

Novel Route to Cationic Palladium(II)–Cyclopentadienyl Complexes Containing Phosphine Ligands and Their Catalytic Activities

 , , , , , , ,
, , , , , , ,  and
and 
Abstract
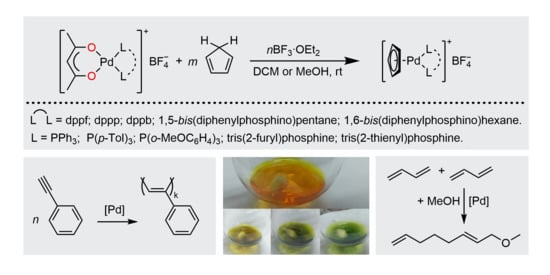
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Palladium(II) Complexes
2.2. X-ray Crystal Structures of 1, 7, and 8

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length of Intermolecular Contact in Å * | ρ(r) | ∇2ρ(r) | λ2 | Hb | V(r) | G(r) | Eint ** |
|---|---|---|---|---|---|---|---|
| 1 | |||||||
| 2.686 | 0.008 | 0.030 | −0.008 | 0.002 | −0.004 | 0.006 | 1.3 |
| 2.881 | 0.006 | 0.020 | −0.006 | 0.001 | −0.003 | 0.004 | 0.9 |
| 2.799 | 0.006 | 0.018 | −0.006 | 0.001 | −0.002 | 0.003 | 0.6 |
| 2.961 | 0.004 | 0.015 | −0.004 | 0.001 | −0.002 | 0.003 | 0.6 |
| 3.322 | 0.002 | 0.008 | −0.002 | 0.000 | −0.001 | 0.001 | 0.3 |
| 2.986 | 0.004 | 0.016 | −0.004 | 0.001 | −0.002 | 0.003 | 0.6 |
| 3.053 | 0.004 | 0.015 | −0.004 | 0.001 | −0.002 | 0.003 | 0.6 |
| 7 | |||||||
| 3.034 | 0.004 | 0.013 | −0.004 | 0.001 | −0.001 | 0.002 | 0.3 |
| 2.966 | 0.004 | 0.015 | −0.004 | 0.001 | −0.002 | 0.003 | 0.6 |
| 3.077 | 0.004 | 0.013 | −0.004 | 0.001 | −0.001 | 0.002 | 0.3 |
| 3.034 | 0.004 | 0.013 | −0.004 | 0.001 | −0.001 | 0.002 | 0.3 |
| 3.077 | 0.004 | 0.013 | −0.004 | 0.001 | −0.001 | 0.002 | 0.3 |
| 3.028 | 0.005 | 0.017 | −0.005 | 0.001 | −0.002 | 0.003 | 0.6 |
| 8 | |||||||
| 2.778 | 0.006 | 0.023 | −0.006 | 0.001 | −0.003 | 0.004 | 0.9 |
| 2.877 | 0.006 | 0.019 | −0.006 | 0.001 | −0.002 | 0.003 | 0.6 |
| 2.662 | 0.009 | 0.031 | −0.009 | 0.001 | −0.005 | 0.006 | 1.6 |
| 2.952 | 0.005 | 0.019 | −0.005 | 0.002 | −0.002 | 0.004 | 0.6 |
| 3.043 | 0.003 | 0.011 | −0.003 | 0.001 | −0.001 | 0.002 | 0.3 |
2.3. Catalytic Studies
3. Materials and Methods
3.1. General Procedures and Materials
3.2. General Procedure for 1,3-Butadiene Telomerization
3.3. General Procedure for Phenylacetylene Polymerization
3.4. Synthesis of Palladium Complexes
3.4.1. Preparation of (η5-Cyclopentadienyl)bis(triphenylphosphine-κP)palladium(II) Tetrafluoroborate, [Pd(η5-C5H5)(PPh3)2]BF4 (1)
3.4.2. Preparation of (η5-Cyclopentadienyl)bis[tris(4-methylphenyl)phosphine-κP]palladium(II) Tetrafluoroborate, [Pd(η5-C5H5)(P(p-MeC6H4)3)2]BF4 (2)
Preparation of 2 in CH2Cl2 as Solvent
Preparation of 2 in MeOH as Solvent
3.4.3. Preparation of (η5-Cyclopentadienyl)bis[tris(2-methoxyphenyl)phosphine-κP]palladium(II) Tetrafluoroborate, [Pd(η5-C5H5)(TOMPP)2]BF4 (TOMPP = Tris(2-methoxyphenyl)phosphine) (3)
3.4.4. Preparation of (η5-Cyclopentadienyl)bis[tris(2-furyl)phosphine-κP]palladium(II) Tetrafluoroborate, [Pd(η5-C5H5)(TFP)2]BF4 (TFP = Tris(2-furyl)phosphine) (4)
3.4.5. Preparation of (η5-Cyclopentadienyl)bis[tris(2-thienyl)phosphine-κP]palladium(II) Tetrafluoroborate, [Pd(η5-C5H5)(TTP)2]BF4 (TTP = Tris(2-thienyl)phosphine) (5)
3.4.6. Preparation of (η5-Cyclopentadienyl)(1,1′-bis(diphenylphosphino)ferrocene-κ2P,P′)palladium(II) Tetrafluoroborate, [Pd(η5-C5H5)(dppf)]BF4 (Dppf = 1,1′-Bis(diphenylphosphino)ferrocene) (6)
3.4.7. Preparation of (η5-Cyclopentadienyl)(1,3-bis(diphenylphosphino)propane-κ2P,P′)palladium(II) Tetrafluoroborate, [Pd(η5-C5H5)(Dppp)]BF4 (Dppp = 1,3-Bis(diphenylphosphino)propane) (7)
3.4.8. Preparation of (η5-Cyclopentadienyl)(1,4-bis(diphenylphosphino)butane-κ2P,P′)palladium(II) Tetrafluoroborate, [Pd(η5-C5H5)(Dppb)]BF4 (Dppb = 1,4-Bis(diphenylphosphino)butane) (8)
3.4.9. Preparation of (η5-Cyclopentadienyl)(1,5-bis(diphenylphosphino)pentane-κ2P,P′)palladium(II) Tetrafluoroborate, [Pd(η5-C5H5)(Dpppt)]BF4 (Dpppt = 1,5-Bis(diphenylphosphino)pentane) (9)
3.4.10. Preparation of {[Pd(η5-C5H5)(Dpphx)]BF4}n (Dpphx = 1,6-Bis(diphenylphosphino)hexane, n = 2, 3) (10) (Mixture of di-μ-(1,6-Bis(diphenylphosphino)hexane-κ2P,P′)-bis[(η5-cyclopentadienyl)palladium(II)] Bis(tetrafluoroborate) and Tri-μ-(1,6-Bis(diphenylphosphino)hexane-κ2P,P′)-tris[(η5-cyclopentadienyl)palladium(II)] Tris(tetrafluoroborate))
3.5. Reaction of [Pd(κ2-O,O′-Acac)(TOMPP)2]BF4 with BF3∙OEt2
3.6. X-ray Crystallographic Studies
3.7. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pfab, W.; Fischer, E.O. Zur Kristallstruktur Der Di-Cyclopentadienyl-Verbindungen Des Zweiwertigen Eisens, Kobalts Und Nickels. Z. Anorg. Allg. Chem. 1953, 274, 316–322. [Google Scholar] [CrossRef]
- Wilkinson, G.; Rosenblum, M.; Whiting, M.C.; Woodward, R.B. The Structure of Iron Bis-Cyclopentadienyl. J. Am. Chem. Soc. 1952, 74, 2125–2126. [Google Scholar] [CrossRef]
