1. Introduction
Biomass is an environmentally friendly and renewable energy source that has the potential to replace traditional petroleum resources. Lignin is the most abundant renewable aromatic polymer in nature [
1], with up to 50 billion tons formed annually through photosynthesis on earth [
2]. Approximately 50 million tons of them were produced as the by-product of traditional pulp and paper industry and biorefineries per year [
3]. However, the complex chemical structure of lignin makes it difficult to be utilized directly. Typically, more than 98% of these lignin waste is directly burned for energy [
4], resulting in a low utilization efficiency. Thus, it is necessary to utilize lignin waste to improve the comprehensive utilization efficiency of biomass resources.
The lignin depolymerization process [
5] can convert lignin into high-quality biofuels and chemicals. It is beneficial to improve the economic efficiency of pulp and bio-refining industries [
6]. Currently, the depolymerization methods of lignin mainly include thermochemical conversion, mechanical depolymerization, catalytic fast pyrolysis (CFP), and biodegradation [
7]. Among them, CFP refers to a process that produces bio-oil, non-condensable gas, and bio-char by heating biomass pellets in the presence of a catalyst in an oxygen-free or anoxic environment [
8]. It is a relatively efficient biomass conversion technology that has been widely used for converting lignin into aromatic compounds [
9]. Development of an efficient catalyst with good performance is the core of this technology. Commonly, catalysts used in CFP technology include metal salts [
10,
11], metal oxides [
12], zeolite molecular sieves [
13,
14], etc. Zeolite molecular sieves have become one of the most promising catalysts in CFP technology due to their excellent hydrothermal stability, controllable acidic strength, shape selectivity, and other properties [
15]. Among them, HZSM-5 has attracted wide attention due to its unique pore structure, excellent shape-selective catalytic ability, and good deoxygenation performance. It has been used for catalytic thermal cracking of lignin into bio-oil rich in monocyclic aromatic hydrocarbon products [
16]. However, due to the hydrogen-deficient and oxygen-rich nature of lignin and its fragments, coke is always generated during pyrolysis. This can cover acidic sites and block pores, leading to a decrease in catalyst activity and selectivity. In addition, lignin pyrolysis products contain a large number of phenolics, which can rapidly deactivate the molecular sieve [
17]. Improper acidity of the molecular sieve is another key reason for both coke formation and low catalytic efficiency [
18]. Although the catalyst activity can be restored through oxidative regeneration, low oil yield from lignin pyrolysis caused by coking is a major obstacle to industrialization of this process [
19,
20]. Therefore, modification of the zeolite catalyst is necessary to obtain a suitable pore structure and an appropriate acid strength and molecular content.
Presently, HZSM-5 modification methods include alkali treatment, impregnation, ion exchange, hydrothermal synthesis, etc. Among these methods, introduction of metals such as Zn [
21], Fe [
22], Ga [
23], Mo [
24], and La [
25] into the molecular sieve to prepare the bimetallic catalyst (Me/HZSM-5) has been reported in catalytic pyrolysis of lignin and lignin derivatives. This proves that the modified catalyst retains the shape-selective catalytic ability of HZSM-5; meanwhile, addition of a metal active site promotes phenol deoxidation, thus, improved the yield of monoaromatics. Moreover, the synergistic catalytic action of metal sites and Brønsted acid sites reduces the coking and deactivation of the catalyst to some extent [
26]. Most of these modified HZSM-5 catalysts were prepared by a impregnation method; the metal active sites were supported on the outer surface of zeolite or in the micropore channels [
27]. This increased the diffusion resistance of reactants and products, and prevented the contact between the raw materials and the active site on the catalyst. The metal loaded on the surface of molecular sieves is prone to detachment, leading to a decrease in the catalyst performance. Compared with the impregnation method, hydrothermal synthesis can give a better dispersion of metals and improve the stability of the catalyst [
28]. Metal species (Sn, Fe, and Zn) have been incorporated with HZSM-5 to generate H[Me, Al]ZSM-5 using this method and it has been used in methanol aromatization reactions. The results have shown that the highly dispersed metals were beneficial to improve both the yield of mono-aromatic hydrocarbons and the catalyst stability [
29,
30].
In recent years, owing to the special electronic structure Nb
5c, metal Nb-modified molecular sieves have attracted great attention for the effective catalytic cracking of C-O bonds [
31,
32]. Yang [
31] et al. synthesized NbAlS-1 by integrating Nb (V) and Al (III) active sites into an MFI-type zeolite framework. NbAlS-1 showed strong water resistance and could quantitatively convert γ-valerolactone aqueous solution to butene at atmospheric pressure. In addition, a synergistic catalytic effect of Nb (v) sites and Brønsted acid sites in promoting C-O bond breaking was found. Wang [
32] et al. prepared a Nb-doped SBA-15 molecular sieve by hydrothermal synthesis and applied it in 2, 5-dimethyltetrahydrofuran hydrodeoxygenation to prepare hexane. This proved that the Nb active sites with a low coordination number could effectively activate a C-O bond in the hydrodeoxidation reaction. However, preparing a H[Nb]ZSM-5 bifunctional catalyst by hydrothermal synthesis and using it in catalytic pyrolysis of lignin has rarely been reported.
In this study, a new type of bifunctional niobium-doped ZSM-5 (H[Nb]ZSM-5) was prepared using the hydrothermal synthesis method. Then, the as-prepared catalyst was characterized by X-ray powder diffraction (XRD), physical adsorption, NH3-TPD, and X-ray photoelectron spectroscopy (XPS). Finally, the lignin was pyrolyzed using pyrolysis gas chromatography-mass spectrometry (Py-GC/MS), and the effect of H[Nb]ZSM-5 catalyst on the production of light aromatic hydrocarbons (benzene-toluene-xylene mixture, BTX) was studied using nest HZSM-5 as the control. The catalyst stability as well as carbon/coke accumulation behavior were evaluated.
3. Materials and Methods
3.1. Experimental Materials
The following reagents were purchased from commercial suppliers: aluminum isopropoxide (Fuchen Co., Ltd., Tianjin, China, 98.5%), sodium hydroxide (Pengkun Co., Ltd., Tianjin, China, 96.0%), tetraethyl orthosilicate (Fuchen Co., Ltd., Tianjin, China, 98.5%), tetrapropylammonium hydroxide (Siyoupu Co., Ltd., Hefei, China, 1 mol/L), niobium oxalate (Haoxuan Co., Ltd., Guangzhou, China, 95.0%), ammonia chloride (Nankai Co., Ltd., Tianjin, China, 99.5%), and alkali lignin (Merck KGaA, Darmstadt, Germany, 99.5%.
