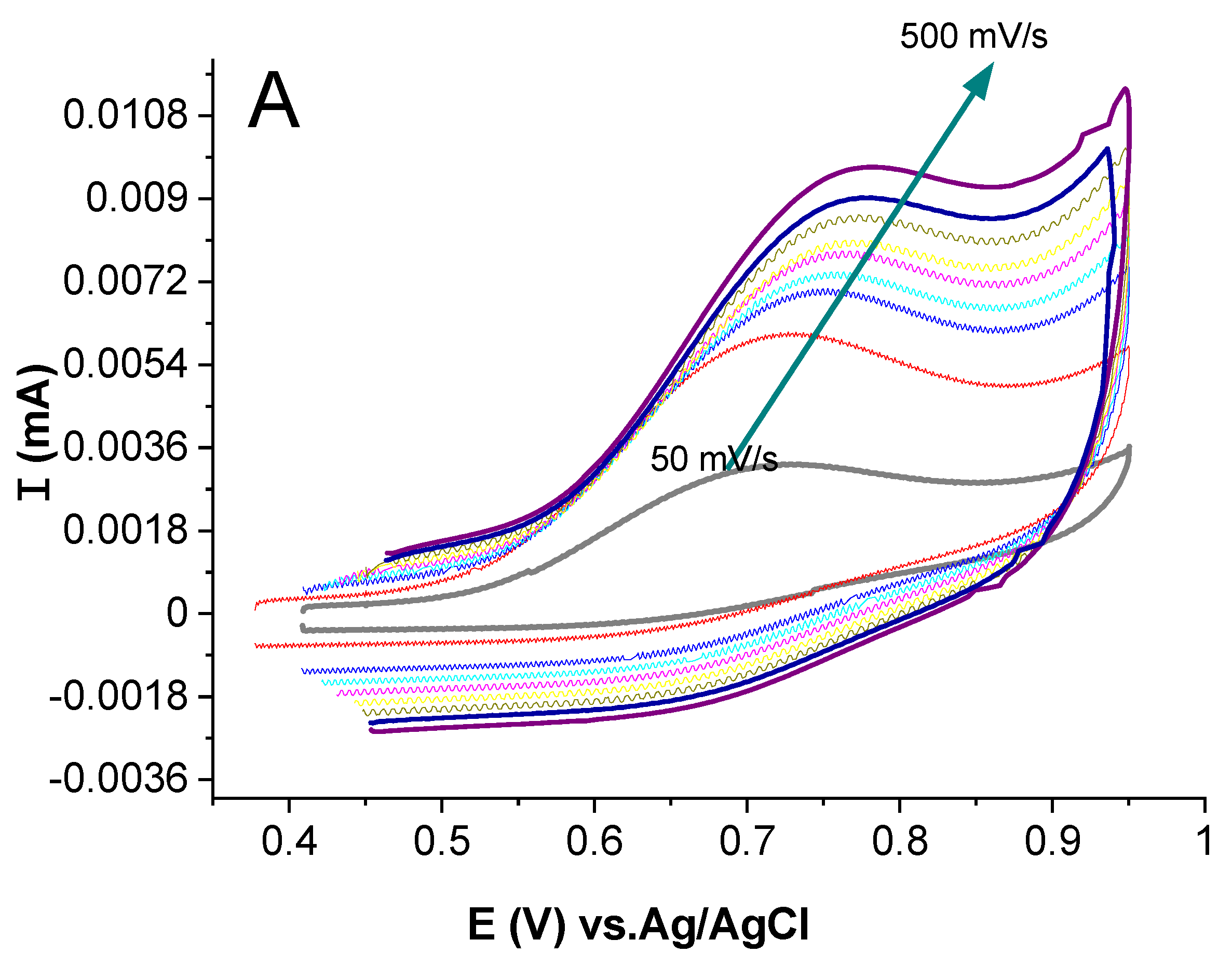
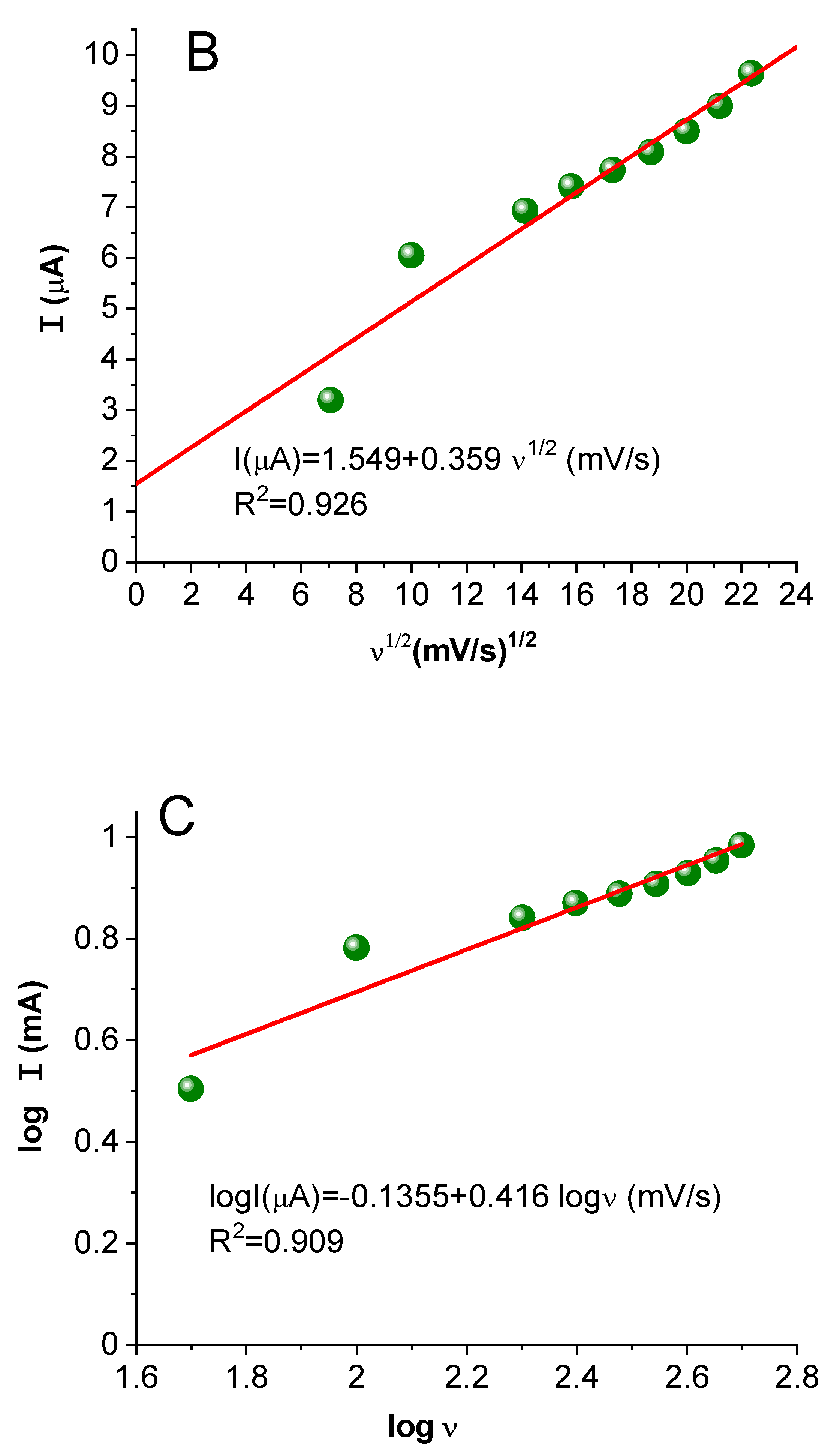
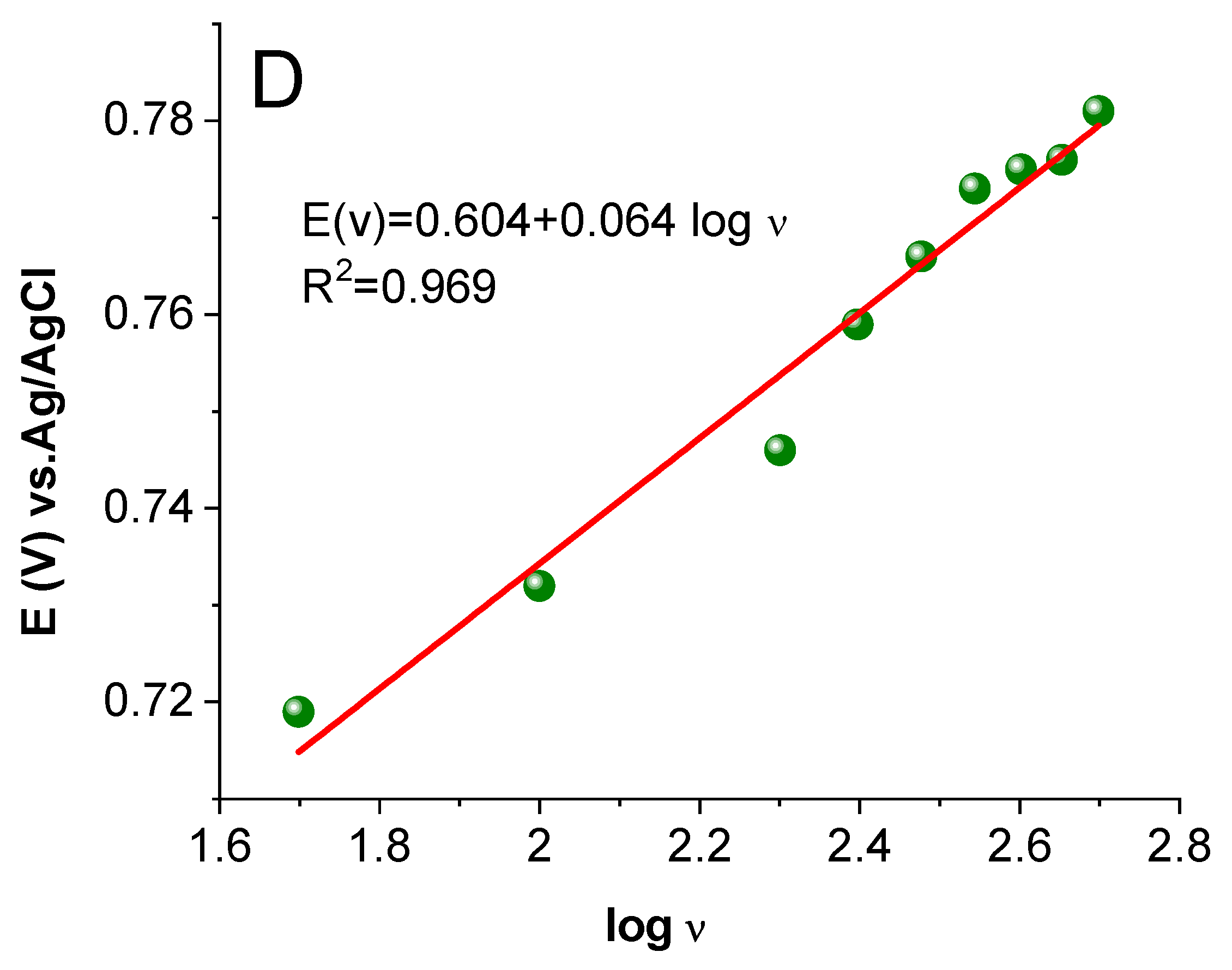
2.3.4. Study of the Effect of Scan Rate Change
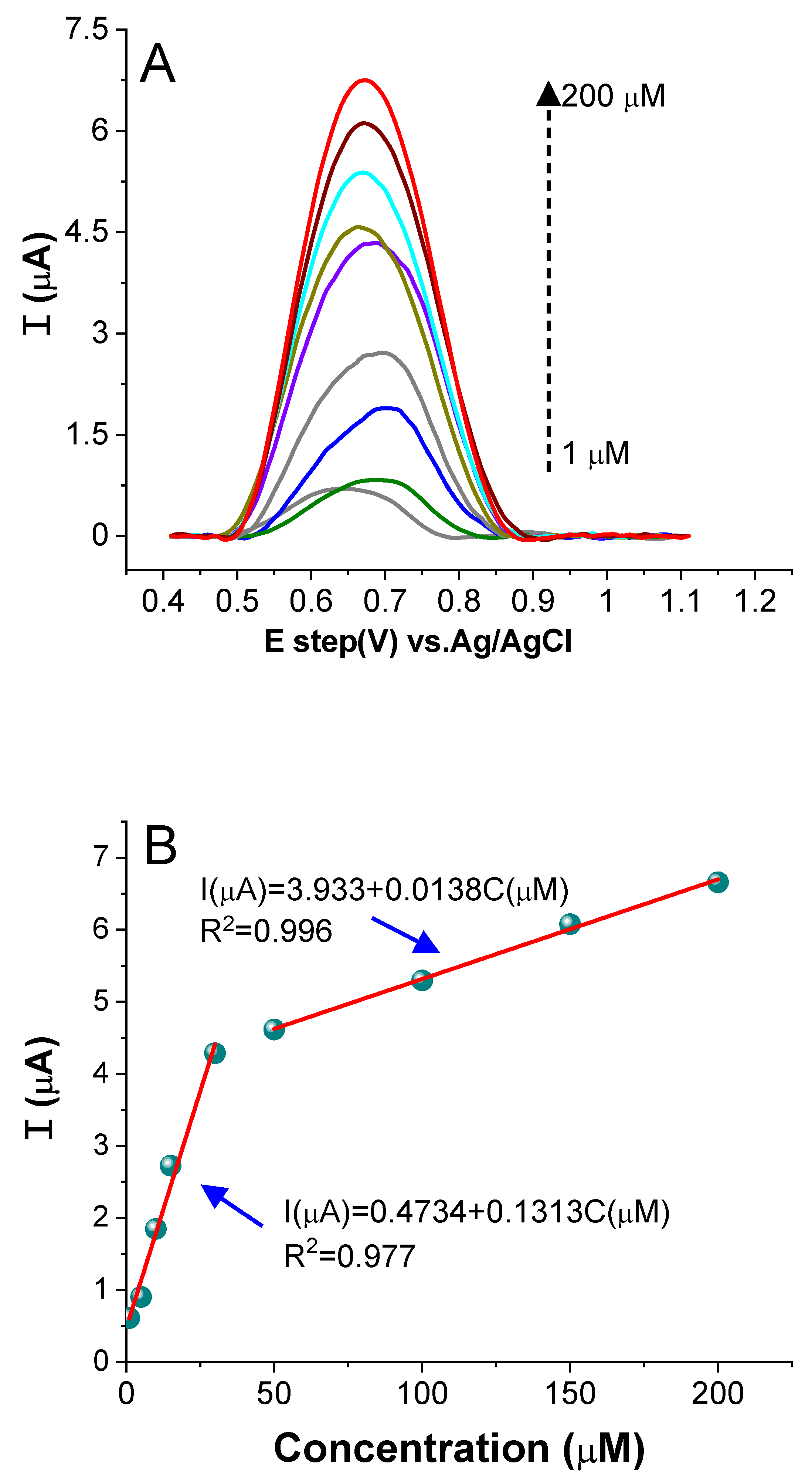
2.3.5. Electrochemical Sensing of CR on the AuNPs/CPE
2.4. Stability, Reproducibility, and Interference Study
2.5. Practical Application of CR Recovery in Real Samples
2.6. Mechanism
3. Experimental Work
3.1. Chemical Reagents
3.2. Instrumentation
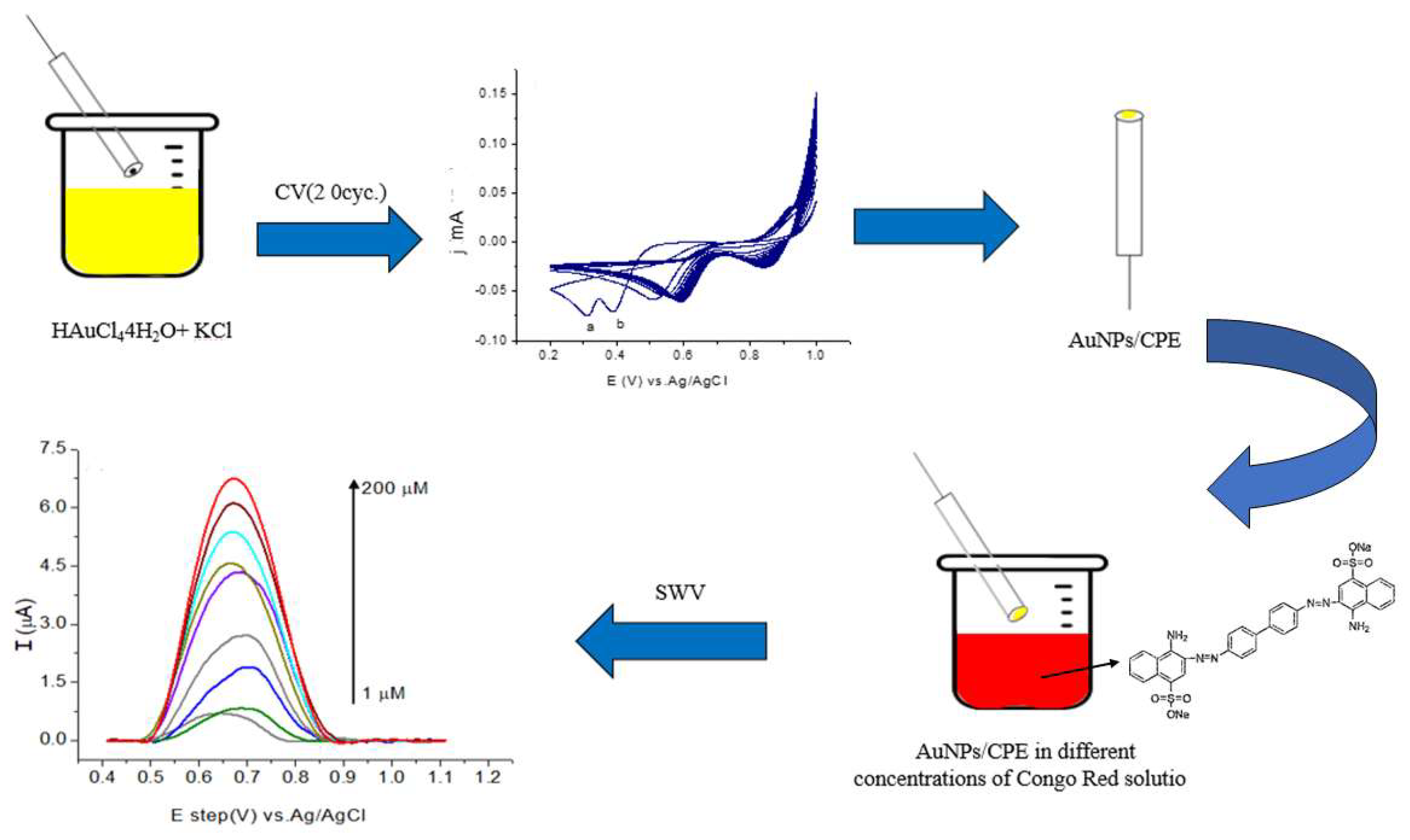
3.3. Preparation of the Modified AuNP/CPE Electrode
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- El Qada, E.N.; Allen, S.J.; Walker, G.M. Adsorption of basic dyes from aqueous solution onto activated carbons. Chem. Eng. J. 2008, 135, 174–184. [Google Scholar] [CrossRef]
- Brüschweiler, B.J.; Merlot, C. Azo dyes in clothing textiles can be cleaved into a series of mutagenic aromatic amines which are not regulated yet. Regul. Toxicol. Pharmacol. 2017, 88, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Banerjee, A.; Halder, U.; Biswas, R.; Bandopadhyay, R. Degradation of Synthetic Azo Dyes of Textile Industry: A Sustainable Approach Using Microbial Enzymes. Water Conserv. Sci. Eng. 2017, 2, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Deng, Q.; Lin, S.; Du, C.; Zhao, S.; Hu, Y.; Yang, Z.; Lyu, Y.; Han, J. A new approach for risk assessment of aggregate dermal exposure to banned azo dyes in textiles. Regul. Toxicol. Pharmacol. 2017, 91, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Kariyajjanavar, P.; Jogttappa, N.; Nayaka, Y.A. Studies on degradation of reactive textile dyes solution by electrochemical method. J. Hazard. Mater. 2011, 190, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Zamora, M.H.; Martínez-Jerónimo, F.; Cristiani-Urbina, E.; Cañizares-Villanueva, R.O. Congo red dye affects survival and reproduction in the cladoceran Ceriodaphnia dubia. Effects of direct and dietary exposure. Ecotoxicology 2016, 25, 1832–1840. [Google Scholar] [CrossRef]