- Trost, B.M.; Ryan, M.C. Indenylmetal Catalysis in Organic Synthesis. Angew. Chem. Int. Ed. 2017, 56, 2862–2879. [Google Scholar] [CrossRef] [PubMed]
- Kharitonov, V.B.; Muratov, D.V.; Loginov, D.A. Cyclopentadienyl Complexes of Group 9 Metals in the Total Synthesis of Natural Products. Coord. Chem. Rev. 2022, 471, 214744. [Google Scholar] [CrossRef]
- Yu, H.; Xu, Y.; Havener, K.; Zhang, M.; Zhang, L.; Wu, W.; Huang, K. Temperature-Controlled Selectivity of Hydrogenation and Hydrodeoxygenation of Biomass by Superhydrophilic Nitrogen/Oxygen Co-Doped Porous Carbon Nanosphere Supported Pd Nanoparticles. Small 2022, 18, 2106893. [Google Scholar] [CrossRef]
- Yu, H.; Xu, Y.; Zhang, M.; Zhang, L.; Wu, W.; Huang, K. Magnetically-Separable Cobalt Catalyst Embedded in Metal Nitrate-Promoted Hierarchically Porous N-Doped Carbon Nanospheres for Hydrodeoxygenation of Lignin-Derived Species. Fuel 2023, 331, 125917. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, L.; Gao, S.; Wang, H.; He, Z.; Xu, Y.; Huang, K. In Situ Encapsulated Ultrafine Pd Nanoparticles in Nitrogen-Doped Porous Carbon Derived from Hyper-Crosslinked Polymers Effectively Catalyse Hydrogenation. J. Catal. 2021, 396, 342–350. [Google Scholar] [CrossRef]
- Van Leeuwen, P.W.N.M.; Chadwick, J.C. Homogeneous Catalysts: Activity, Stability, Deactivation; Wiley-VCH: Weinheim, Germany, 2011; ISBN 978-3-527-32329-6. [Google Scholar]
- Kaminsky, W. Discovery of Methylaluminoxane as Cocatalyst for Olefin Polymerization. Macromolecules 2012, 45, 3289–3297. [Google Scholar] [CrossRef]
- Kaminsky, W. Polyolefins: 50 Years after Ziegler and Natta II; Kaminsky, W., Ed.; Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2013; Volume 258, ISBN 978-3-642-40804-5. [Google Scholar]
- Alt, H.G.; Köppl, A. Effect of the Nature of Metallocene Complexes of Group IV Metals on Their Performance in Catalytic Ethylene and Propylene Polymerization. Chem. Rev. 2000, 100, 1205–1222. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, X.; Wang, Y.; Liu, T.; Li, H.; Zhang, Y.; Wang, L.; Wang, X.; Zhao, S.; Luo, Y. Ancillary Ligand Effects on α-Olefin Polymerization Catalyzed by Zirconium Metallocene: A Computational Study. RSC Adv. 2022, 12, 21111–21121. [Google Scholar] [CrossRef]
- Poli, R. Monocyclopentadienyl Halide Complexes of the D- and f-Block Elements. Chem. Rev. 1991, 91, 509–551. [Google Scholar] [CrossRef]
- Bielinski, E.A.; Dai, W.; Guard, L.M.; Hazari, N.; Takase, M.K. Synthesis, Properties, and Reactivity of Palladium and Nickel NHC Complexes Supported by Combinations of Allyl, Cyclopentadienyl, and Indenyl Ligands. Organometallics 2013, 32, 4025–4037. [Google Scholar] [CrossRef]
- Butler, I.R. Transition Metal Complexes of Cyclopentadienyl Ligands. In Organometallic Chemistry; Royal Society of Chemistry: Cambridge, UK, 2001; pp. 442–478. [Google Scholar]
- Chalkley, M.J.; Guard, L.M.; Hazari, N.; Hofmann, P.; Hruszkewycz, D.P.; Schmeier, T.J.; Takase, M.K. Synthesis, Electronic Structure, and Reactivity of Palladium(I) Dimers with Bridging Allyl, Cyclopentadienyl, and Indenyl Ligands. Organometallics 2013, 32, 4223–4238. [Google Scholar] [CrossRef]
- Cross, R.J.; Wardle, R. Cyclopentadienyls of Palladium and Platinum. J. Chem. Soc. A Inorg. Phys. Theor. 1971, 2000–2007. [Google Scholar] [CrossRef]
- Felkin, H.; Kevin Turner, G. Dimeric Palladium (I) Complexes Containing the Bridging Cyclopentadienyl Group. J. Organomet. Chem. 1977, 129, 429–436. [Google Scholar] [CrossRef]
- Grushin, V.V.; Bensimon, C.; Alper, H. Condensation of Cyclopentadiene with Bridging Hydroxo Organopalladium and -Platinum Dimers: A Novel Simple Entry to.Eta.5-Cyclopentadienyl Complexes of Palladium and Platinum. Organometallics 1993, 12, 2737–2740. [Google Scholar] [CrossRef]
- Gubin, S.P.; Rubezhov, A.Z.; Winch, B.L.; Nesmeyanov, A.N. Cleavage of the C5H5-Palladium Bond in Cyclopentadienylallylpalladium. Tetrahedron Lett. 1964, 5, 2881–2887. [Google Scholar] [CrossRef]
- McClellan, W.R.; Hoehn, H.H.; Cripps, H.N.; Muetterties, E.L.; Howk, B.W. π-Allyl Derivatives of Transition Metals. J. Am. Chem. Soc. 1961, 83, 1601–1607. [Google Scholar] [CrossRef]
- Shaw, B.L. Allyl(Cyclopentadienyl)Palladium(II). Proc. Chem. Soc. 1960, 7, 247. [Google Scholar] [CrossRef]
- Smidt, J.; Jira, R. Verbindung des Cyclopentadiens mit Palladium. Angew. Chem. 1959, 71, 651. [Google Scholar] [CrossRef]
- Werner, H.; Kraus, H.-J.; Schubert, U.; Ackermann, K.; Hofmann, P. Strukturdynamische organometall-komplexe. J. Organomet. Chem. 1983, 250, 517–536. [Google Scholar] [CrossRef]