3.2. Catalyst Synthesis
H[Nb]ZSM-5 and HZSM-5 were prepared using a hydrothermal method according to the literature [
31]. In this study, however, niobium oxalate was used to replace the expensive and unstable niobium (V) ethanolate. In a typical synthesis, aluminium isopropoxide was first dissolved in deionized water, into which tetrapropylammonium hydroxide (TPAOH) solution as the structure-directing agent was added. The mixture was stirred at room temperature for 2 h, then niobium oxalate was added and the mixture was stirred for another 2 h. Next, tetraethyl orthosilicate was added dropwise and the mixture was stirred for another 2 h, which resulted in a gel with a chemical composition of 1Si: 0.09Al: 0.03Nb: 0.25TPAOH: 15H
2O. The gel was then transferred to a Teflon-lined stainless-steel autoclave, which was sealed and heated to specific temperatures (110 °C, 130 °C, 150 °C, 160 °C, 170 °C, 180 °C) for certain times (24 h, 48 h, 72 h, 96 h). After centrifugation, the solid products were washed with deionized water, dried overnight at 80 °C and calcined at 550 °C under air flow for 6 h. The as-prepared solid (Na[Nb]ZSM-5) was ion exchanged with 1 mol/L ammonium chloride solution three times, followed by suction filtration, washing, drying, and roasting at 550 °C for 6 h to obtain hydrogen-type ZSM-5 molecular sieve. Throughout the entire process, sodium hydroxide was added dropwise at appropriate times to adjust the pH value. The as-prepared solid (Na[Nb]ZSM-5) was ion exchanged three times with a 1 mol/L ammonium chloride solution, followed by filtration, washing, drying, and finally calcining at 550 °C for 6 h to obtain a hydrogen-type ZSM-5 molecular sieve (H[Nb]ZSM-5). HZSM-5 sample was synthesized by the same procedure, but without the addition of niobium oxalate.
3.3. Catalyst Characterization
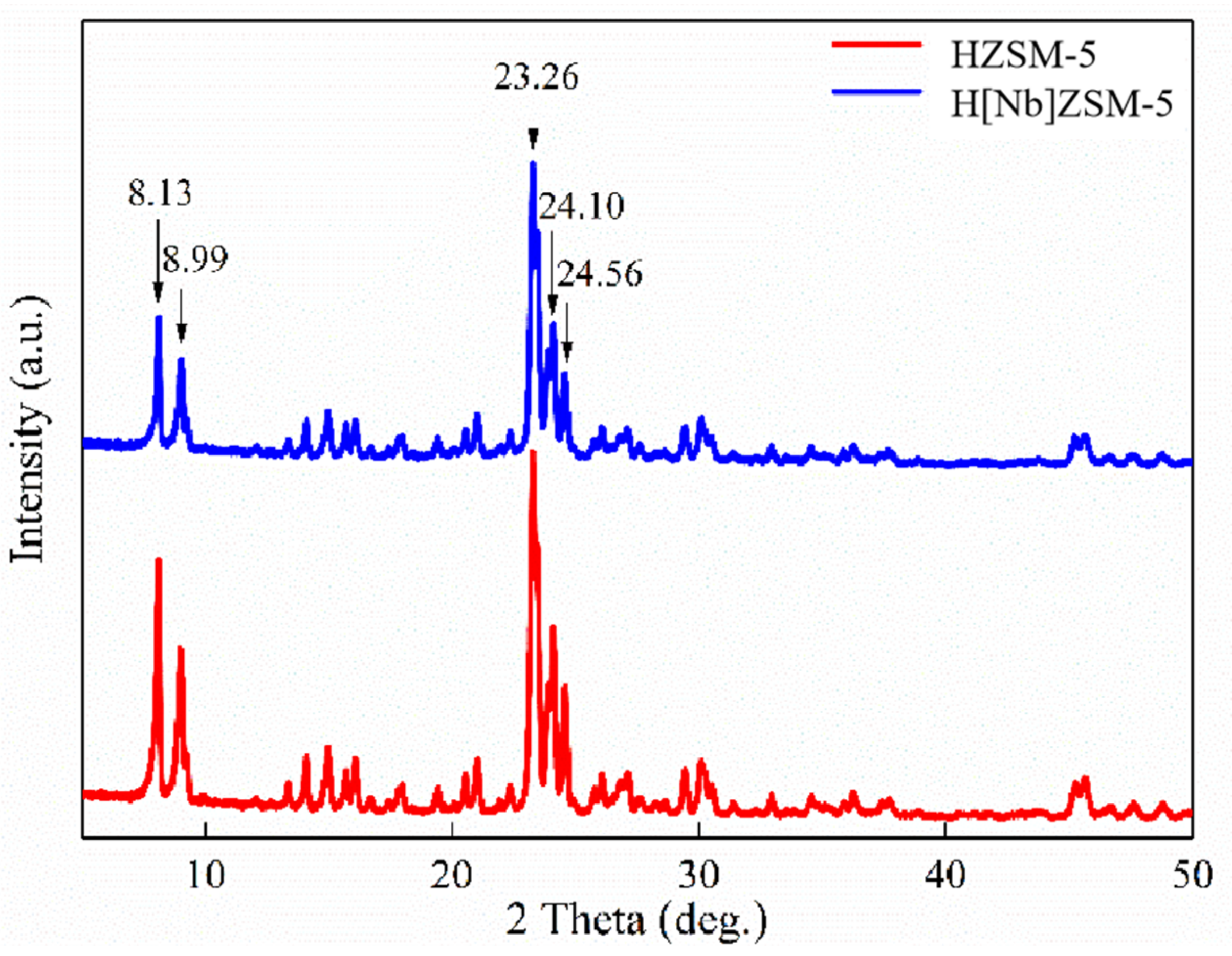
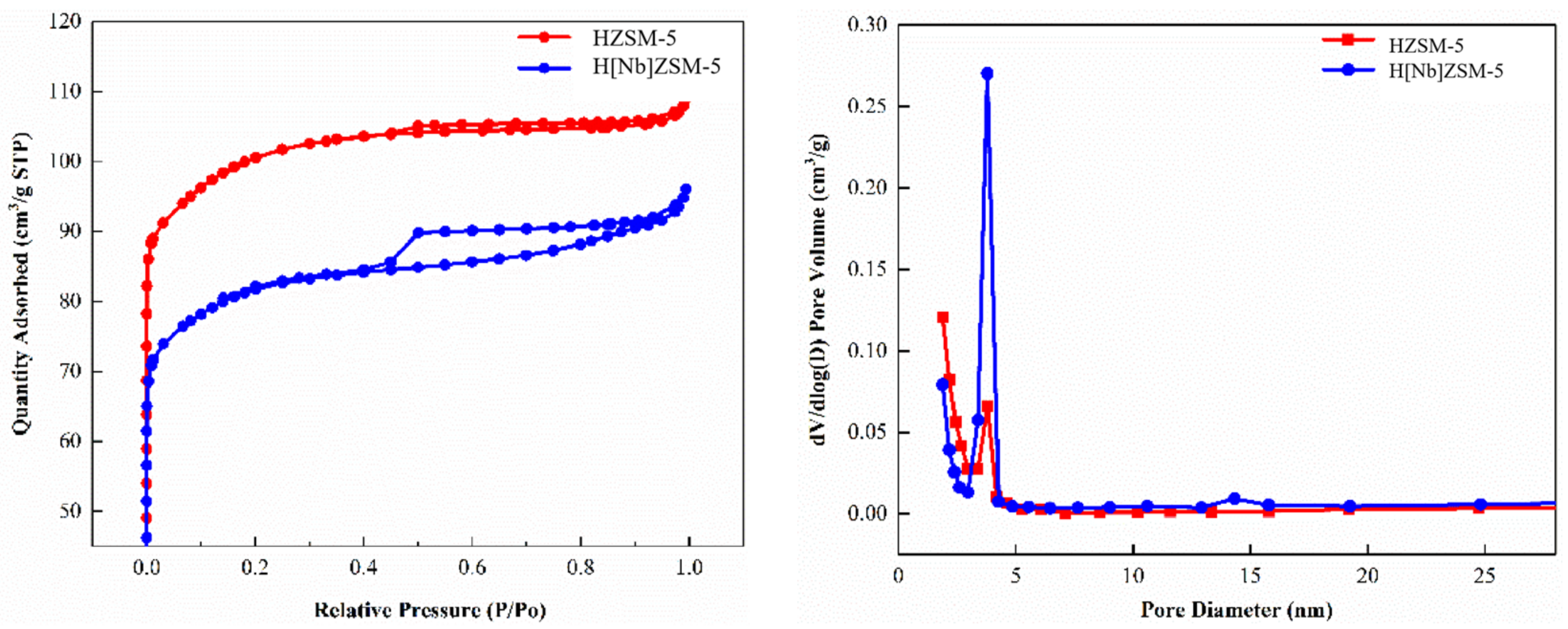
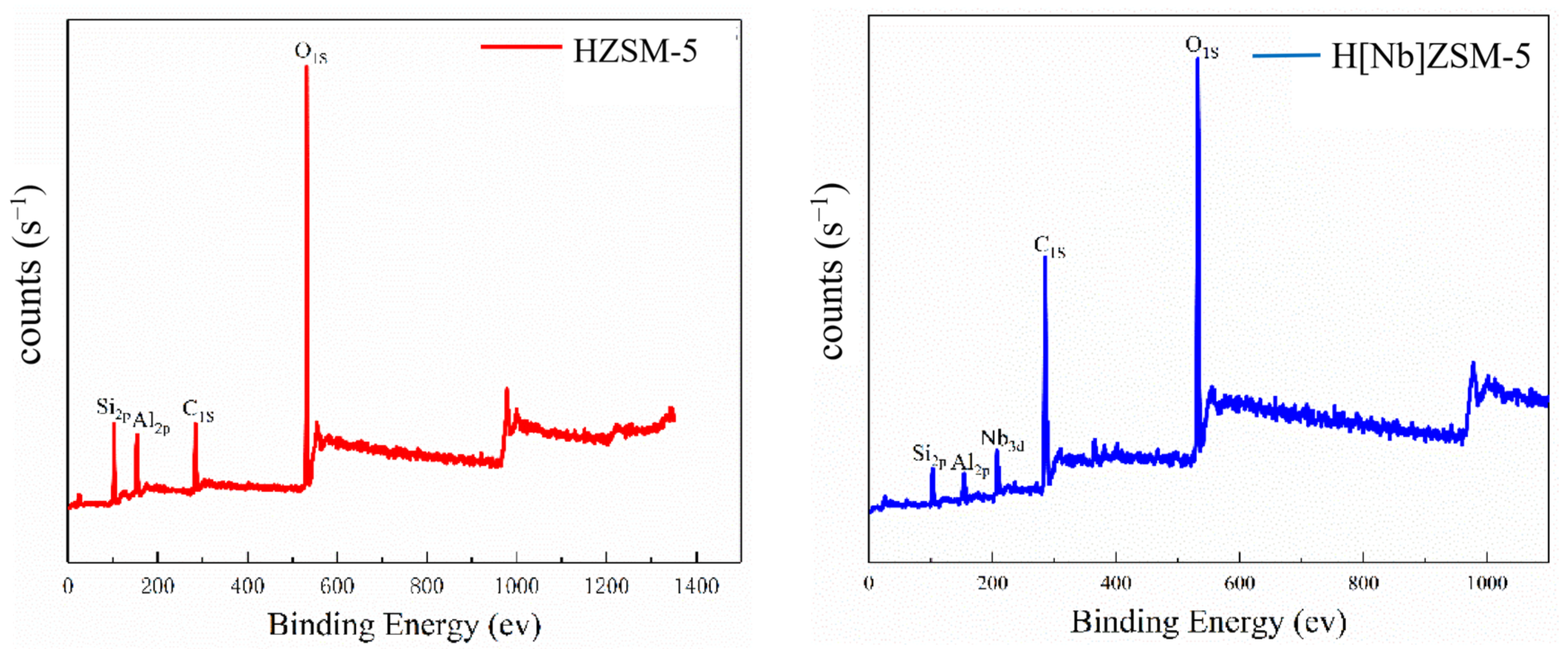
The crystal structures of the catalysts were examined using a D/max 6100 X-ray diffractometer (Shimadzu Corporation, Kyoto, Japan) with Cu Ka radiation (λ = 1.5406 Å) at 40 kV and 30 mA at a scanning rate of 8°/min from 5 to 50°. The relative crystallinity of the catalyst was calculated according to the American Society for Testing and Materials (ASTM) standard D5758-01 [
