
Recent Progress in the Integration of CO2 Capture and Utilization

Abstract
:1. Introduction
2. Integration of CO2 Capture and Utilization
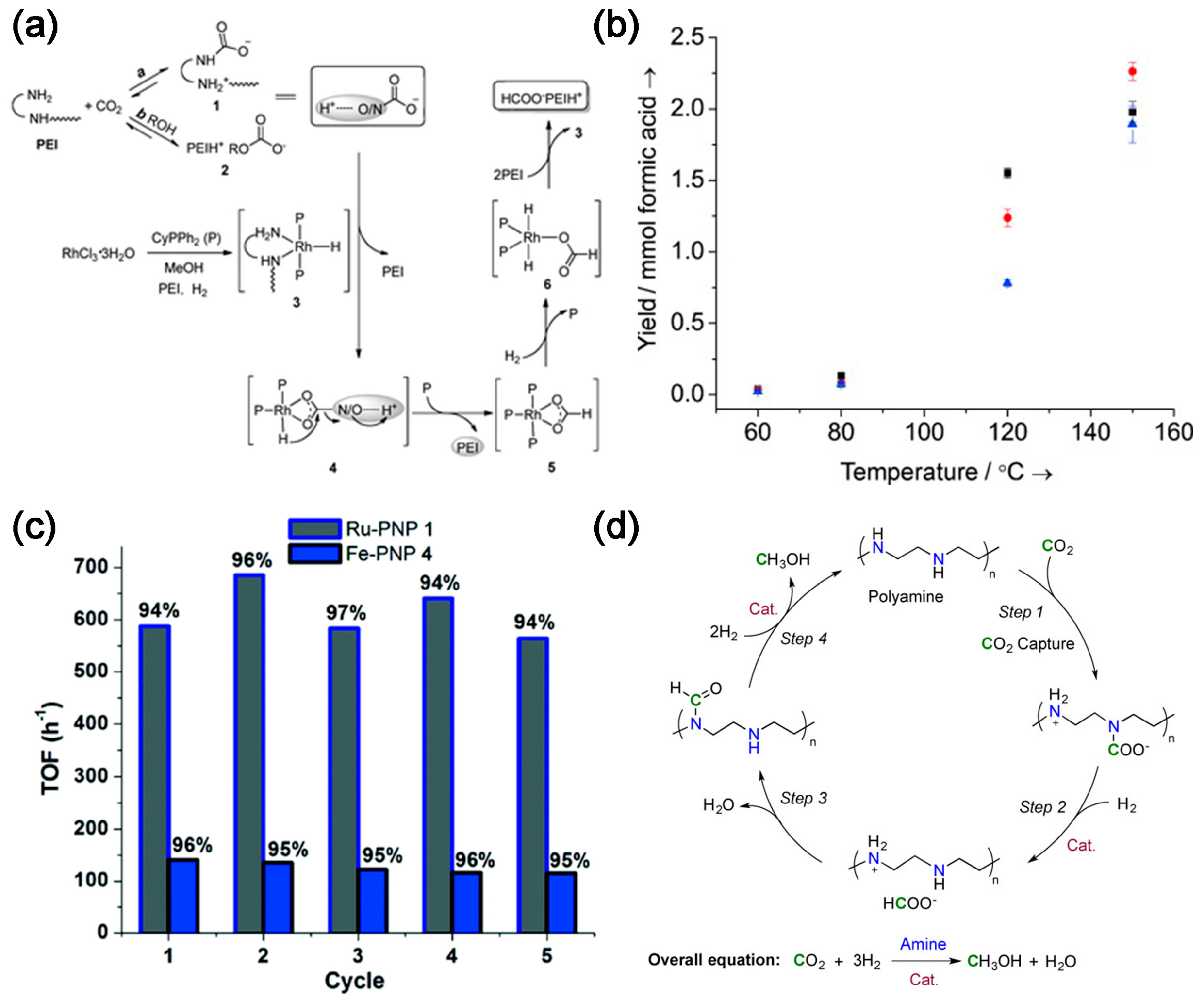
2.1. Integration of CO2 Absorption and Conversion
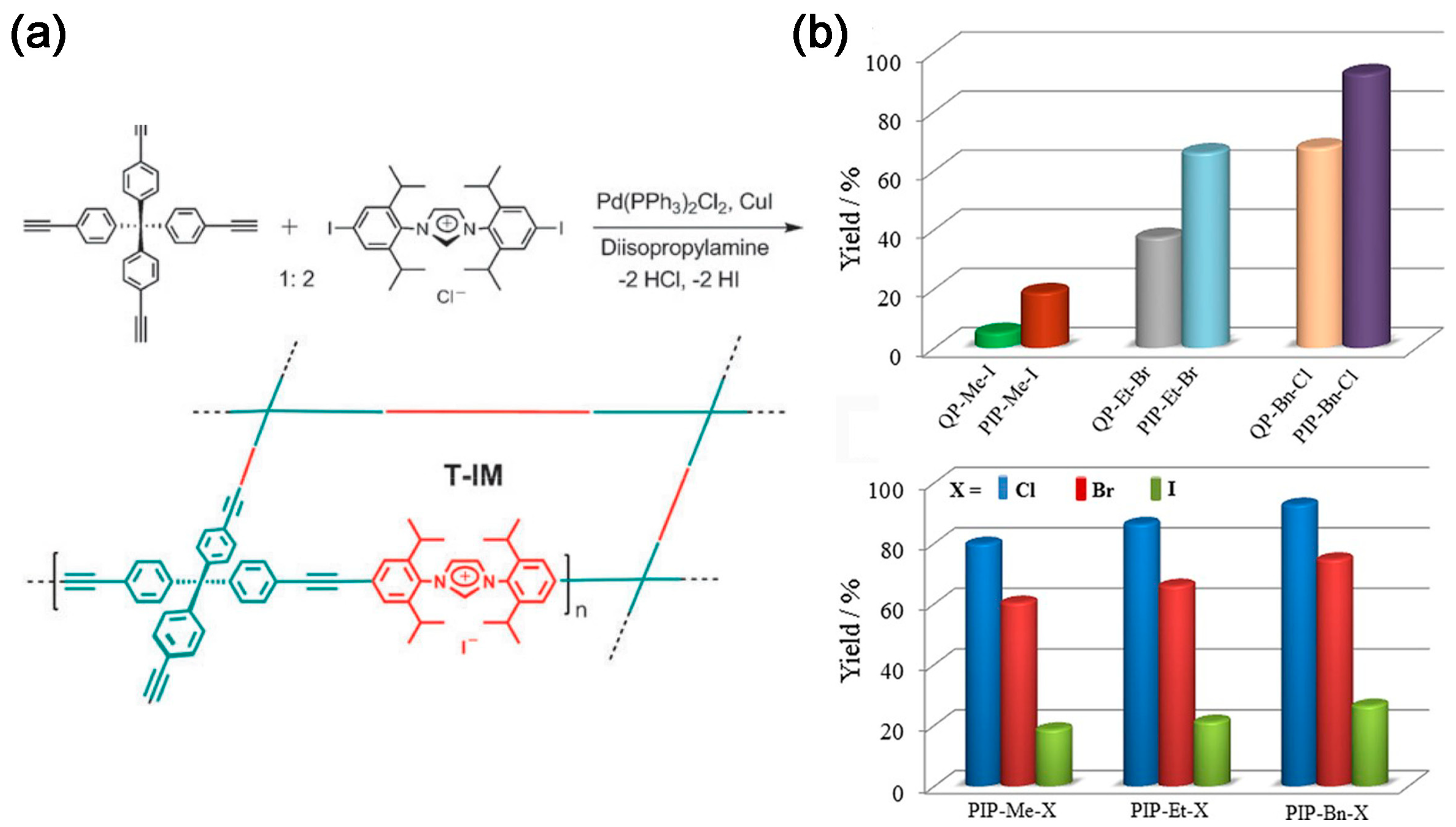
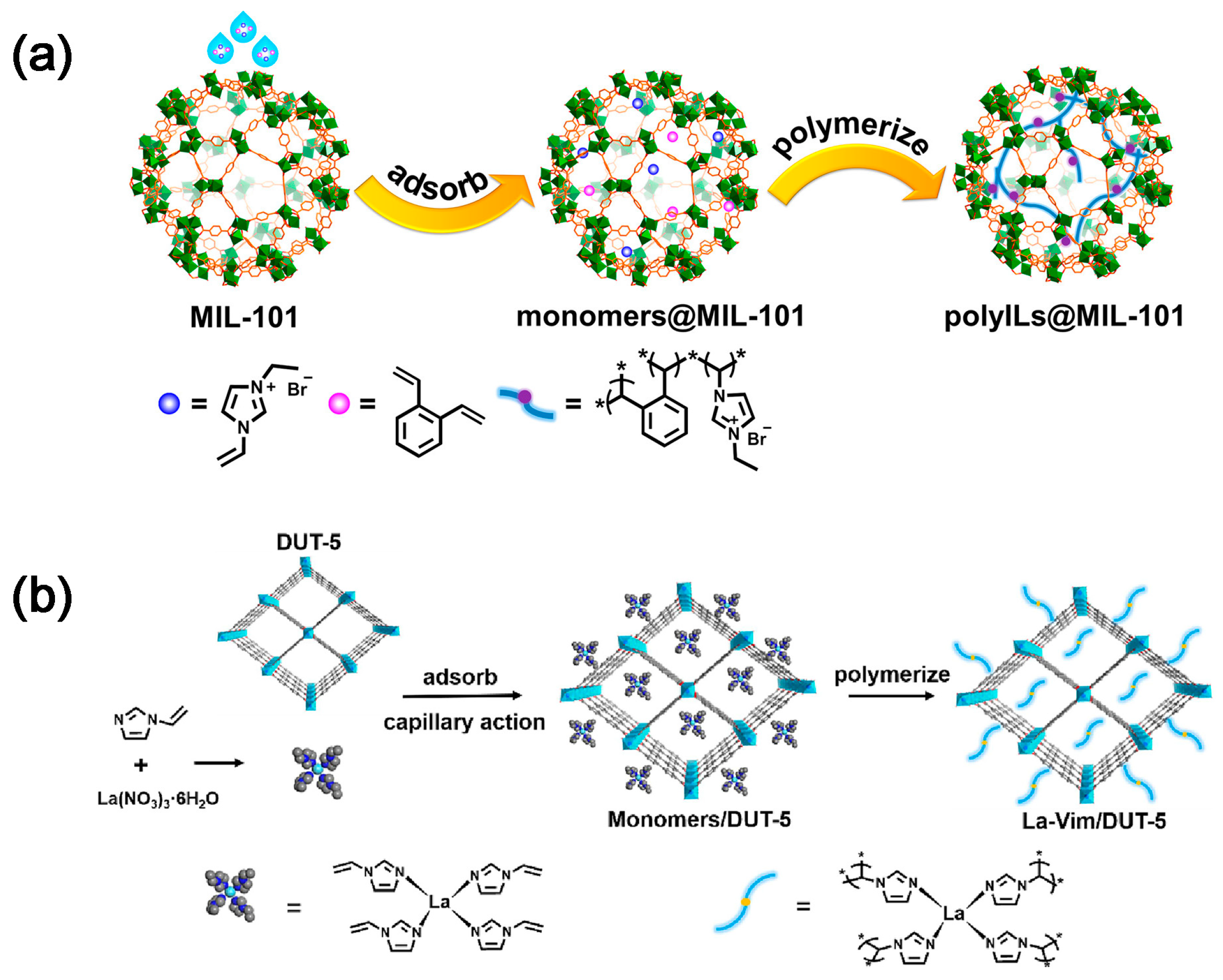
2.2. CO2 Adsorption and Conversion Integration
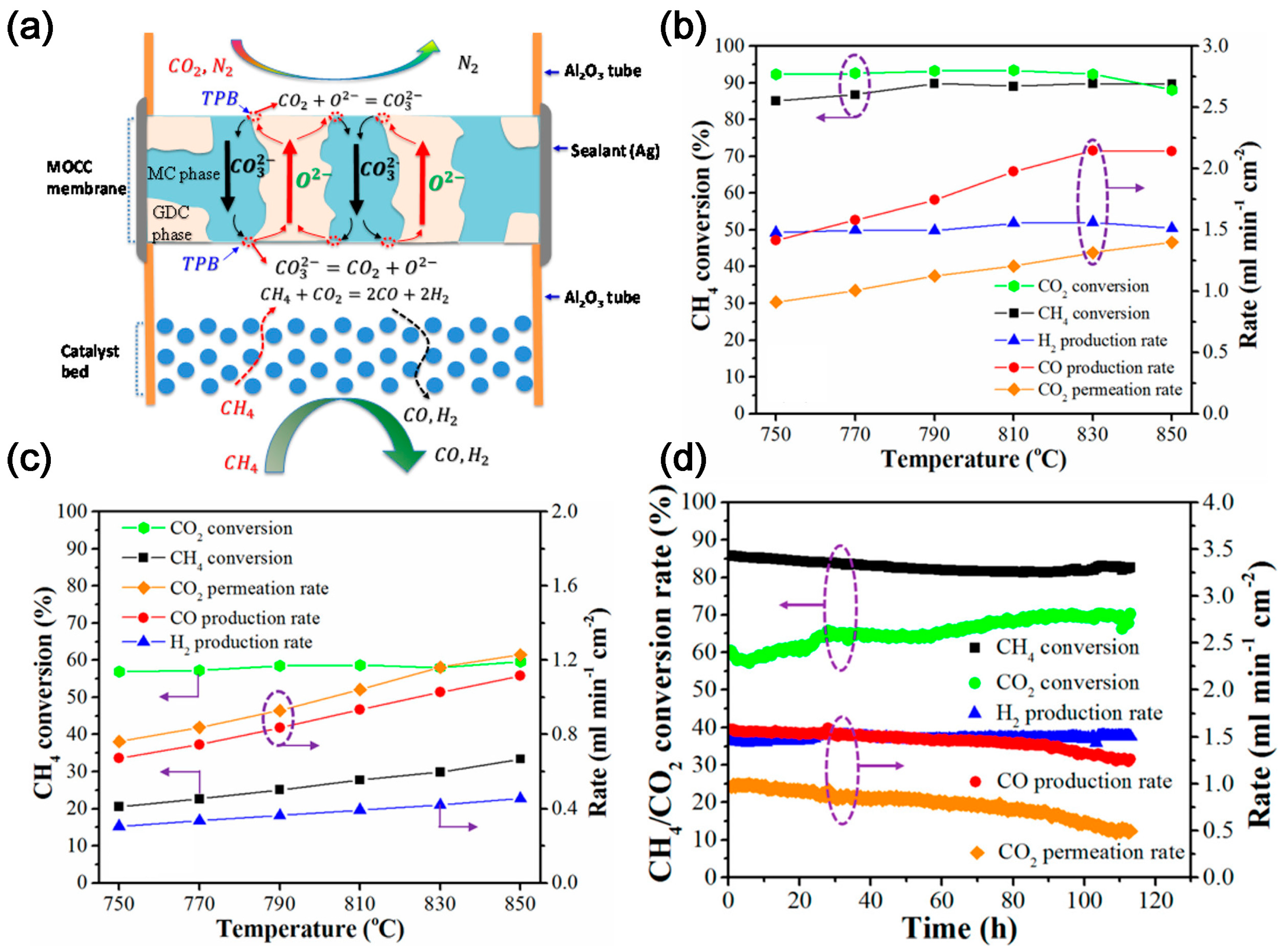
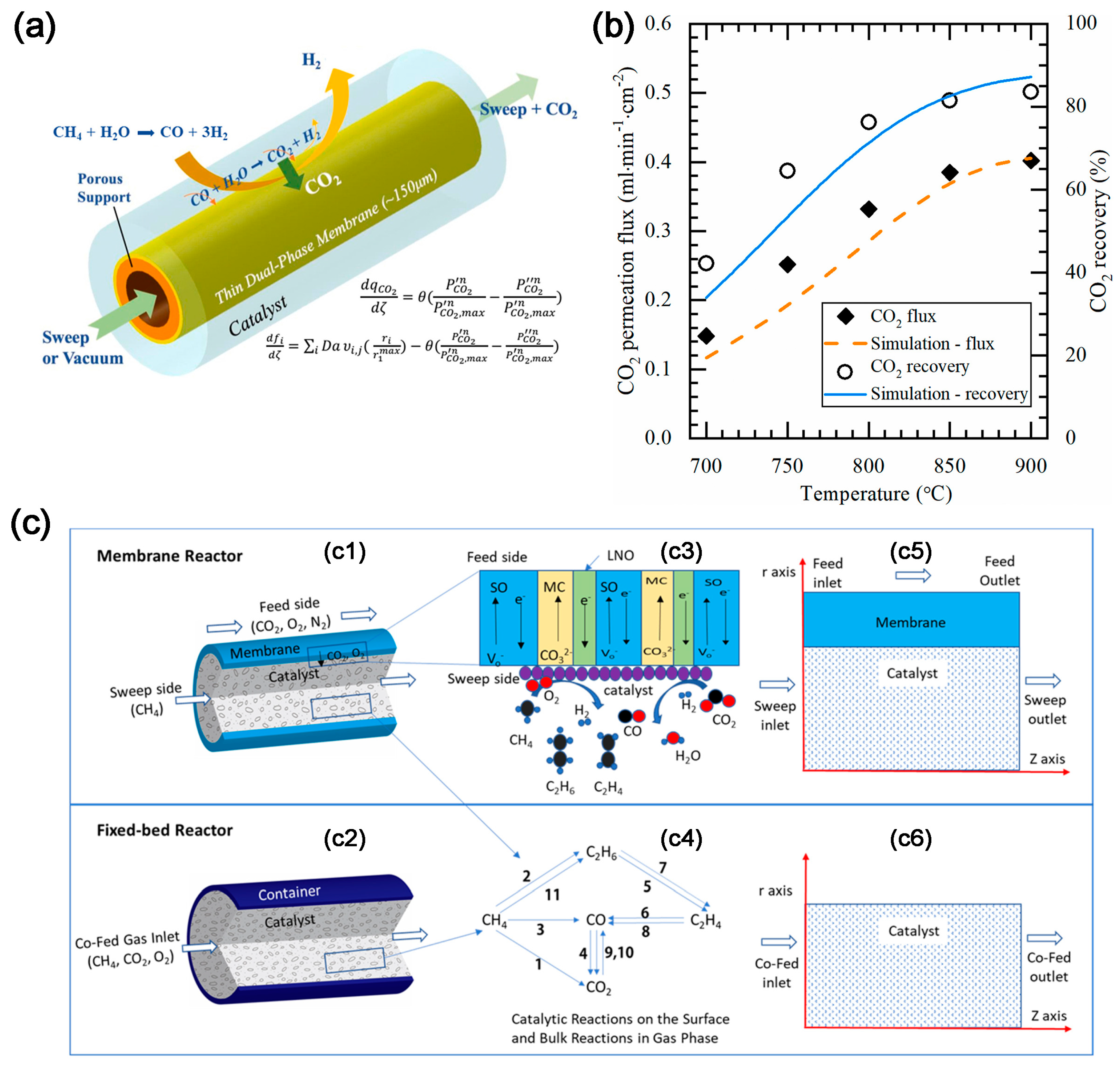
2.3. CO2 Electrochemical Membrane Separation and Conversion Integration

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membrane | Catalyst | Reaction | Ref. |
|---|---|---|---|
| La0.6Sr0.4Co0.8Fe0.2O3-δ Li-Na-K | 10 wt%Ni-/γ-Al2O3 | DMR | [29] |
| Ce0.8Gd0.2O1.9 Li-Na | Ni-MgO-1 wt% Pt LaNi0.6Fe0.4O3-δ | DMR | [31] |
| Ag Li-Na | Ni-MgO-1 wt% Pt | DOMR | [32] |
| NiO-SDC Li-Na | Ni-MgO-1 wt% Pt | DOMR | [40] |
| Ce0.8Gd0.2O1.9 Li-Na | 5 wt% Cr2O3- ZSM-5 | Ethane-to-Ethylene | [33] |
| Ce0.9Pr0.1O2-δ-Pr0.6Sr0.4Fe0.5Co0.5O3-δ Li-Na-K | 10 wt%Ni-/γ-Al2O3 | DOMR | [41] |
| LNO/SDC Li-Na | LNO/LCNO | RWGS | [34] |
| BYS-SDC Li-Na-K | Ni-based catalyst (HiFUEL R110) | SMR | [36] |
| γ-LiAlO2-Ag Li-Na-K | γ-LiAlO2-Ag | Syngas production | [42] |
| NiO-SDC Li-Na | 2%Mn-5%Na2WO4/SiO2 | OCM | [39] |
2.4. CO2 Capture and Conversion over Dual-Function Materials (DFMs)
3. Conclusions, Challenges and Opportunities
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Song, K.S.; Fritz, P.W.; Coskun, A. Porous organic polymers for CO2 capture, separation and conversion. Chem. Soc. Rev. 2022, 51, 9831–9852. [Google Scholar] [CrossRef]
- Zhang, P.; Tong, J.; Huang, K.; Zhu, X.; Yang, W. The current status of high temperature electrochemistry-based CO2 transport membranes and reactors for direct CO2 capture and conversion. Prog. Energy Combust. Sci. 2021, 82, 100888. [Google Scholar] [CrossRef]