- Torregrosa, R.R.P. (η3-Allyl)(η5-Cyclopentadienyl)Palladium. In Encyclopedia of Reagents for Organic Synthesis; Charette, A., Bode, J., Rovis, T., Shenvi, R., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2013; p. rn01566. ISBN 978-0-471-93623-7. [Google Scholar]
- Bachechi, F.; Lehmann, R.; Venanzi, L.M. Crystal and Molecular Structure of [Pd(η5-C5H5)(Bis-1,2-Diphenylphosphinoethane)][CF3SO3]. J. Crystallogr. Spectrosc. Res. 1988, 18, 721–728. [Google Scholar] [CrossRef]
- Cross, R.J.; Hoyle, R.W.; Kennedy, A.R.; Manojlović-Muir, L.; Muir, K.W. Distortion of Cyclopentadienyl Rings in η5-Cyclopentadienyl-Palladium Complexes: Crystal Structures of [Pd(C5H5)Cl(PMe2Ph)] and [Pd(C5H5)(Ph2PCH2CH2PPh2)][PF6]. J. Organomet. Chem. 1994, 468, 265–271. [Google Scholar] [CrossRef]
- Fallis, S.; Rodriguez, L.; Anderson, G.K.; Rath, N.P. Synthesis and Reactions of Cationic Palladium and Platinum Cyclopentadienyl Complexes. Molecular Structure of (H5-Cyclopentadienyl)[1, 2-Bis-(Diphenylphosphino)Ethane]Platinum(II) Triflate. Organometallics 1993, 12, 3851–3855. [Google Scholar] [CrossRef]
- Gusev, O.V.; Morozova, L.N.; Peganova, T.A.; Petrovskii, P.V.; Ustynyuk, N.A.; Maitlis, P.M. Synthesis of η5-1,2,3,4,5-Pentamethylcyclopentadienyl-Platinum Complexes. J. Organomet. Chem. 1994, 472, 359–363. [Google Scholar] [CrossRef]
- Gusev, O.V.; Morozova, L.N.; Peterleitner, M.G.; Peregudova, S.M.; Petrovskii, P.V.; Ustynyuk, N.A.; Maitlis, P.M. Synthesis of Palladium Cyclopentadienyl Complexes. Decamethylpalladocene Dication [Pd(η5-C5Me5)]2+. J. Organomet. Chem. 1996, 509, 95–99. [Google Scholar] [CrossRef]
- Johnson, B.F.G.; Lewis, J.; White, D.A. Reactions of Co-Ordinated Ligands. Part V. Reactions of Triphenylmethyl Tetrafluoroborate and Fluoroboric Acid with a Variety of Enyl Metal Complexes. J. Chem. Soc. A Inorg. Phys. Theor. 1970, 1738–1745. [Google Scholar] [CrossRef]
- Kurosawa, H.; Majima, T.; Asada, N. Synthesis, Structures, Stabilities, and Reactions of Cationic Olefin Complexes of Palladium(II) Containing the.Eta.5-Cyclopentadienyl Ligand. J. Am. Chem. Soc. 1980, 102, 6996–7003. [Google Scholar] [CrossRef]
- Maitlis, P.M.; Efraty, A.; Games, M.L. Cyclobutadiene-Metal Complexes. J. Organomet. Chem. 1964, 2, 284–286. [Google Scholar] [CrossRef]
- Manojlović-Muir, L.; Cross, R.J.; Hoyle, R.W. Structure of (η5-Cyclopentadienyl)Bis(Dimethylphenylphosphine)Palladium(II) Perchlorate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1993, 49, 1603–1606. [Google Scholar] [CrossRef]
- Roberts, N.K.; Skelton, B.W.; White, A.H.; Wild, S.B. Cationic η5-Cyclopentadienylpalladium(II Complexes. Compounds of Type [Pd(η5-C5H5)L2]PF6 Containing Tertiary Phosphines, Arsines, and Stibines. Crystal and Molecular Structure of [Pd(η5-C5H5)(SbPh3)2]PF6 ·CH2Cl2. J. Chem. Soc. Dalton Trans. 1982, 12, 2093–2097. [Google Scholar] [CrossRef]
- White, D.A. Cationic Complexes of Cycloocta-1,5-Diene with Palladium(II) and Platjnum(II). Synth. React. Inorg. Met.-Org. Chem. 1971, 1, 133–139. [Google Scholar] [CrossRef]
- Pike, R.D. Thallium: Organometallic Chemistry—Based in Part on the Article Thallium: Organometallic Chemistry by William S. Rees, Jr. & Gertrud Kräuter Which Appeared in the Encyclopedia of Inorganic Chemistry, First Edition. In Encyclopedia of Inorganic and Bioinorganic Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2011. [Google Scholar]
- Suslov, D.S.; Bykov, M.V.; Abramov, P.A.; Pahomova, M.V.; Ushakov, I.A.; Voronov, V.K.; Tkach, V.S. [Pd(Acac)(MeCN)2]BF4: Air-Tolerant, Activator-Free Catalyst for Alkene Dimerization and Polymerization. RSC Adv. 2015, 5, 104467–104471. [Google Scholar] [CrossRef]
- Guibert, I.; Neibecker, D.; Tkatchenko, I. Stoicheiometric Dimerisation of Methyl Acrylate Mediated by Pd(Acac)2·HBF4 Systems and Its Relevance to the Mechanism of Catalytic Dimerisation of Acrylates (Hacac = MeCOCH2COMe). J. Chem. Soc. Chem. Commun. 1989, 1850–1852. [Google Scholar] [CrossRef]
- Bermeshev, M.V.; Chapala, P.P. Addition Polymerization of Functionalized Norbornenes as a Powerful Tool for Assembling Molecular Moieties of New Polymers with Versatile Properties. Prog. Polym. Sci. 2018, 84, 1–46. [Google Scholar] [CrossRef]
- Mehler, C.; Risse, W. Addition Polymerization of Norbornene Catalyzed by Palladium(2+) Compounds. A Polymerization Reaction with Rare Chain Transfer and Chain Termination. Macromolecules 1992, 25, 4226–4228. [Google Scholar] [CrossRef]