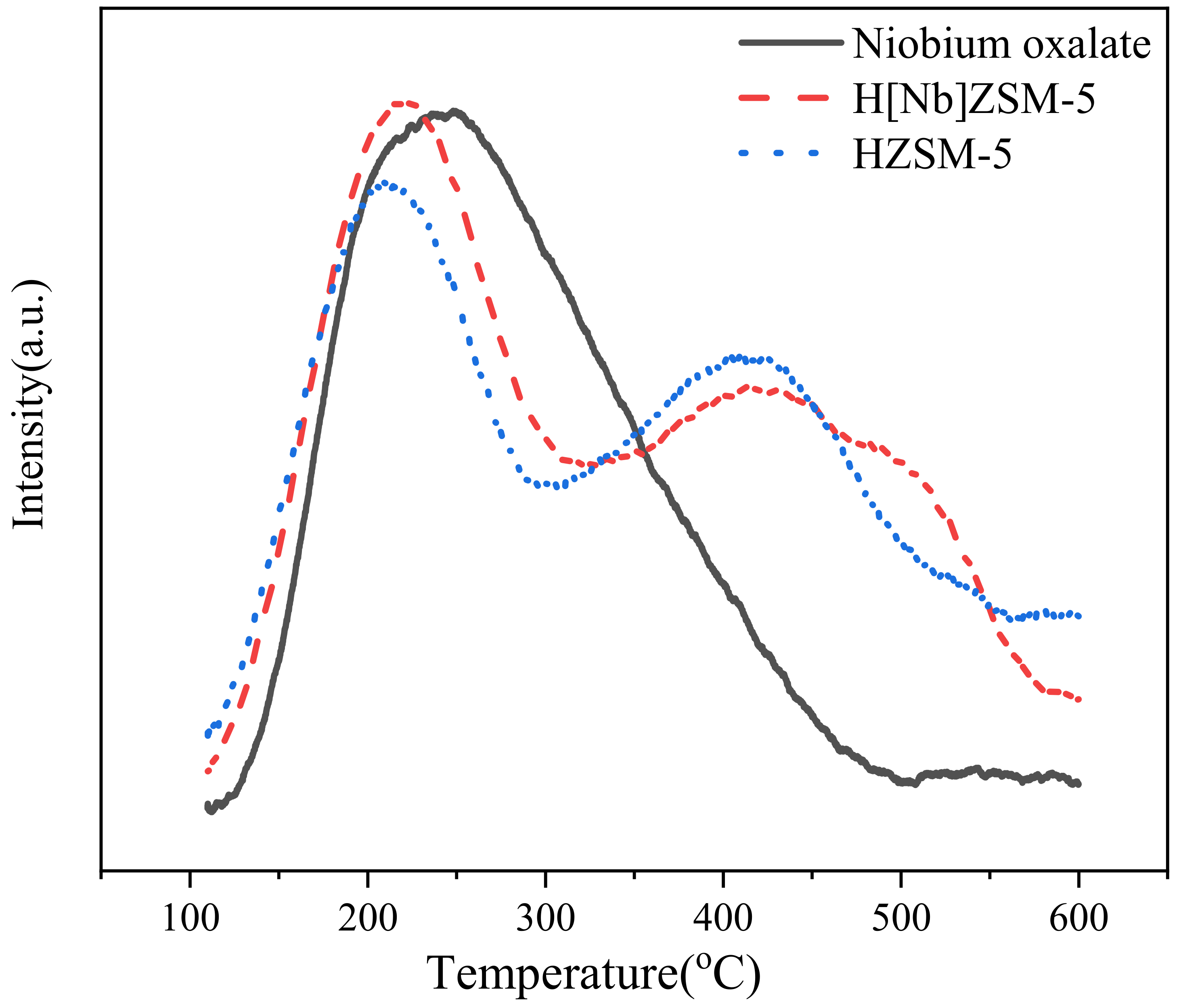
43]. This test method provides a number that is the ratio of the intensity of portions of the XRD pattern of the sample to the intensity of the corresponding portion of the pattern of a reference zeolite. This intensity ratio, expressed as a percentage, is then labeled relative crystallinity. In this study, the relative crystallinity refers to the ratio of integral peak area of sample to that of the reference HZSM-5 in the range of 2θ = 22.5–25°, and calculated based on the three main diffraction peaks at 23.26°, 24.10°, and 24.56°. X-ray photoelectron spectrometer (XPS) analysis was performed on a Thermo Scientific K-Alpha (Thermo Fisher Scientific, Waltham, MA, USA) using Mg Ka as the photon source. The anode voltage was set at 15 kV and the anode current at 10 mA. The binding energies were calibrated by using the C1s peak at 284.8 eV as a reference. Field emission scanning electron microscopy and energy dispersive X-ray spectroscopy (FESEM-EDX) (Apreo S HiVac/EDX, Thermo Scientific Co., Ltd., Waltham, MA, USA) were