- Yamada, H. Amine-based capture of CO2 for utilization and storage. Polym. J. 2021, 53, 93–102. [Google Scholar] [CrossRef]
- Maina, J.W.; Pringle, J.M.; Razal, J.M.; Nunes, S.; Vega, L.; Gallucci, F.; Dumée, L.F. Strategies for integrated capture and conversion of CO2 from dilute flue gases and the atmosphere. ChemSusChem 2021, 14, 1805–1820. [Google Scholar] [CrossRef] [PubMed]
- Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G.A.; Prakash, G.S. Conversion of CO2 from air into methanol using a polyamine and a homogeneous ruthenium catalyst. J. Am. Chem. Soc. 2016, 138, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Goeppert, A.; Prakash, G.S. Combined CO2 capture and hydrogenation to methanol: Amine immobilization enables easy recycling of active elements. ChemSusChem 2019, 12, 3172–3177. [Google Scholar] [CrossRef]
- Hanusch, J.M.; Kerschgens, I.P.; Huber, F.; Neuburger, M.; Gademann, K. Pyrrolizidines for direct air capture and CO2 conversion. Chem. Commun. 2019, 55, 949–952. [Google Scholar] [CrossRef]
- Guan, C.; Pan, Y.; Ang, E.P.L.; Hu, J.; Yao, C.; Huang, M.-H.; Li, H.; Lai, Z.; Huang, K.-W. Conversion of CO2 from air into formate using amines and phosphorus-nitrogen PN 3P-Ru (ii) pincer complexes. Green Chem. 2018, 20, 4201–4205. [Google Scholar] [CrossRef]
- Tailor, R.; Abboud, M.; Sayari, A. Supported polytertiary amines: Highly efficient and selective SO2 adsorbents. Environ. Sci. Technol. 2014, 48, 2025–2034. [Google Scholar] [CrossRef]
- Yang, S.; Zhan, L.; Xu, X.; Wang, Y.; Ling, L.; Feng, X. Graphene-based porous silica sheets impregnated with polyethyleneimine for superior CO2 capture. Adv. Mater. 2013, 25, 2130–2134. [Google Scholar] [CrossRef]
- Goeppert, A.; Czaun, M.; May, R.B.; Prakash, G.S.; Olah, G.A.; Narayanan, S. Carbon dioxide capture from the air using a polyamine based regenerable solid adsorbent. J. Am. Chem. Soc. 2011, 133, 20164–20167. [Google Scholar] [CrossRef]