- Mehler, C.; Risse, W. Pd(II)-Catalyzed Polymerization of Norbornene Derivatives. Die Makromol. Chem. Rapid Commun. 1992, 13, 455–459. [Google Scholar] [CrossRef]
- Hausoul, P.J.C. Palladium-Catalyzed Telomerization of 1,3-Butadiene with Biomass-Based Oxygenates. Ph.D. Thesis, Utrecht University Repository, Utrecht, The Netherlands, 2013. [Google Scholar]
- Allman, T.; Goel, R.G. The Basicity of Phosphines. Can. J. Chem. 1982, 60, 716–722. [Google Scholar] [CrossRef]
- Hirsivaara, L.; Guerricabeitia, L.; Haukka, M.; Suomalainen, P.; Laitinen, R.H.; Pakkanen, T.A.; Pursiainen, J. M(CO)6 (M=Cr, Mo, W) Derivatives of (o-Anisyl)Diphenylphosphine, Bis(o-Anisyl)Phenylphosphine Tris(o-Anisyl)Phosphine and (p-Anisyl)Bis(o-Anisyl)Phosphine. Inorg. Chim. Acta 2000, 307, 48–57. [Google Scholar] [CrossRef]
- Amatore, C.; Jutand, A.; Meyer, G.; Atmani, H.; Khalil, F.; Chahdi, F.O. Comparative Reactivity of Palladium(0) Complexes Generated in Situ in Mixtures of Triphenylphosphine or Tri-2-Furylphosphine and Pd(Dba)2. Organometallics 1998, 17, 2958–2964. [Google Scholar] [CrossRef]
- Monkowius, U.; Noga, S.; Schmidbaur, H. Ligand Properties of Tri(2-Thienyl)- and Tri(2-Furyl)Phosphine and -Arsine (2-C4H3E)3P/As (E = O, S) in Gold(I) Complexes Uwe. Z. Nat. 2003, 58b, 751–758. [Google Scholar] [CrossRef]
- Nataro, C.; Fosbenner, S.M. Synthesis and Characterization of Transition-Metal Complexes Containing 1,1′-Bis(Diphenylphosphino)Ferrocene. J. Chem. Educ. 2009, 86, 1412. [Google Scholar] [CrossRef]
- Dean, P.A.W. Nuclear Magnetic Resonance Studies of the Solvation of Phosphorus(V) Selenides, 1,2-Bis(Diphenylphosphino)Ethane, and Tris(Dimethylamino)Phosphine Telluride by Sulfur Dioxide. Can. J. Chem. 1979, 57, 754–761. [Google Scholar] [CrossRef]
- Suslov, D.S.; Bykov, M.V.; Pakhomova, M.V.; Abramov, Z.D.; Ratovskii, G.V.; Ushakov, I.A.; Borodina, T.N.; Smirnov, V.I.; Tkach, V.S. [Pd(Acac)(PR3)(PhCN)][BF4] and [Pd(Acac)(S)2][BF4] (R = Phenyl, 2-Methoxyphenyl; S = Benzonitrile, Pyridine): Synthesis, Characterization, Reactivity and Catalytic Behavior. Crystal Structure of Pd(κ2-O,O′-Acac)(κ1-C-Acac)(P(2-MeOC6H4)3). J. Mol. Struct. 2020, 1217, 128425. [Google Scholar] [CrossRef]
- Suslov, D.S.; Bykov, M.V.; Abramov, Z.D.; Ushakov, I.A.; Borodina, T.N.; Smirnov, V.I.; Ratovskii, G.V.; Tkach, V.S. Cationic Palladium(II)–Acetylacetonate Complexes Containing Phosphine and Aminophosphine Ligands and Their Catalytic Activities in Telomerization of 1,3-Butadiene with Methanol. J. Organomet. Chem. 2020, 923, 121413. [Google Scholar] [CrossRef]
- Suslov, D.S.; Bykov, M.V.; Pakhomova, M.V.; Abramov, P.A.; Ushakov, I.A.; Tkach, V.S. Cationic Acetylacetonate Palladium Complexes/Boron Trifluoride Etherate Catalyst Systems for Hydroamination of Vinylarenes Using Arylamines. Catal. Commun. 2017, 94, 69–72. [Google Scholar] [CrossRef]
- Suslov, D.S.; Bykov, M.V.; Belova, M.V.; Abramov, P.A.; Tkach, V.S. Palladium(II)–Acetylacetonate Complexes Containing Phosphine and Diphosphine Ligands and Their Catalytic Activities in Telomerization of 1,3-Dienes with Diethylamine. J. Organomet. Chem. 2014, 752, 37–43. [Google Scholar] [CrossRef]
- Tkach, V.S.; Suslov, D.S.; Myagmarsuren, G.; Ratovskii, G.V.; Rohin, A.V.; Tuczek, F.; Shmidt, F.K. An Effective Route for the Synthesis of Cationic Palladium Complexes of General Formula [(Acac)PdL1L2]+A−. J. Organomet. Chem. 2008, 693, 2069–2073. [Google Scholar] [CrossRef]
- Hausoul, P.J.C.; Parvulescu, A.N.; Lutz, M.; Spek, A.L.; Bruijnincx, P.C.A.; Klein Gebbink, R.J.M.; Weckhuysen, B.M. Mechanistic Study of the Pd/TOMPP-Catalyzed Telomerization of 1,3-Butadiene with Biomass-Based Alcohols: On the Reversibility of Phosphine Alkylation. ChemCatChem 2011, 3, 845–852. [Google Scholar] [CrossRef]
- De Pater, J.J.M.; Tromp, D.S.; Tooke, D.M.; Spek, A.L.; Deelman, B.-J.; van Koten, G.; Elsevier, C.J. Palladium(0)-Alkene Bis(Triarylphosphine) Complexes as Catalyst Precursors for the Methoxycarbonylation of Styrene. Organometallics 2005, 24, 6411–6419. [Google Scholar] [CrossRef]
- Loos, M.; Gerber, C.; Corona, F.; Hollender, J.; Singer, H. Accelerated Isotope Fine Structure Calculation Using Pruned Transition Trees. Anal. Chem. 2015, 87, 5738–5744. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Katkova, S.A.; Mikherdov, A.S.; Kinzhalov, M.A.; Novikov, A.S.; Zolotarev, A.A.; Boyarskiy, V.P.; Kukushkin, V.Y. (Isocyano Group π-Hole)···[d-MII] Interactions of (Isocyanide)[MII] Complexes, in Which Positively Charged Metal Centers (d8-M = Pt, Pd) Act as Nucleophiles. Chem. Eur. J. 2019, 25, 8590–8598. [Google Scholar] [CrossRef]