- Hernández-Zamora, M.; Martínez-Jerónimo, F. Congo red dye diversely affects organisms of different trophic levels: A comparative study with microalgae, cladocerans, and zebrafish embryos. Environ. Sci. Pollut. Res. 2019, 26, 11743–11755. [Google Scholar] [CrossRef]
- Liu, L.; Mi, Z.; Wang, J.; Liu, Z.; Feng, F. A label-free fluorescent sensor based on yellow-green emissive carbon quantum dots for ultrasensitive detection of congo red and cellular imaging. Microchem. J. 2021, 168, 106420. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, S.; Wang, G.; Zhao, J.; Guo, Z. A novel bifunctional molecularly imprinted polymer for determination of Congo red in food. RSC Adv. 2015, 5, 22811–22817. [Google Scholar] [CrossRef]
- Morris, B. The components of the Wired Spanning Forest are recurrent. Probab. Theory Relat. Fields 2003, 125, 259–265. [Google Scholar] [CrossRef]
- Sahraei, R.; Farmany, A.; Mortazavi, S.; Noorizadeh, H. Spectrophotometry determination of Congo red in river water samples using nanosilver. Toxicol. Environ. Chem. 2012, 94, 1886–1892. [Google Scholar] [CrossRef]
- Jin, L.-N.; Qian, X.-Y.; Wang, J.-G.; Aslan, H.; Dong, M. MIL-68 (In) nano-rods for the removal of Congo red dye from aqueous solution. J. Colloid Interface Sci. 2015, 453, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Shetti, N.P.; Malode, S.J.; Malladi, R.S.; Nargund, S.L.; Shukla, S.S.; Aminabhavi, T.M. Electrochemical detection and degradation of textile dye Congo red at graphene oxide modified electrode. Microchem. J. 2019, 146, 387–392. [Google Scholar] [CrossRef]
- Ahmad, A.; Senapati, S.; Khan, M.I.; Kumar, R.; Sastry, M. Extracellular Biosynthesis of Monodisperse Gold Nanoparticles by a Novel Extremophilic Actinomycete, Thermomonospora sp. Langmuir 2003, 19, 3550–3553. [Google Scholar] [CrossRef]
- Ahmad, A.; Senapati, S.; Khan, M.I.; Kumar, R.; Sastry, M. Extra-/Intracellular Biosynthesis of Gold Nanoparticles by an Alkalotolerant Fungus, Trichothecium sp. J. Biomed. Nanotechnol. 2005, 1, 47–53. [Google Scholar] [CrossRef]