- Li, Y.-N.; He, L.-N.; Liu, A.-H.; Lang, X.-D.; Yang, Z.-Z.; Yu, B.; Luan, C.-R. In situ hydrogenation of captured CO2 to formate with polyethyleneimine and Rh/monophosphine system. Green Chem. 2013, 15, 2825–2829. [Google Scholar] [CrossRef]
- McNamara, N.D.; Hicks, J.C. CO2 capture and conversion with a multifunctional polyethyleneimine-tethered iminophosphine iridium catalyst/adsorbent. ChemSusChem 2014, 7, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Kothandaraman, J.; Goeppert, A.; Czaun, M.; Olah, G.A.; Prakash, G.S. CO2 capture by amines in aqueous media and its subsequent conversion to formate with reusable ruthenium and iron catalysts. Green Chem. 2016, 18, 5831–5838. [Google Scholar] [CrossRef]
- Rezayee, N.M.; Huff, C.A.; Sanford, M.S. Tandem amine and ruthenium-catalyzed hydrogenation of CO2 to methanol. J. Am. Chem. Soc. 2015, 137, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xing, G.; Chen, W.; Chen, L. Porous organic polymers: A promising platform for efficient photocatalysis. Mater. Chem. Front. 2020, 4, 332–353. [Google Scholar] [CrossRef]
- Cho, H.C.; Lee, H.S.; Chun, J.; Lee, S.M.; Kim, H.J.; Son, S.U. Tubular microporous organic networks bearing imidazolium salts and their catalytic CO2 conversion to cyclic carbonates. Chem. Commun. 2011, 47, 917–919. [Google Scholar] [CrossRef]
- Wang, J.; Sng, W.; Yi, G.; Zhang, Y. Imidazolium salt-modified porous hypercrosslinked polymers for synergistic CO2 capture and conversion. Chem. Commun. 2015, 51, 12076–12079. [Google Scholar] [CrossRef]
- Sun, Q.; Jin, Y.; Aguila, B.; Meng, X.; Ma, S.; Xiao, F.S. Porous ionic polymers as a robust and efficient platform for capture and chemical fixation of atmospheric CO2. ChemSusChem 2017, 10, 1160–1165. [Google Scholar]