- Abramov, P.A.; Novikov, A.S.; Sokolov, M.N. Interactions of Aromatic Rings in the Crystal Structures of Hybrid Polyoxometalates and Ru Clusters. CrystEngComm 2021, 23, 6409–6417. [Google Scholar] [CrossRef]
- Baykov, S.V.; Filimonov, S.I.; Rozhkov, A.V.; Novikov, A.S.; Ananyev, I.V.; Ivanov, D.M.; Kukushkin, V.Y. Reverse Sandwich Structures from Interplay between Lone Pair−π-Hole Atom-Directed C⋯dz2[M] and Halogen Bond Interactions. Cryst. Growth Des. 2020, 20, 995–1008. [Google Scholar] [CrossRef]
- Baykov, S.V.; Mikherdov, A.S.; Novikov, A.S.; Geyl, K.K.; Tarasenko, M.V.; Gureev, M.A.; Boyarskiy, V.P. π–π Noncovalent Interaction Involving 1,2,4- and 1,3,4-Oxadiazole Systems: The Combined Experimental, Theoretical, and Database Study. Molecules 2021, 26, 5672. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Kirina, Y.V.; Novikov, A.S.; Starova, G.L.; Kukushkin, V.Y. Efficient π-Stacking with Benzene Provides 2D Assembly of Trans-[PtCl2(p-CF3C6H4CN)2]. J. Mol. Struct. 2016, 1104, 19–23. [Google Scholar] [CrossRef]
- Lukoyanov, A.N.; Fomenko, I.S.; Gongola, M.I.; Shul’pina, L.S.; Ikonnikov, N.S.; Shul’pin, G.B.; Ketkov, S.Y.; Fukin, G.K.; Rumyantcev, R.V.; Novikov, A.S.; et al. Novel Oxidovanadium Complexes with Redox-Active R-Mian and R-Bian Ligands: Synthesis, Structure, Redox and Catalytic Properties. Molecules 2021, 26, 5706. [Google Scholar] [CrossRef]
- Mukhacheva, A.A.; Komarov, V.Y.; Kokovkin, V.V.; Novikov, A.S.; Abramov, P.A.; Sokolov, M.N. Unusual π–π Interactions Directed by the [{(C 6H6)Ru} 2W8O 30(OH)2 ] 6− Hybrid Anion. CrystEngComm 2021, 23, 4125–4135. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Krykova, M.A.; Ivanov, D.M.; Novikov, A.S.; Sinelshchikova, A.A.; Volostnykh, M.V.; Konovalov, M.A.; Grigoriev, M.S.; Gorbunova, Y.G.; Kukushkin, V.Y. Reverse Arene Sandwich Structures Based upon π-Hole⋯[MII] (d 8 M=Pt, Pd) Interactions, Where Positively Charged Metal Centers Play the Role of a Nucleophile. Angew. Chem. Int. Ed. 2019, 58, 4164–4168. [Google Scholar] [CrossRef]
- Rozhkov, A.V.; Novikov, A.S.; Ivanov, D.M.; Bolotin, D.S.; Bokach, N.A.; Kukushkin, V.Y. Structure-Directing Weak Interactions with 1,4-Diiodotetrafluorobenzene Convert One-Dimensional Arrays of [MII(acac)2] Species into Three-Dimensional Networks. Cryst. Growth Des. 2018, 18, 3626–3636. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From Weak to Strong Interactions: A Comprehensive Analysis of the Topological and Energetic Properties of the Electron Density Distribution Involving X–H⋯F–Y Systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. Van Der Waals Volumes and Radii of Metals in Covalent Compounds. J. Phys. Chem. 1966, 70, 3006–3007. [Google Scholar] [CrossRef]
- Alvarez, S. A Cartography of the van Der Waals Territories. Dalton Trans. 2013, 42, 8617. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Faßbach, T.A.; Vorholt, A.J.; Leitner, W. The Telomerization of 1,3-Dienes—A Reaction Grows Up. ChemCatChem 2019, 11, 1153–1166. [Google Scholar] [CrossRef]
- Behr, A.; Becker, M.; Beckmann, T.; Johnen, L.; Leschinski, J.; Reyer, S. Telomerization: Advances and Applications of a Versatile Reaction. Angew. Chem. Int. Ed. Engl. 2009, 48, 3598–3614. [Google Scholar] [CrossRef]
- Van Leeuwen, P.W.N.M.; Clément, N.D.; Tschan, M.J.-L. New Processes for the Selective Production of 1-Octene. Coord. Chem. Rev. 2011, 255, 1499–1517. [Google Scholar] [CrossRef]
- Benvenuti, F.; Carlini, C.; Marchionna, M.; Patrini, R.; Raspolli Galletti, A.M.; Sbrana, G. Homogeneous Telomerization of 1,3-Butadiene with Alcohols in the Presence of Palladium Catalysts Modified by Hybrid Chelate Ligands. J. Mol. Catal. A Chem. 1999, 140, 139–155. [Google Scholar] [CrossRef]
- Patrini, R.; Lami, M.; Marchionna, M.; Benvenuti, F.; Raspolli Galletti, A.M.; Sbrana, G. Selective Synthesis of Octadienyl and Butenyl Ethers via Reaction of 1,3-Butadiene with Alcohols Catalyzed by Homogeneous Palladium Complexes. J. Mol. Catal. A Chem. 1998, 129, 179–189. [Google Scholar] [CrossRef]
- Takahashi, S.; Shibano, T.; Hagihara, N. The Dimerization of Butadiene by Palladium Complex Catalysts. Tetrahedron Lett. 1967, 8, 2451–2453. [Google Scholar] [CrossRef]
- Vollmüller, F.; Mägerlein, W.; Klein, S.; Krause, J.; Beller, M. Palladium-Catalyzed Reactions for the Synthesis of Fine Chemicals, Highly Efficient Palladium-Catalyzed Telomerization of Butadiene with Methanol. Adv. Synth. Catal. 2001, 343, 29–33. [Google Scholar] [CrossRef]