used to study the crystal morphology, size, and elemental composition. The ratios of Nb/Al/Si were quantified using multiple regions over a sample with an Octane Elect Super detector. Nitrogen adsorption experiments were performed at 77.3 K using an Accelerated Surface Area and Porosimetry System (ASAP 2460, Micromeritics, Lutherville Timonium, MD, USA). Before the analysis, the samples were degassed at 250 ℃ for 12 h under vacuum. The specific surface area, micropore-specific surface area, and pore size distribution of the samples were obtained using multi-point BET, T-plot, and BJH methods, respectively. The acidity was evaluated using temperature-programmed desorption of ammonia (NH
3-TPD) with an AutoChem-II-2920 chemisorption apparatus (Micromeritics, Lutherville Timonium, MD, USA). Typically, 0.1 g of catalyst sample was loaded in the quartz U-tube reactor and pre-treated under He flow at 500 °C for 1 h to remove all moisture. After cooling down to 40 °C, the sample was flushed with a NH
3/He gas mixture (30 vol%) at a flow rate of 30 mL/min for 1 h to adsorb NH
3. Then, the catalyst was purged with helium at 100 °C for 1 h to remove physisorbed NH
3 from the catalyst surface. Next, the temperature rose to 600 °C at a rate of 10 °C/min while the TPD profile was recorded.
3.4. CFP of Lignin
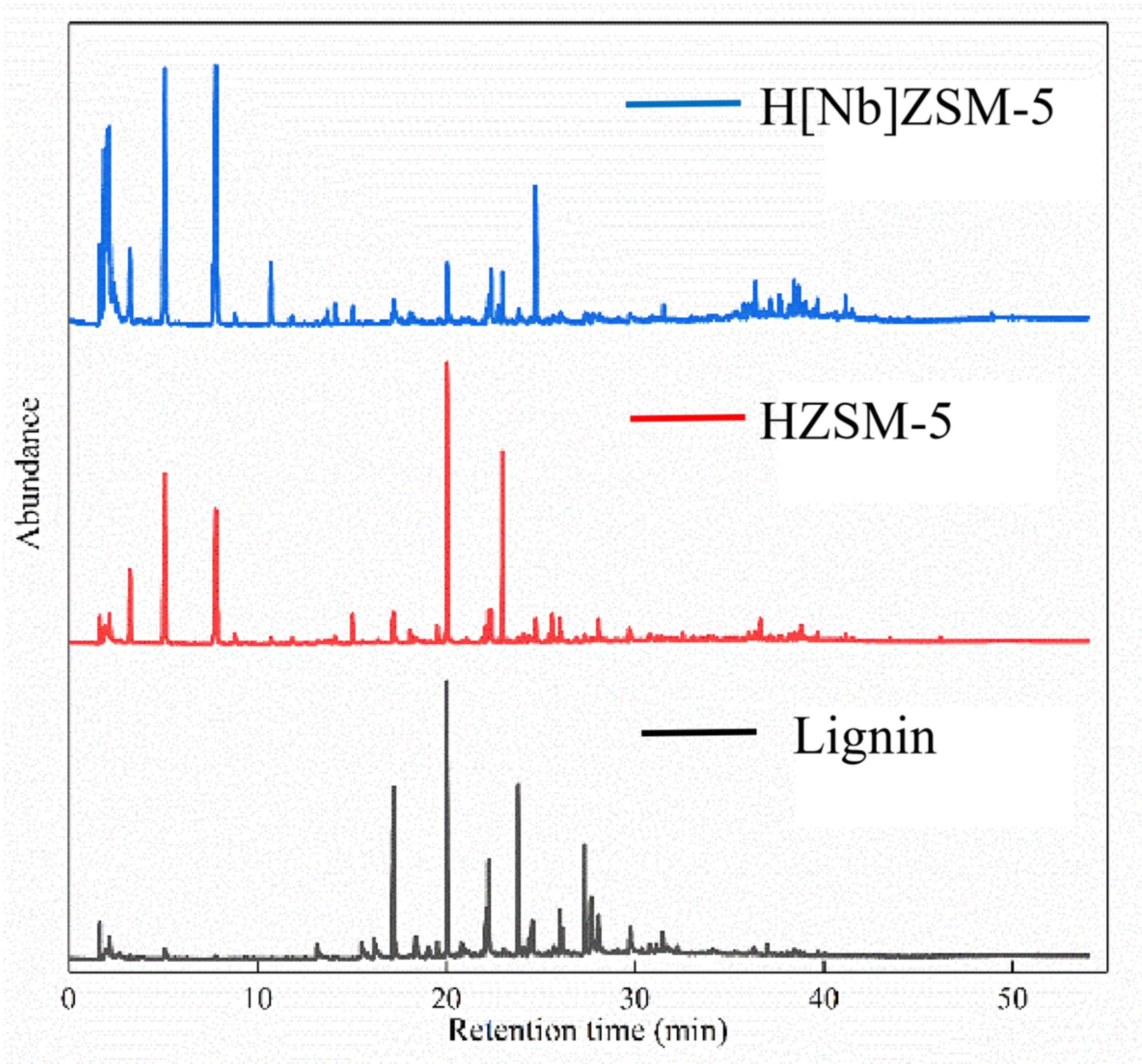
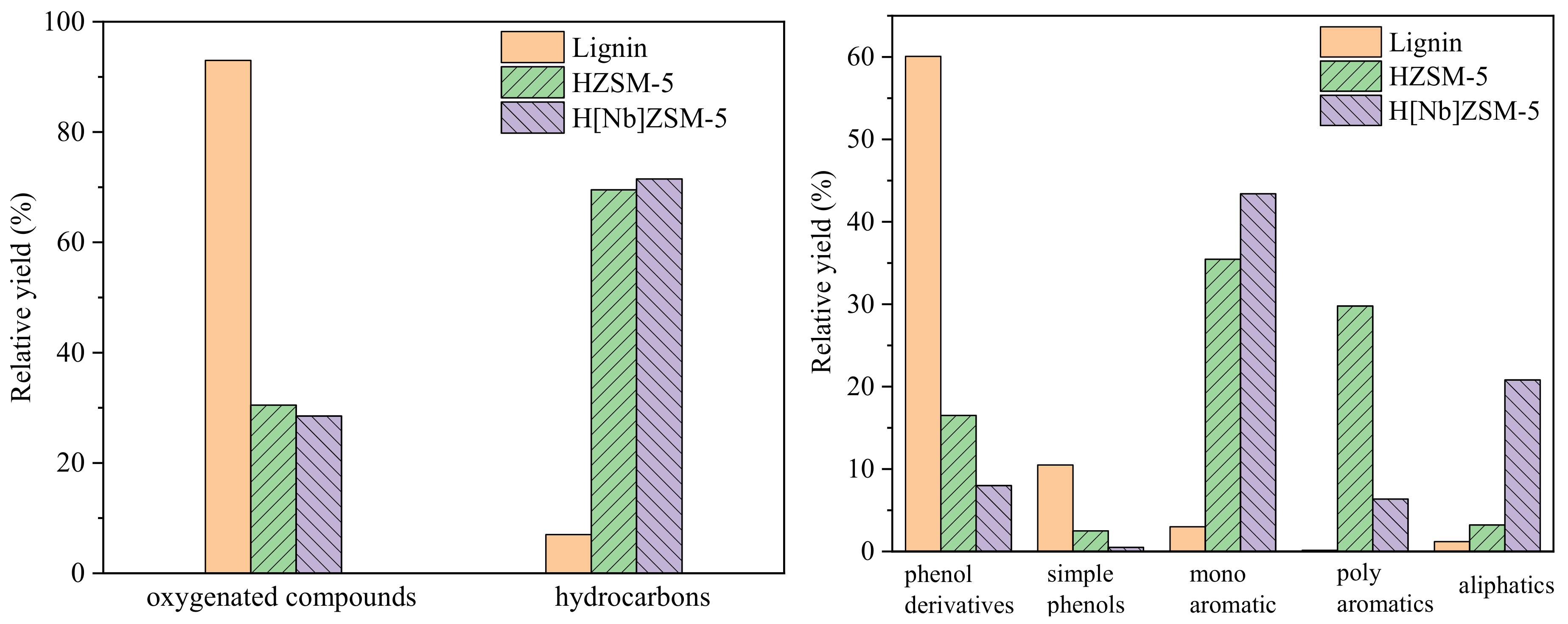
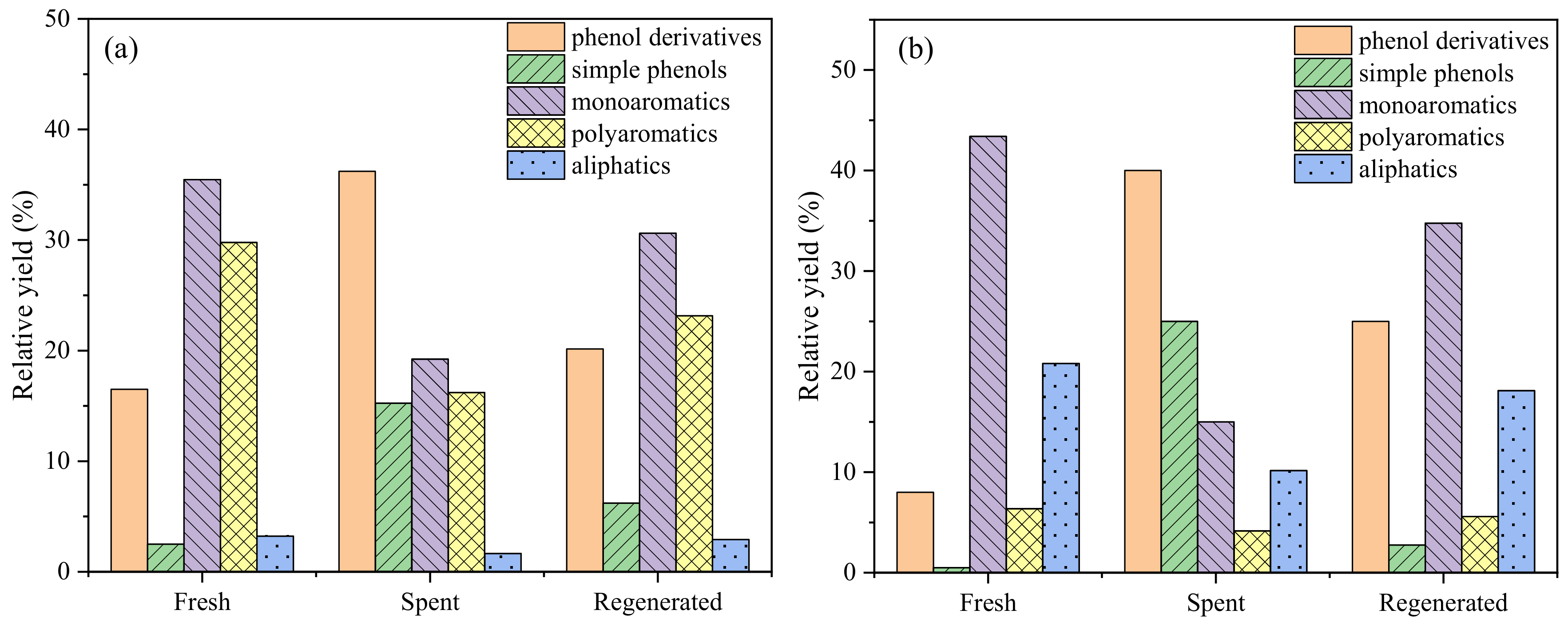
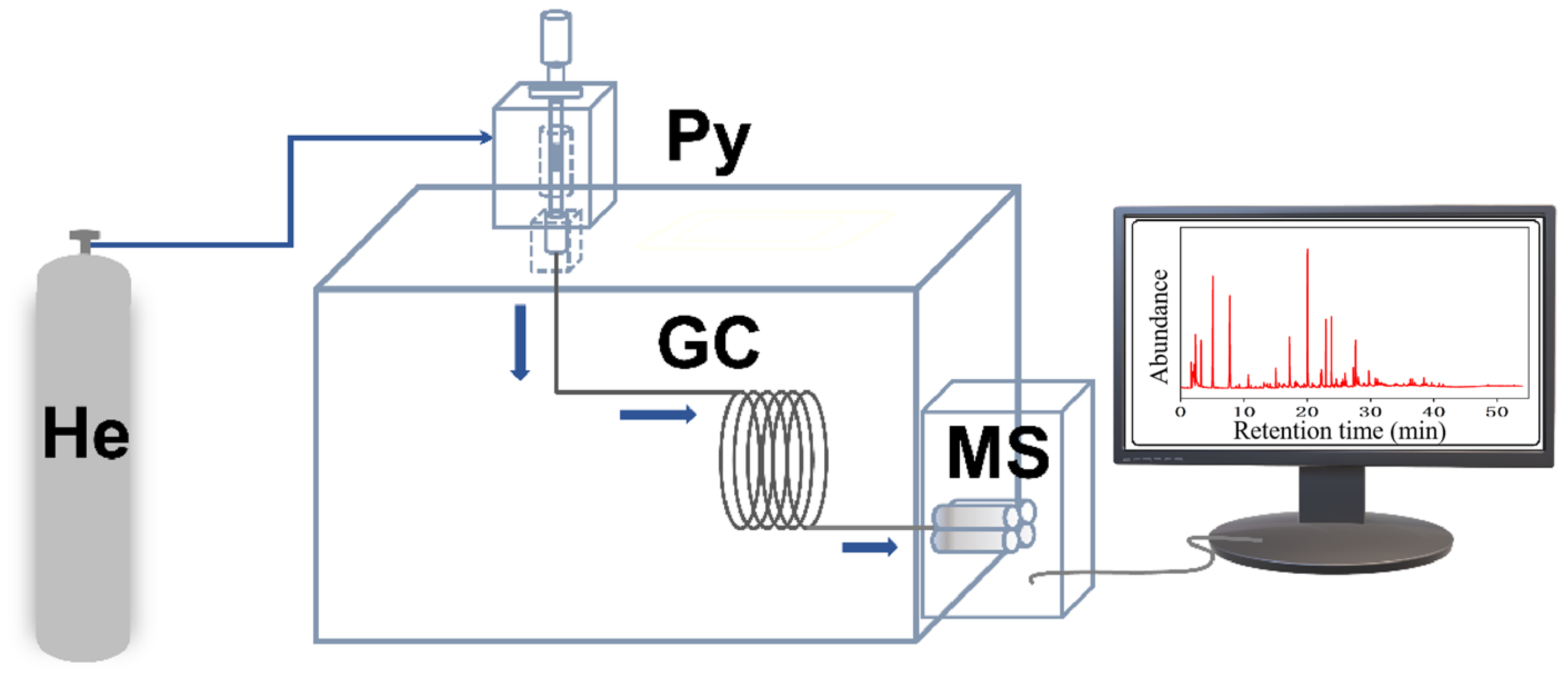
Lignin, obtained from Sigma-Aldrich (USA, CAS# 8068-05-1), had the follwing elements by wt.%: C, 63.55; H, 5.49; O, 29.15; S, 1.49; N, 0.32. Catalytic fast pyrolysis of lignin was performed by coupling a pyrolyzer (CDS5200, CDS Company, Blythewood, SC, USA) to a GC–MS system (6890N/5973, Agilent Company, Santa Clara, CA, USA) (
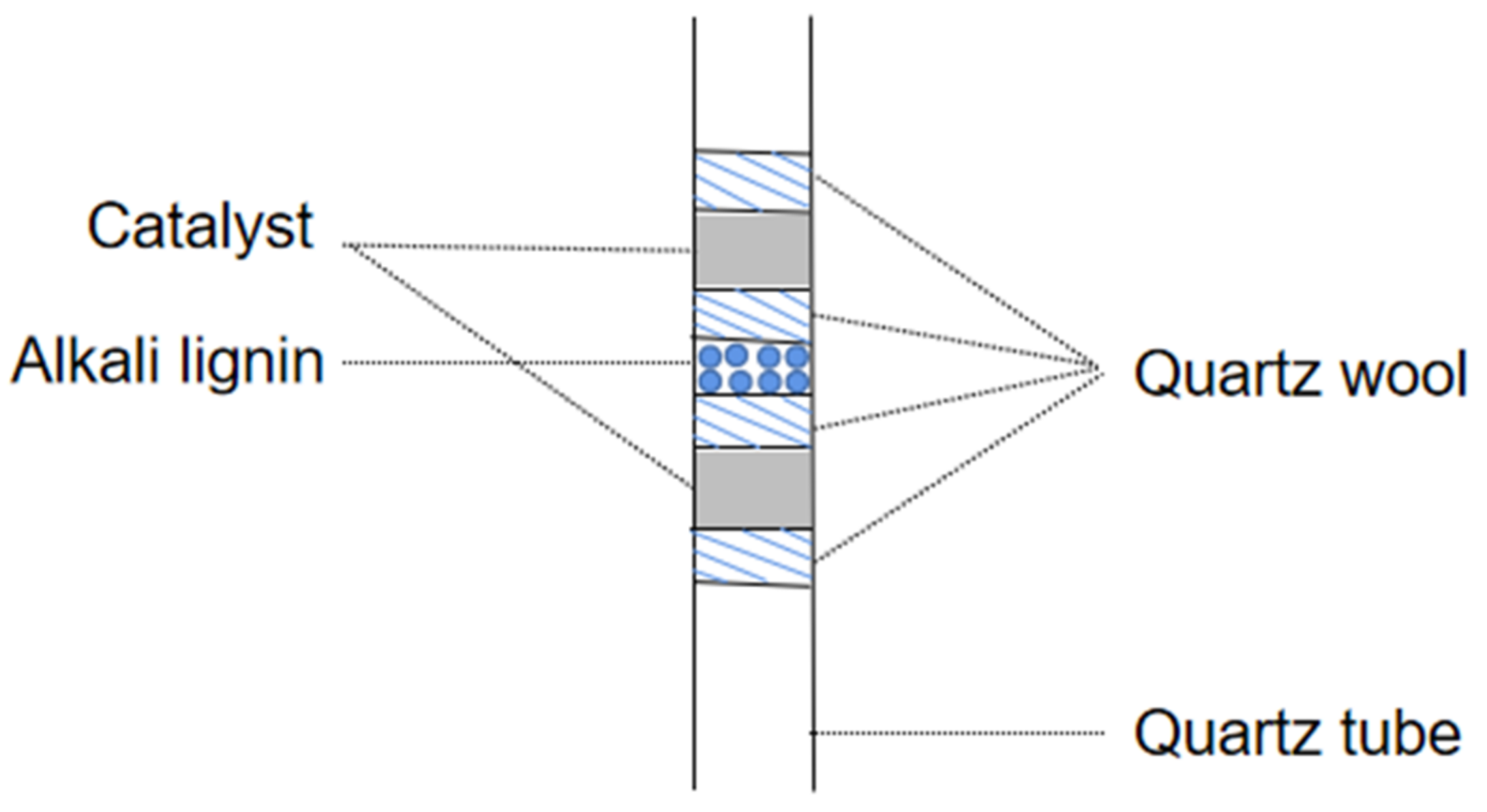
Figure 12). All pyrolyses were carried out at 650 °C, 60 s with a heating rate of 20 °C/ms. The sample was placed in the middle of the pyrolysis tube and the catalyst was next to the sample, with a sample to catalyst w:w ratio of 1:20 at both ends of the tube (
Figure 13). The arrangement of sample between two catalyst layers guarantees that all vapors produced by pyrolysis of the sample pass through the catalyst bed.