- Philip, D. Honey mediated green synthesis of silver nanoparticles. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010, 75, 1078–1081. [Google Scholar] [CrossRef]
- Thakkar, K.N.; Mhatre, S.S.; Parikh, R.Y. Biological synthesis of metallic nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 257–262. [Google Scholar] [CrossRef]
- Ganash, A.; Almonshi, R. Effects of types and the ratio of poly(o-phenetidine)/TiO2 nanocomposite as anticorrosive coating. Polym. Bull. 2017, 75, 3859–3881. [Google Scholar] [CrossRef]
- Ganash, A.A.; Aljubairy, R.A. Efficient electrochemical detection of hazardous para-nitrophenol based on a carbon paste electrode modified with green synthesized gold/iron oxide nanocomposite. Chem. Pap. 2022, 76, 3169–3183. [Google Scholar] [CrossRef]
- Ganash, A.A.; Alghamdi, R.A. Fabrication of a novel polyaniline/green-synthesized, silver-nanoparticle-modified carbon paste electrode for electrochemical sensing of lead ions. J. Chin. Chem. Soc. 2021, 68, 2312–2325. [Google Scholar] [CrossRef]
- Haiss, W.; Thanh, N.T.K.; Aveyard, J.; Fernig, D.G. Determination of Size and Concentration of Gold Nanoparticles from UV−Vis Spectra. Anal. Chem. 2007, 79, 4215–4221. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Liao, F.; Molesa, S.; Redinger, D.; Subramanian, V. Plastic-Compatible Low Resistance Printable Gold Nanoparticle Conductors for Flexible Electronics. J. Electrochem. Soc. 2003, 150, G412–G417. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Lee, D.Y. Near-Infrared-Responsive Cancer Photothermal and Photodynamic Therapy Using Gold Nanoparticles. Polymers 2018, 10, 961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, S.; Esterle, A.; Sharma, N.C.; Sahi, S.V. Yucca-derived synthesis of gold nanomaterial and their catalytic potential. Nanoscale Res. Lett. 2014, 9, 627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sneha, K.; Sathishkumar, M.; Kim, S.; Yun, Y.-S. Counter ions and temperature incorporated tailoring of biogenic gold nanoparticles. Process. Biochem. 2010, 45, 1450–1458. [Google Scholar] [CrossRef]