- Smith, J.G. Organic Chemistry, 3rd ed.; McGraw-Hill: Singapore, 2011. [Google Scholar]
- Dai, Z.; Sun, Q.; Liu, X.; Bian, C.; Wu, Q.; Pan, S.; Wang, L.; Meng, X.; Deng, F.; Xiao, F.-S. Metalated porous porphyrin polymers as efficient heterogeneous catalysts for cycloaddition of epoxides with CO2 under ambient conditions. J. Catal. 2016, 338, 202–209. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, R.; Xu, Q.; Zhang, W.; Zhou, X.; Ji, H. State-of-the-art aluminum porphyrin-based heterogeneous catalysts for the chemical fixation of CO2 into cyclic carbonates at ambient conditions. ChemCatChem 2017, 9, 767–773. [Google Scholar] [CrossRef]
- Zhai, G.; Liu, Y.; Lei, L.; Wang, J.; Wang, Z.; Zheng, Z.; Wang, P.; Cheng, H.; Dai, Y.; Huang, B. Light-promoted CO2 conversion from epoxides to cyclic carbonates at ambient conditions over a Bi-based metal–organic framework. ACS Catal. 2021, 11, 1988–1994. [Google Scholar] [CrossRef]
- Ding, M.; Jiang, H.-L. Incorporation of imidazolium-based poly (ionic liquid)s into a metal–organic framework for CO2 capture and conversion. ACS Catal. 2018, 8, 3194–3201. [Google Scholar] [CrossRef]
- Dai, W.; Li, Q.; Long, J.; Mao, P.; Xu, Y.; Yang, L.; Zou, J.; Luo, X. Hierarchically mesoporous imidazole-functionalized covalent triazine framework: An efficient metal-and halogen-free heterogeneous catalyst towards the cycloaddition of CO2 with epoxides. J. CO2 Util. 2022, 62, 102101. [Google Scholar] [CrossRef]
- Liu, F.; Duan, X.; Dai, X.; Du, S.; Ma, J.; Liu, F.; Liu, M. Metal-decorated porous organic frameworks with cross-linked pyridyl and triazinyl as efficient platforms for CO2 activation and conversion under mild conditions. Chem. Eng. J. 2022, 445, 136687. [Google Scholar] [CrossRef]