- Mesnager, J.; Kuntz, E.; Pinel, C. Isolated-Palladium Complexes for Catalyzed Telomerization of Butadiene with Methanol in the Presence of Water. J. Organomet. Chem. 2009, 694, 2513–2518. [Google Scholar] [CrossRef]
- Hausoul, P.J.C.; Parvulescu, A.N.; Lutz, M.; Spek, A.L.; Bruijnincx, P.C.A.; Weckhuysen, B.M.; Klein Gebbink, R.J.M. Facile Access to Key Reactive Intermediates in the Pd/PR3-Catalyzed Telomerization of 1,3-Butadiene. Angew. Chem. Int. Ed. 2010, 49, 7972–7975. [Google Scholar] [CrossRef]
- Vollmüller, F.; Krause, J.; Klein, S.; Mägerlein, W.; Beller, M. Palladium-Catalyzed Reactions for the Synthesis of Fine Chemicals, 14([≠]) Control of Chemo- and Regioselectivity in the Palladium-Catalyzed Telomerization of Butadiene with Methanol—Catalysis and Mechanism. Eur. J. Inorg. Chem. 2000, 8, 1825–1832. [Google Scholar] [CrossRef]
- Tschan, M.J.-L.; García-Suárez, E.J.; Freixa, Z.; Launay, H.H.; Hagen, H.; Benet-Buchholz, J.; van Leeuwen, P.W.N.M.; García-Suárez, E.J.; Freixa, Z.; Launay, H.H.; et al. Efficient Bulky Phosphines for the Selective Telomerization of 1,3-Butadiene with Methanol. J. Am. Chem. Soc. 2010, 132, 6463–6473. [Google Scholar] [CrossRef]
- Briggs, J.R.; Hagen, H.; Julka, S.; Patton, J.T. Palladium-Catalyzed 1,3-Butadiene Telomerization with Methanol. Improved Catalyst Performance Using Bis-o-Methoxy Substituted Triarylphosphines. J. Organomet. Chem. 2011, 696, 1677–1686. [Google Scholar] [CrossRef]
- Tschan, M.J.-L.; López-Valbuena, J.-M.; Freixa, Z.; Launay, H.; Hagen, H.; Benet-Buchholz, J.; van Leeuwen, P.W.N.M. Large P−P Distance Diphosphines and Their Monophosphine Analogues as Ligands in the Palladium-Catalyzed Telomerization of 1,3-Butadiene and Methanol. Organometallics 2011, 30, 792–799. [Google Scholar] [CrossRef]
- Jackstell, R.; Harkal, S.; Jiao, H.; Spannenberg, A.; Borgmann, C.; Röttger, D.; Nierlich, F.; Elliot, M.; Niven, S.; Cavell, K.; et al. An Industrially Viable Catalyst System for Palladium-Catalyzed Telomerizations of 1,3-Butadiene with Alcohols. Chem. Eur. J. 2004, 10, 3891–3900. [Google Scholar] [CrossRef] [PubMed]
- Jackstell, R.; Frisch, A.; Beller, M.; Röttger, D.; Malaun, M.; Bildstein, B. Efficient Telomerization of 1,3-Butadiene with Alcohols in the Presence of in Situ Generated Palladium(0)Carbene Complexes. J. Mol. Catal. A Chem. 2002, 185, 105–112. [Google Scholar] [CrossRef]
- Clement, N.D.; Routaboul, L.; Grotevendt, A.; Jackstell, R.; Beller, M. Development of Palladium-Carbene Catalysts for Telomerization and Dimerization of 1,3-Dienes: From Basic Research to Industrial Applications. Chem. Eur. J. 2008, 14, 7408–7420. [Google Scholar] [CrossRef] [PubMed]
- Benvenuti, F.; Carlini, C.; Lami, M.; Marchionna, M.; Patrini, R.; Raspolli Galletti, A.M.; Sbrana, G. Telomerization of 1,3-Butadiene with Alcohols Catalyzed by Homogeneous Palladium(0) Complexes in the Presence of Mono- and Diphosphine Ligands. J. Mol. Catal. A Chem. 1999, 144, 27–40. [Google Scholar] [CrossRef]
- Tolman, C.A. Steric Effects of Phosphorus Ligands in Organometallic Chemistry and Homogeneous Catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Darkwa, J. Palladium Catalyzed Phenylacetylene Polymerization to Low Molecular Weight Cis-Transoidal and Trans-Cisoidal Poly(Phenylacetylene)s: A Perspective. Polym. Rev. 2017, 57, 52–64. [Google Scholar] [CrossRef]
- Chen, Z.-H.; Daugulis, O.; Brookhart, M. Polymerization of Terminal Acetylenes by a Bulky Monophosphine-Palladium Catalyst. Organometallics 2023, 42, 235–239. [Google Scholar] [CrossRef]
- Kishimoto, Y.; Eckerle, P.; Miyatake, T.; Kainosho, M.; Ono, A.; Ikariya, T.; Noyori, R. Well-Controlled Polymerization of Phenylacetylenes with Organorhodium(I) Complexes: Mechanism and Structure of the Polyenes. J. Am. Chem. Soc. 1999, 121, 12035–12044. [Google Scholar] [CrossRef]
- Onishi, N.; Shiotsuki, M.; Masuda, T.; Sano, N.; Sanda, F. Polymerization of Phenylacetylenes Using Rhodium Catalysts Coordinated by Norbornadiene Linked to a Phosphino or Amino Group. Organometallics 2013, 32, 846–853. [Google Scholar] [CrossRef]
- Shiotsuki, M.; Sanda, F.; Masuda, T. Polymerization of Substituted Acetylenes and Features of the Formed Polymers. Polym. Chem. 2011, 2, 1044–1058. [Google Scholar] [CrossRef]
- Huber, J.; Mecking, S. Aqueous Poly(Arylacetylene) Dispersions. Macromolecules 2010, 43, 8718–8723. [Google Scholar] [CrossRef]
- Li, K.; Mohlala, M.S.; Segapelo, T.V.; Shumbula, P.M.; Guzei, I.A.; Darkwa, J. Bis(Pyrazole)- and Bis(Pyrazolyl)-Palladium Complexes as Phenylacetylene Polymerization Catalysts. Polyhedron 2008, 27, 1017–1023. [Google Scholar] [CrossRef]