- León, E.R.; Rodríguez-Vázquez, B.E.; Martínez-Higuera, A.; Rodríguez-Beas, C.; Larios-Rodríguez, E.; Navarro, R.E.; López-Esparza, R.; Iñiguez-Palomares, R.A. Synthesis of Gold Nanoparticles Using Mimosa tenuiflora Extract, Assessments of Cytotoxicity, Cellular Uptake, and Catalysis. Nanoscale Res. Lett. 2019, 14, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etesami, M.; Karoonian, F.S.; Mohamed, N. Electrochemical Deposition of Gold Nanoparticles on Pencil Graphite by Fast Scan Cyclic Voltammetry. J. Chin. Chem. Soc. 2011, 58, 688–693. [Google Scholar] [CrossRef]
- Lee, K.X.; Shameli, K.; Miyake, M.; Kuwano, N.; Khairudin, N.B.B.A.; Mohamad, S.E.B.; Yew, Y.P. Green Synthesis of Gold Nanoparticles Using Aqueous Extract of Garcinia mangostana Fruit Peels. J. Nanomater. 2016, 2016, 8489094. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Schwartzberg, A.M.; Xu, K.; Gu, C.; Zhang, J.Z. Electrical and thermal conductivities of gold and silver nanoparticles in solutions and films and electrical field enhanced Surface-Enhanced Raman Scattering (SERS). In Proceedings of SPIE—The International Society for Optical Engineering; Society of Photo-optical Instrumentation Engineers: Bellingham, WA, USA, 2005. [Google Scholar] [CrossRef]
- Adebayo, M.A.; Adebomi, J.I.; Abe, T.O.; Areo, F.I. Removal of aqueous Congo red and malachite green using ackee apple seed–bentonite composite. Colloid Interface Sci. Commun. 2020, 38, 100311. [Google Scholar] [CrossRef]
- Jiang, M.-Q.; Jin, X.-Y.; Lu, X.-Q.; Chen, Z.-L. Adsorption of Pb(II), Cd(II), Ni(II) and Cu(II) onto natural kaolinite clay. Desalination 2010, 252, 33–39. [Google Scholar] [CrossRef]
- Al-Qasmi, N.; Soomro, M.T.; Aslam, M.; Rehman, A.U.; Ali, S.; Danish, E.Y.; Ismail, I.M.; Hameed, A. The efficacy of the ZnO:α-Fe2O3 composites modified carbon paste electrode for the sensitive electrochemical detection of loperamide: A detailed investigation. J. Electroanal. Chem. 2016, 783, 112–124. [Google Scholar] [CrossRef]