- Ma, P.; Ding, M.; Zhang, Y.; Rong, W.; Yao, J. Integration of lanthanide-imidazole containing polymer with metal-organic frameworks for efficient cycloaddition of CO2 with epoxides. Sep. Purif. Technol. 2023, 313, 123498. [Google Scholar] [CrossRef]
- Rui, Z.; Ji, H.; Lin, Y.S. Modeling and analysis of ceramic-carbonate dual-phase membrane reactor for carbon dioxide reforming with methane. Int. J. Hydrogen Energy 2011, 36, 8292–8300. [Google Scholar] [CrossRef]
- Anderson, M.; Lin, Y.S. Carbon dioxide separation and dry reforming of methane for synthesis of syngas by a dual-phase membrane reactor. AlChE J. 2013, 59, 2207–2218. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef]
- Zhang, P.; Tong, J.; Huang, K. Combining electrochemical CO2 capture with catalytic dry methane reforming in a single reactor for low-cost syngas production. ACS Sustain. Chem. Eng. 2016, 4, 7056–7065. [Google Scholar] [CrossRef]
- Zhang, P.; Tong, J.; Huang, K. Dry-oxy methane reforming with mixed e(-)/CO32- conducting membranes. ACS Sustain. Chem. Eng. 2017, 5, 5432–5439. [Google Scholar]
- Zhang, P.; Tong, J.; Huang, K. Role of CO2 in catalytic ethane-to-ethylene conversion using a high-temperature CO2 transport membrane reactor. ACS Sustain. Chem. Eng. 2019, 7, 6889–6897. [Google Scholar] [CrossRef]
- Chen, T.J.; Wang, Z.G.; Liu, L.N.; Pati, S.; Wai, M.H.; Kawi, S. Coupling CO2 separation with catalytic reverse water-gas shift reaction via ceramic-carbonate dual-phase membrane reactor. Chem. Eng. J. 2020, 379, 122182. [Google Scholar] [CrossRef]
- Xing, W.; Peters, T.; Fontaine, M.-L.; Evans, A.; Henriksen, P.P.; Norby, T.; Bredesen, R. Steam-promoted CO2 flux in dual-phase CO2 separation membranes. J. Membr. Sci. 2015, 482, 115–119. [Google Scholar] [CrossRef]
- Wu, H.C.; Rui, Z.B.; Lin, J.Y.S. Hydrogen production with carbon dioxide capture by dual-phase ceramic-carbonate membrane reactor via steam reforming of methane. J. Membr. Sci. 2020, 598, 117780. [Google Scholar] [CrossRef]