- Suslov, D.S.; Pakhomova, M.V.; Bykov, M.V.; Ushakov, I.A.; Tkach, V.S. Polymerization of Phenylacetylene by Cationic Acetylacetonate Palladium Complexes. Catal. Commun. 2019, 119, 16–21. [Google Scholar] [CrossRef]
- Shiotsuki, M.; Takahashi, K.; Rodriguez Castanon, J.; Sanda, F. Synthesis of Block Copolymers Using End-Functionalized Polyacetylenes as Macroinitiators. Polym. Chem. 2018, 9, 3855–3863. [Google Scholar] [CrossRef]
- Li, K.; Wei, G.; Darkwa, J.; Pollack, S.K. Polymerization of Phenylacetylene Catalyzed by Diphosphinopalladium(II) Complexes. Macromolecules 2002, 35, 4573–4576. [Google Scholar] [CrossRef]
- Simionescu, C.I.; Percec, V.; Dumitrescu, S. Polymerization of Acetylenic Derivatives. XXX. Isomers of Polyphenylacetylene. J. Polym. Sci. Polym. Chem. Ed. 1977, 15, 2497–2509. [Google Scholar] [CrossRef]
- Castanon, J.R.; Sano, N.; Shiotsuki, M.; Sanda, F. New Approach to the Polymerization of Disubstituted Acetylenes by Bulky Monophosphine-Ligated Palladium Catalysts. ACS Macro Lett. 2014, 3, 51–54. [Google Scholar] [CrossRef]
- Rodriguez-Castanon, J.; Murayama, Y.; Sano, N.; Sanda, F. Polymerization of a Disubstituted Acetylene Using Palladium Catalysts. Chem. Lett. 2015, 44, 1200–1201. [Google Scholar] [CrossRef]
- Li, M.; Chen, C. Polymerization of Disubstituted Acetylenes by Monodentate NHC-Pd Catalysts. Polym. Chem. 2015, 6, 7127–7132. [Google Scholar] [CrossRef]
- Zou, W.; Pang, W.; Chen, C. Redox Control in Palladium Catalyzed Norbornene and Alkyne Polymerization. Inorg. Chem. Front. 2017, 4, 795–800. [Google Scholar] [CrossRef]
- Jesus Rodriguez, C.; Kuwata, K.; Shiotsuki, M.; Sanda, F. Synthesis of End-Functionalized Polyacetylenes That Contain Polar Groups by Employing Well-Defined Palladium Catalysts. Chem. Eur. J. 2012, 18, 14085–14093. [Google Scholar] [CrossRef] [PubMed]
- Pelagatti, P.; Carcelli, M.; Pelizzi, C.; Costa, M. Polymerisation of Phenylacetylene in Water Catalysed by Pd(NN′O)Cl Complexes. Inorg. Chim. Acta 2003, 342, 323–326. [Google Scholar] [CrossRef]
- Yoshida, I.; Kobayashi, H.; Ueno, K. Differential Thermal Analysis of Some Divalent Metal Chelates of 1,5-Dialkylpentane-2,4-Diones. J. Inorg. Nucl. Chem. 1973, 35, 4061–4070. [Google Scholar] [CrossRef]
- Klosin, J.; Abboud, K.A.; Jones, W.M. Bis(Triphenylphosphine)Palladium Cycloheptadienynylium Tetrafluoroborate: A Palladium Complex of Tropyne. Organometallics 1996, 15, 2465–2468. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Frisch, M.J.; Frisch, G.W.; Trucks, H.B.; Schlegel, G.E.; Scuseria, M.A.; Robb, J.R.; Cheeseman, G.; Scalmani, V.; Barone, B.; Mennucci, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]










| Free Ligand | δ 31P{1H} Chemical Shift, ppm | Complex | δ 31P{1H} Chemical Shift, ppm | Δ(31P{1H}) (δcomplex − δligand), ppm |
|---|---|---|---|---|
| PPh3 | −6.3 [44] | 1 | 33.8 | 40.1 |
| P(p-Tol)3 | −8.3 [44] | 2 | 32.9 | 41.2 |
| TOMPP | −37.9 [45] | 3 | 8.2 | 46.1 |
| TFP | −75.2 [46] | 4 | −24.0 | 51.2 |
| TTP | −45.6 [47] | 5 | −1.8 | 43.8 |
| dppf | −16.8 [48] | 6 | 38.6 | 55.4 |
| dppp | −17.7 [49] | 7 | 16.7 | 34.4 |
| dppb | −16.3 [49] | 8 | 33.1 | 49.4 |
| dppent | −16.5 [49] | 9 | 19.1 | 35.6 |
| dpphex | −16.4 [49] | 10 | 28.6 | 45.0 |
| Complex | Cp | Phosphine Ligand |
|---|---|---|
| 1a | 5.51 (t, J = 2.1 Hz, 5H) | 7.45–7.38 (m, 6H, HPh), 7.38 –7.28 (m, 24H, HPh) |
| 2 | 5.49 (t, J = 2.1 Hz, 5H) | 7.25–7.16 (m, 12H, HPh), 7.16–7.08 (m, 12H, HPh), 2.35 (s, 18H, CH3) |
| 3 | 5.12 (t, J = 2.2 Hz, 5H) | 7.61–7.05 (m, 12H, HPh), 7.02–6.73 (m, 12H, HPh), 3.75–2.84 (m, br., 18H, CH3O) |
| 4 | 6.04 (t, J = 2.3 Hz, 5H) | 7.79–7.74 (m, 6H, HFur5) (Fur = furyl), 6.84–6.78 (m, 6H, HFur3), 6.58–6.48 (m, 6H, HFur4) |
| 5 | 5.80 (t, J = 2.2 Hz, 5H) | 7.88–7.82 (m, 6H, HThi5) (Thi = thienyl), 7.38–7.32 (m, 6H, HThi3), 7.14 (t, J = 4.3 Γц, 6H, HThi4) |
| 6 | 5.45 (t, J = 2.1 Hz, 5H) | 7.70–7.60 (m, 12H, HPh), 7.59–7.49 (m, 8H, HPh,meta), 4.59–4.52 (m, 4H, HCp′(dppf),β), 4.39–4.34 (m, 4H, HCp′(dppf),α). |
| 7 | 5.51 (t, J = 2.1 Hz, 5H) | 7.61–7.42 (m, 20H, HPh), 2.78–2.57 (m, CH2, 4H), 2.17 (s, CH2). |
| 8 | 5.39 (t, J = 2.1 Hz, 5H) | 7.68–7.49 (m, 20H, HPh), 2.60 (s, br, 4H, CH2), 1.83–1.63 (m, 4H, CH2). |
| 9 | 5.72 (t, J = 2.0 Hz, 0.7H, minor isomer), 5.44 (t, J = 2.0 Hz, 4H, major isomer), | 7.61–7.26 (m, 20H, HPh), 2.54 (tt, J = 9.6, 4.8 Γц, 4H, CH2), 2.30–2.10 (m, 2H, CH2) 1.84–1.69 (m, 4H, CH2). |