- Vieira, S.S.; Magriotis, Z.M.; Santos, N.A.; Cardoso, M.D.G.; Saczk, A.A. Macauba palm (Acrocomia aculeata) cake from biodiesel processing: An efficient and low cost substrate for the adsorption of dyes. Chem. Eng. J. 2011, 183, 152–161. [Google Scholar] [CrossRef]
- Laviron, E. The use of linear potential sweep voltammetry and of a.c. voltammetry for the study of the surface electrochemical reaction of strongly adsorbed systems and of redox modified electrodes. J. Electroanal. Chem. Interfacial Electrochem. 1979, 100, 263–270. [Google Scholar] [CrossRef]
- Rhieu, S.Y.; Reipa, V. Tuning the Size of Gold Nanoparticles with Repetitive Oxidation-reduction Cycles. Am. J. Nanomater. 2015, 3, 15–21. [Google Scholar] [CrossRef]
- El-Deab, M.S.; Ohsaka, T. Hydrodynamic voltammetric studies of the oxygen reduction at gold nanoparticles-electrodeposited gold electrodes. Electrochim. Acta 2002, 47, 4255–4261. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, G. Electrochemical Deposition of Gold Nanoparticles on Reduced Graphene Oxide by Fast Scan Cyclic Voltammetry for the Sensitive Determination of As(III). Nanomaterials 2018, 9, 41. [Google Scholar] [CrossRef] [Green Version]
- Gunawardena, G.; Hills, G.; Montenegro, I.; Scharifker, B. Electrochemical nucleation. J. Electroanal. Chem. Interfacial Electrochem. 1982, 138, 225–239. [Google Scholar] [CrossRef]
- Grujicic, D.; Pesic, B. Electrodeposition of copper: The nucleation mechanisms. Electrochim. Acta 2002, 47, 2901–2912. [Google Scholar] [CrossRef]
- Hezard, T.; Fajerwerg, K.; Evrard, D.; Collière, V.; Behra, P.; Gros, P. Gold nanoparticles electrodeposited on glassy carbon using cyclic voltammetry: Application to Hg(II) trace analysis. J. Electroanal. Chem. 2012, 664, 46–52. [Google Scholar] [CrossRef]
- Finot, M.; Braybrook, G.D.; McDermott, M.T. Characterization of electrochemically deposited gold nanocrystals on glassy carbon electrodes. J. Electroanal. Chem. 1999, 466, 234–241. [Google Scholar] [CrossRef]