- Ovalle-Encinia, O.; Wu, H.C.; Chen, T.J.; Lin, J.Y.S. CO2-permselective membrane reactor for steam reforming of methane. J. Membr. Sci. 2022, 641, 119914. [Google Scholar] [CrossRef]
- Li, X.; Huang, K.; Van Dam, N.; Jin, X. Performance projection of a high-temperature CO2 transport membrane reactor for combined CO2 capture and methane-to-ethylene conversion. J. Electrochem. Soc. 2022, 169, 053501. [Google Scholar]
- Zhang, K.; Sun, S.; Huang, K. Oxidative coupling of methane (OCM) conversion into C2 products through a CO2/O2 co-transport membrane reactor. J. Membr. Sci. 2022, 661, 120915. [Google Scholar] [CrossRef]
- Zhang, P.; Tong, J.; Huang, K. Self-formed, mixed-conducting, triple-phase membrane for efficient CO2/O2 capture from flue gas and in situ dry-oxy methane reforming. ACS Sustain. Chem. Eng. 2018, 6, 14162–14169. [Google Scholar] [CrossRef]
- Fabian-Anguiano, J.A.; Mendoza-Serrato, C.G.; Gomez-Yanez, C.; Zeifert, B.; Ma, X.; Ortiz-Landeros, J. Simultaneous CO2 and O2 separation coupled to oxy-dry reforming of CH4 by means of a ceramic-carbonate membrane reactor for in situ syngas production. Chem. Eng. Sci. 2019, 210, 115250. [Google Scholar]
- Fabian-Anguiano, J.A.; Ramirez-Moreno, M.J.; Balmori-Ramirez, H.; Romero-Serrano, J.A.; Romero-Ibarra, I.C.; Ma, X.; Ortiz-Landeros, J. Syngas production with CO2 utilization through the oxidative reforming of methane in a new cermet-carbonate packed-bed membrane reactor. J. Membr. Sci. 2021, 637, 119607. [Google Scholar] [CrossRef]
- Shao, B.; Zhang, Y.; Sun, Z.; Li, J.; Gao, Z.; Xie, Z.; Hu, J.; Liu, H. CO2 capture and in-situ conversion: Recent progresses and perspectives. Green Chem. Eng. 2022, 3, 189–198. [Google Scholar] [CrossRef]
- Sun, S.; Sun, H.; Williams, P.T.; Wu, C. Recent advances in integrated CO2 capture and utilization: A review. Sustain. Energy Fuels 2021, 5, 4546–4559. [Google Scholar] [CrossRef]