| 10 | 5.78 (t, J = 2.0 Hz, 3H, major isomer), 5.76–5.69 (m, 1.5H, minor isomer), | 7.72–7.21 (m, 20H, HPh), 2.34–2.06 (m, 3H, CH2), 1.99–1.46 (m, 3H, CH2), 1.41–1.21 (m, 6H, CH2). |
| Entry [a] | Pd | Conv. BD % [b] | Selectivity, mol% | Chemo,mol% [c] | n/iso [d] | E/Z [e] | TON [f] | ||
|---|---|---|---|---|---|---|---|---|---|
| OCT | 3-MOD | 1-MOD | |||||||
| 1 | 1 | 67.4 | 13.5 | 5.0 | 81.5 | 86.5 | 16 | 10 | 8100 |
| 4 | 2 | 66.8 | 13.0 | 5.6 | 81.5 | 87.1 | 15 | 10 | 8000 |
| 5 | 3 | 25.9 | 4.7 | 3.3 | 92.0 | 95.3 | 28 | 18 | 3100 |
| 6 | 4 | 8.7 | 4.8 | 4.4 | 90.8 | 95.2 | 21 | 12 | 1050 |
| 7 | 5 | 18.0 | 5.0 | 4.9 | 90.1 | 95.0 | 18 | 10 | 2150 |
| 8 | 6 | 0.3 | — | — | — | — | — | — | — |
| 9 | 7 | 0.2 | — | — | — | — | — | — | — |
| 10 | 8 | 0.3 | — | — | — | — | — | — | — |
| 11 | 9 | 0.3 | — | — | — | — | — | — | — |
| 12 | 10 | 0.2 | — | — | — | — | — | — | — |
| 13 [g] | 1 | 49.2 | 12.8 | 4.9 | 81.5 | 86.4 | 17 | 12 | 11,800 |
| 14 [h] | 1 | 49.3 | 16.6 | 4.5 | 77.8 | 82.3 | 17 | 11 | 23,700 |
| 15 [i] | 1 | 3.5 | 11.4 | 4.5 | 69.9 | 74.4 | 15 | 10 | 4200 |
| Entry [a] | Complex | Time (h) | Solvent | t (°C) | Yield (%) | TON [b] | Activity [c] | Sel [d] (%) | Cis [e] (%) | Polymer | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mw (/103) [f] | Mw/ Mn[f] | ||||||||||
| 1 | 1 | 24 | — | 25 | 0.3 | 3 | 13 | 63 | 90 | 2.6 | 1.6 |
| 2 | 2 | 24 | — | 25 | 0.2 | 2 | 9 | 65 | 94 | 2.2 | 1.5 |
| 3 | 3 | 24 | — | 25 | 26.2 | 262 | 1113 | 78 | 94 | 17.2 | 2.1 |
| 4 | 4 | 24 | — | 25 | 0 | 0 | 0 | — | — | — | |
| 5 | 5 | 24 | — | 25 | 0 | 0 | 0 | — | — | — | |
| 6 | 6 | 24 | — | 25 | 1.3 | 13 | 55 | 92 | 92 | 19.0 | 2.1 |
| 7 | 7 | 24 | — | 25 | 0 | 0 | 0 | — | — | — | |
| 8 | 8 | 24 | — | 25 | 0 | 0 | 0 | — | — | — | |
| 9 | 9 | 24 | — | 25 | 0 | 0 | 0 | — | — | — | |
| 10 | 10 | 24 | — | 25 | 0 | 0 | 0 | — | — | — | |
| 11 [h] | 3 | 24 | — | 4 | 2.7 | 14 | 60 | >99 | 90 | 21.4 | 2.1 |
| 12 [h] | 3 | 24 | — | 25 | 53.4 | 267 | 1140 | 79 | 94 | 15.5 | 2.0 |
| 13 [h] | 3 | 24 | — | 50 | 91.3 | 457 | 1940 | 47 | 98 | 7.9 | 1.6 |
| 14 [h] | 3 | 24 | — | 60 | 84.8 | 424 | 1800 | 38 | 98 | 2.0 | 1.5 |
| 15 [h] | 3 | 1 | — | 25 | 2.2 | 11 | 1219 | 99 | n.d. | 16.5 | 2.1 |
| 16 [h] | 3 | 2 | — | 25 | 5.2 | 26 | 1320 | 99 | 94 | 17.9 | 1.9 |
| 17 [h] | 3 | 3 | — | 25 | 7.0 | 35 | 1120 | 99 | 95 | 17.3 | 1.9 |
| 18 [h] | 3 | 4 | — | 25 | 12.3 | 62 | 1580 | 99 | 94 | 19.1 | 2.0 |
| 19 [h] | 3 | 5 | — | 25 | 25.2 | 126 | 2570 | 98 | 95 | 16.2 | 2.0 |
| 20 [h] | 3 | 24 | — | 25 | 53.9 | 270 | 1150 | 79 | 94 | 13.4 | 2.0 |
| 21 [h] | 3 | 48 | — | 25 | 57.0 | 285 | 605 | 67 | 96 | 16.4 | 2.0 |
| 22 [h] | 3 | 5 | pentane | 25 | 0.4 | 2 | 40 | 95 | 87 | 14.9 | 2.4 |
| 23 [h] | 3 | 5 | benzene | 25 | 2.0 | 10 | 200 | 98 | 90 | 8.5 | 1.9 |
| 24 [h] | 3 | 5 | THF | 25 | 87.0 | 435 | 8890 | 94 | 92 | 23.2 | 2.6 |
| 25 [h] | 3 | 5 | MeCN | 25 | 4.0 | 20 | 410 | 96 | 90 | 2.3 | 1.4 |
| 26 [h] | 3 | 5 | CH2Cl2 | 25 | 19.3 | 97 | 1980 | 94 | 90 | 16.1 | 2.2 |
| 27 [h] | 3 | 5 | DCE [g] | 25 | 25.6 | 128 | 2610 | 90 | 87 | 18.3 | 2.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suslov, D.S.; Bykov, M.V.; Pakhomova, M.V.; Orlov, T.S.; Abramov, Z.D.; Suchkova, A.V.; Ushakov, I.A.; Abramov, P.A.; Novikov, A.S. Novel Route to Cationic Palladium(II)–Cyclopentadienyl Complexes Containing Phosphine Ligands and Their Catalytic Activities. Molecules 2023, 28, 4141. https://doi.org/10.3390/molecules28104141
Suslov DS, Bykov MV, Pakhomova MV, Orlov TS, Abramov ZD, Suchkova AV, Ushakov IA, Abramov PA, Novikov AS. Novel Route to Cationic Palladium(II)–Cyclopentadienyl Complexes Containing Phosphine Ligands and Their Catalytic Activities. Molecules. 2023; 28(10):4141. https://doi.org/10.3390/molecules28104141
Chicago/Turabian StyleSuslov, Dmitry S., Mikhail V. Bykov, Marina V. Pakhomova, Timur S. Orlov, Zorikto D. Abramov, Anastasia V. Suchkova, Igor A. Ushakov, Pavel A. Abramov, and Alexander S. Novikov. 2023. "Novel Route to Cationic Palladium(II)–Cyclopentadienyl Complexes Containing Phosphine Ligands and Their Catalytic Activities" Molecules 28, no. 10: 4141. https://doi.org/10.3390/molecules28104141
APA StyleSuslov, D. S., Bykov, M. V., Pakhomova, M. V., Orlov, T. S., Abramov, Z. D., Suchkova, A. V., Ushakov, I. A., Abramov, P. A., & Novikov, A. S. (2023). Novel Route to Cationic Palladium(II)–Cyclopentadienyl Complexes Containing Phosphine Ligands and Their Catalytic Activities. Molecules, 28(10), 4141. https://doi.org/10.3390/molecules28104141