- Daneshgar, P.; Norouzi, P.; Ganjali, M.R.; Dinarvand, R.; Moosavi-Movahedi, A.A. Determination of Diclofenac on a Dysprosium Nanowire-Modified Carbon Paste Electrode Accomplished in a Flow Injection System by Advanced Filtering. Sensors 2009, 9, 7903–7918. [Google Scholar] [CrossRef] [PubMed]
- Thanh, N.M.; Luyen, N.D.; Toan, T.T.T.; Phong, N.H.; Van Hop, N. Voltammetry Determination of Pb(II), Cd(II), and Zn(II) at Bismuth Film Electrode Combined with 8-Hydroxyquinoline as a Complexing Agent. J. Anal. Methods Chem. 2019, 2019, 4593135. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Ahmad, S. Recent development in hybrid conducting polymers: Synthesis, applications and future prospects. J. Ind. Eng. Chem. 2018, 60, 53–84. [Google Scholar] [CrossRef]
- Xu, Q.; Chen, L.-L.; Lu, G.-J.; Hu, X.-Y.; Li, H.-B.; Ding, L.; Wang, Y. Electrochemical preparation of poly(bromothymol blue) film and its analytical application. J. Appl. Electrochem. 2010, 41, 143–149. [Google Scholar] [CrossRef]
- Gonçalves, L.M.; Batchelor-McAuley, C.; Barros, A.A.; Compton, R.G. Electrochemical Oxidation of Adenine: A Mixed Adsorption and Diffusion Response on an Edge-Plane Pyrolytic Graphite Electrode. J. Phys. Chem. C 2010, 114, 14213–14219. [Google Scholar] [CrossRef]
- Hasnat, M.; Ahamad, N.; Uddin, S.N.; Mohamed, N. Silver modified platinum surface/H+ conducting Nafion membrane for cathodic reduction of nitrate ions. Appl. Surf. Sci. 2012, 258, 3309–3314. [Google Scholar] [CrossRef]
- Fletcher, S. Tafel slopes from first principles. J. Solid State Electrochem. 2008, 13, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yu, S.; He, W.; Uyama, H.; Xie, Q.; Zhang, L.; Yang, F. Electrochemical sensor based on carbon-supported NiCoO2 nanoparticles for selective detection of ascorbic acid. Biosens. Bioelectron. 2014, 55, 446–451. [Google Scholar] [CrossRef]
- Malode, S.J.; Shetti, N.; Nandibewoor, S.T. Voltammetric behavior of theophylline and its determination at multi-wall carbon nanotube paste electrode. Colloids Surf. B Biointerfaces 2012, 97, 1–6. [Google Scholar] [CrossRef]