- Zhang, W.; Ma, D.; Pérez-Ramírez, J.; Chen, Z. Recent progress in materials exploration for thermocatalytic, photocatalytic, and integrated photothermocatalytic CO2-to-fuel conversion. Adv. Energy Sustain. Res. 2022, 3, 2100169. [Google Scholar] [CrossRef]
- Kim, S.M.; Abdala, P.M.; Broda, M.; Hosseini, D.; Copéret, C.; Müller, C. Integrated CO2 capture and conversion as an efficient process for fuels from greenhouse gases. ACS Catal. 2018, 8, 2815–2823. [Google Scholar] [CrossRef]
- Tian, S.; Yan, F.; Zhang, Z.; Jiang, J. Calcium-looping reforming of methane realizes in situ CO2 utilization with improved energy efficiency. Sci. Adv. 2019, 5, eaav5077. [Google Scholar] [CrossRef]
- Hu, J.; Hongmanorom, P.; Chirawatkul, P.; Kawi, S. Efficient integration of CO2 capture and conversion over a Ni supported CeO2-modified CaO microsphere at moderate temperature. Chem. Eng. J. 2021, 426, 130864. [Google Scholar] [CrossRef]
- Wang, G.; Guo, Y.; Yu, J.; Liu, F.; Sun, J.; Wang, X.; Wang, T.; Zhao, C. Ni-CaO dual function materials prepared by different synthetic modes for integrated CO2 capture and conversion. Chem. Eng. J. 2022, 428, 132110. [Google Scholar] [CrossRef]
- Sun, H.; Wang, J.; Zhao, J.; Shen, B.; Shi, J.; Huang, J.; Wu, C. Dual functional catalytic materials of Ni over Ce-modified CaO sorbents for integrated CO2 capture and conversion. Appl. Catal. B Environ. 2019, 244, 63–75. [Google Scholar] [CrossRef]
- Duyar, M.S.; Trevino, M.A.A.; Farrauto, R.J. Dual function materials for CO2 capture and conversion using renewable H2. Appl. Catal. B Environ. 2015, 168, 370–376. [Google Scholar] [CrossRef]
- Arellano-Treviño, M.A.; He, Z.; Libby, M.C.; Farrauto, R.J. Catalysts and adsorbents for CO2 capture and conversion with dual function materials: Limitations of Ni-containing DFMs for flue gas applications. J. CO2 Util. 2019, 31, 143–151. [Google Scholar] [CrossRef]