The pyrolysis volatiles were transferred by high-purity helium gas into the GC/MS instrument through a transmission line, which was maintained at 285 °C. A 50:1 split ratio of the carrier gas (helium) was employed. The compressible cracking products were separated by HP-5MS quartz column (30 m × 0.25 mm × 0.25 um film thickness). The GC injector was kept at 280 °C. The GC oven was programmed to hold at 40 °C for 3 min, then heat to 270 °C for 5 min at a rate of 5 °C/min, and finally heat to 280 °C for 5 min. The mass spectrometry detector was operated in electron ionization mode (70 eV) over the m/z range from 20 to 550 amu. The ion source was kept at 230 °C. Identification of the GC-sensitive compounds was carried out by comparing with spectra of the NIST mass spectral library. Each experiment was repeated at least twice under the same condition to ensure accuracy.
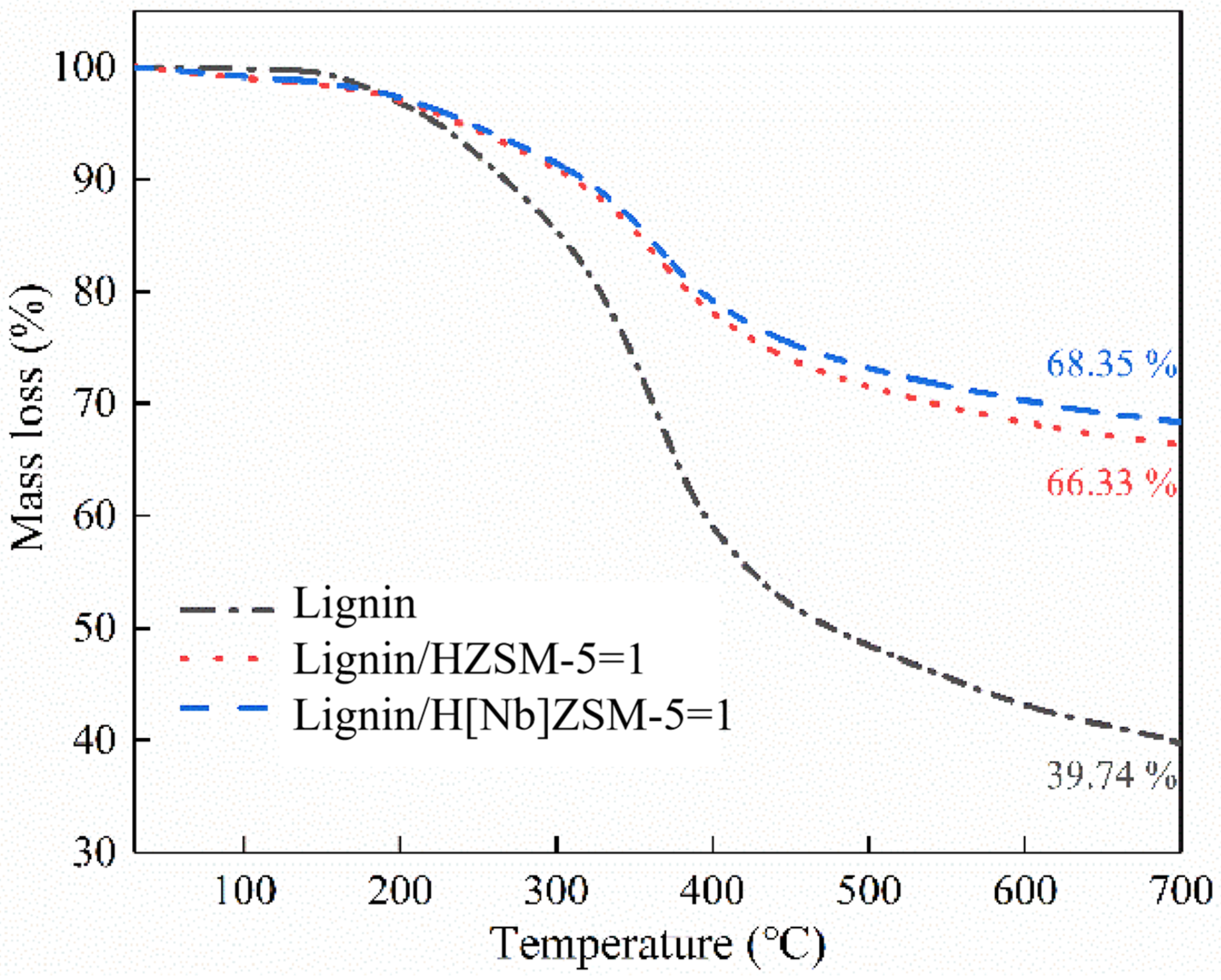
A TG 209 F1 Libra thermogravimetric analyzer (NETZSCH, Selb, Germany) was used to heat 5–10 mg samples (uniformly mixed, 1:1 mass ratio of alkali lignin and catalyst) from 30 °C to 700 °C at a heating rate of 10 °C/min in argon atmosphere. TG curves were analyzed to evaluate the influence of catalyst on the amount of residual carbon from alkali lignin pyrolysis.