- Desimoni, E.; Brunetti, B. Presenting Analytical Performances of Electrochemical Sensors. Some Suggestions. Electroanalysis 2013, 25, 1645–1651. [Google Scholar] [CrossRef]
- Iwunze, M.O. Electrooxidation of congo red at glassy carbon electrode in aqueous solution. Res. Rev. Electrochem. 2010, 2, 19–24. [Google Scholar]
- Valera, D.; Sánchez, M.; Domínguez, J.R.; Alvarado, J.; Espinoza-Montero, P.J.; Carrera, P.; Bonilla, P.; Manciati, C.; González, G.; Fernández, L. Electrochemical determination of lead in human blood serum and urine by anodic stripping voltammetry using glassy carbon electrodes covered with Ag–Hg and Ag–Bi bimetallic nanoparticles. Anal. Methods 2018, 10, 4114–4121. [Google Scholar] [CrossRef]
- Tufa, L.T.; Siraj, K.; Soreta, T.R. Electrochemical determination of lead using bismuth modified glassy carbon electrode. Russ. J. Electrochem. 2013, 49, 59–66. [Google Scholar] [CrossRef]
- Vu, H.D.; Nguyen, L.H.; Nguyen, T.D.; Nguyen, H.B.; Nguyen, T.L.; Tran, D.L. Anodic stripping voltammetric determination of Cd2+ and Pb2+ using interpenetrated MWCNT/P1,5-DAN as an enhanced sensing interface. Ionics 2014, 21, 571–578. [Google Scholar] [CrossRef]
- Fichorova, R.N.; Richardson-Harman, N.; Alfano, M.; Belec, L.; Carbonneil, C.; Chen, S.; Cosentino, L.; Curtis, K.; Dezzutti, C.S.; Donoval, B.; et al. Biological and Technical Variables Affecting Immunoassay Recovery of Cytokines from Human Serum and Simulated Vaginal Fluid: A Multicenter Study. Anal. Chem. 2008, 80, 4741–4751. [Google Scholar] [CrossRef]
- Ilieva, D.; Surleva, A.; Murariu, M.; Drochioiu, G.; Abdullah, M.M.A.B. Evaluation of ICP-OES Method for Heavy Metal and Metalloids Determination in Sterile Dump Material. Solid State Phenom. 2018, 273, 159–166. [Google Scholar] [CrossRef]
- Bonancêa, C.E.; Nascimento, G.D.; de Souza, M.; Temperini, M.L.A.; Corio, P. Substrate development for surface-enhanced Raman study of photocatalytic degradation processes: Congo red over silver modified titanium dioxide films. Appl. Catal. B Environ. 2006, 69, 34–42. [Google Scholar] [CrossRef]
- Tomkiewicz, M.; Klein, M.P. Photochemically induced dynamic nuclear polarization in hydrosol suspensions of Congo Red. J. Am. Chem. Soc. 1973, 95, 3132–3136. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Linear Range (µM) | LOD (µM) | Ref. No. |
|---|---|---|---|
| Electro-oxidation at GCE | ----- | 1.07 | [52] |
| DPV at GO/GCE | 0.01–0.2 | 0.24 | [13] |
| UV-Visible absorption spectra | 1.15–345 | 0.86 | [11] |
| SPMIP-HPLC | ----- | 0.1 × 10−3 | [9] |
| Fluorescent sensor | 0.72–71.8 71.8–244 | 0.05 0.04 | [8] |
| SWV at AuNPs/CPE | 1–30 50–200 | 0.07 0.70 | This work |
| Sample | Cadd [μM] | Cfound [μM] | Recovery [%] |
|---|---|---|---|
| Jelley | 30 | 27.08 | 90.27 |
| 200 | 198.12 | 99.06 | |
| Wastewater | 30 | 30.99 | 103.29 |
| 200 | 212.39 | 106.19 | |
| Soil | 30 | 28.32 | 94.41 |
| 200 | 211.59 | 105.79 | |
| Tap water | 30 | 28.29 | 94.33 |
| 200 | 180.29 | 90.15 | |
| Candy | 30 | 26.53 | 88.44 |
| 200 | 212.68 | 106.34 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganash, A.; Alshammari, S.; Ganash, E. Development of a Novel Electrochemical Sensor Based on Gold Nanoparticle-Modified Carbon-Paste Electrode for the Detection of Congo Red Dye. Molecules 2023, 28, 19. https://doi.org/10.3390/molecules28010019
Ganash A, Alshammari S, Ganash E. Development of a Novel Electrochemical Sensor Based on Gold Nanoparticle-Modified Carbon-Paste Electrode for the Detection of Congo Red Dye. Molecules. 2023; 28(1):19. https://doi.org/10.3390/molecules28010019
Chicago/Turabian StyleGanash, Aisha, Sahar Alshammari, and Entesar Ganash. 2023. "Development of a Novel Electrochemical Sensor Based on Gold Nanoparticle-Modified Carbon-Paste Electrode for the Detection of Congo Red Dye" Molecules 28, no. 1: 19. https://doi.org/10.3390/molecules28010019
APA StyleGanash, A., Alshammari, S., & Ganash, E. (2023). Development of a Novel Electrochemical Sensor Based on Gold Nanoparticle-Modified Carbon-Paste Electrode for the Detection of Congo Red Dye. Molecules, 28(1), 19. https://doi.org/10.3390/molecules28010019