3.5. Catalyst Stability Evaluation
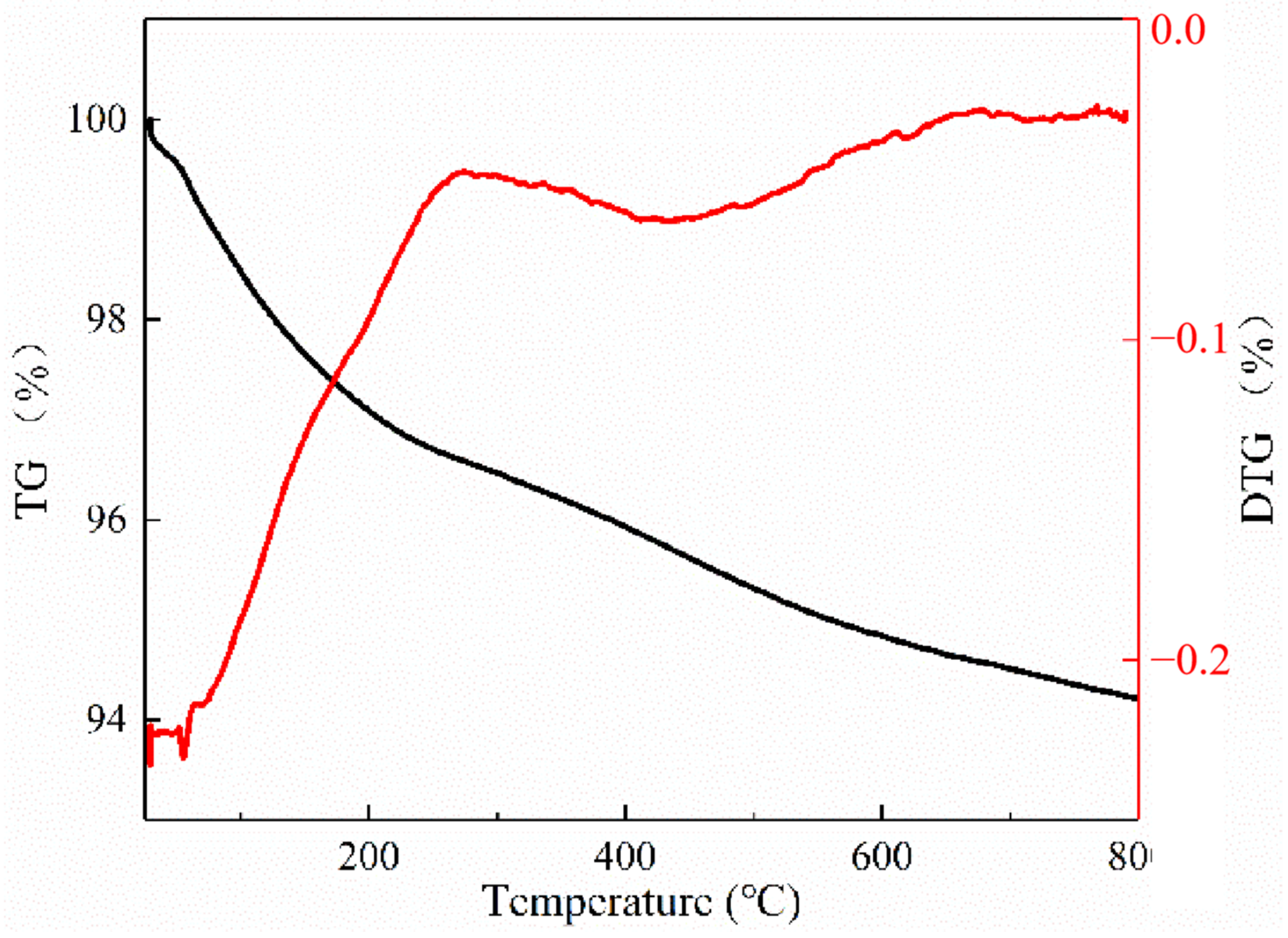
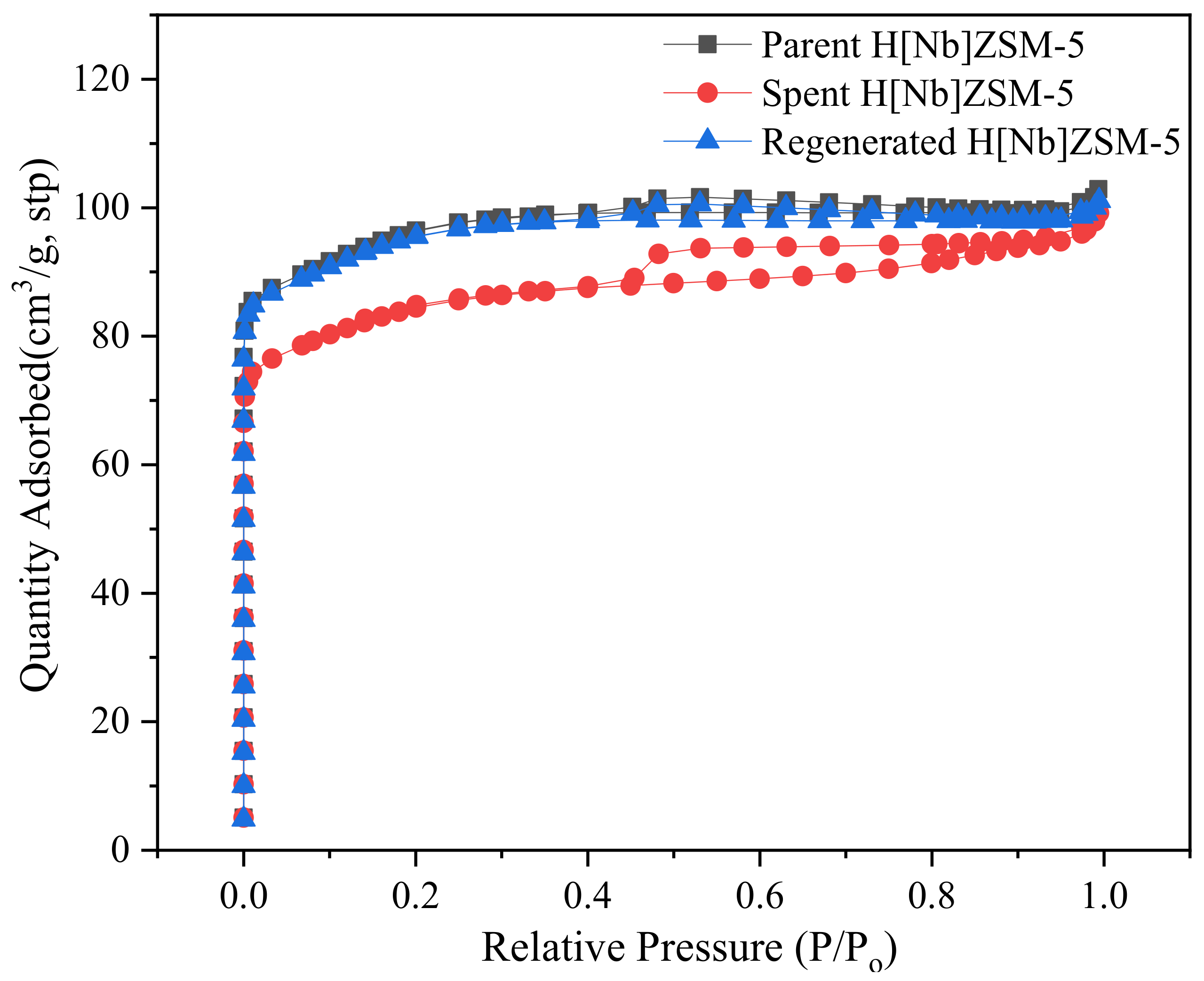
Thermal gravimetric analysis (TGA) was used to determine the coke behaviors of the spent catalyst. Samples weighing 10 mg were heated from 30 °C to 700 °C at a heating rate of 40 °C/min in oxygen atmosphere with a flow of 20 mL/min. The nitrogen adsorption experiment of the catalyst was also performed to investigate the effects of carbon accumulation on the catalyst-specific surface area and pore structure. The specific experimental conditions were consistent with the nitrogen adsorption experiment described in
Section 3.3. For the calculation of the amount of carbon accumulation [
44], it was assumed that N
2 molecules could pass through the interconnected three-dimensional zeolite channels and fully contact the residual micropore volume of the catalyst after the reaction. The amount of internal coke was calculated based on the reduction of the micropore volume (measured by BET analysis method, cm
3/g). The formula is as follows:
The external coke quantity (i.e., the amount of coke deposited on the external surface of the catalyst) was obtained by subtracting the internal coke quantity from the total coke quantity (determined by thermogravimetric analysis curve, mg
coke/mg
cat). The formula is as follows:
In the two formulas above, QMIC represents the internal coke quantity; QEC is the external coke quantity; QTC is the total coke quantity; VM,F, VM,R are the micropore volumes before and after the catalyst reaction, which were obtained from the nitrogen adsorption experiment; and dC refers to the coke density, which was 1.22 g/cm3 (based on the C/H ratio of 1.25).
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}