- Porta, A.; Visconti, C.G.; Castoldi, L.; Matarrese, R.; Jeong-Potter, C.; Farrauto, R.; Lietti, L. Ru-Ba synergistic effect in dual functioning materials for cyclic CO2 capture and methanation. Appl. Catal. B Environ. 2021, 283, 119654. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.; González-Velasco, J. Mechanism of the CO2 storage and in situ hydrogenation to CH4. Temperature and adsorbent loading effects over Ru-CaO/Al2O3 and Ru-Na2CO3/Al2O3 catalysts. Appl. Catal. B Environ. 2019, 256, 117845. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.; González-Velasco, J. Ni loading effects on dual function materials for capture and in-situ conversion of CO2 to CH4 using CaO or Na2CO3. J. CO2 Util. 2019, 34, 576–587. [Google Scholar] [CrossRef]
- Al-Mamoori, A.; Rownaghi, A.A.; Rezaei, F. Combined capture and utilization of CO2 for syngas production over dual-function materials. ACS Sustain. Chem. Eng. 2018, 6, 13551–13561. [Google Scholar] [CrossRef]
- Duyar, M.S.; Wang, S.; Arellano-Trevino, M.A.; Farrauto, R.J. CO2 utilization with a novel dual function material (DFM) for capture and catalytic conversion to synthetic natural gas: An update. J. CO2 Util. 2016, 15, 65–71. [Google Scholar] [CrossRef]
- Garbarino, G.; Bellotti, D.; Riani, P.; Magistri, L.; Busca, G. Methanation of carbon dioxide on Ru/Al2O3 and Ni/Al2O3 catalysts at atmospheric pressure: Catalysts activation, behaviour and stability. Int. J. Hydrogen Energy 2015, 40, 9171–9182. [Google Scholar] [CrossRef]
- Wang, S.; Schrunk, E.T.; Mahajan, H.; Farrauto, R.J. The role of ruthenium in CO2 capture and catalytic conversion to fuel by dual function materials (DFM). Catalysts 2017, 7, 88. [Google Scholar] [CrossRef]
- Wang, S.; Farrauto, R.J.; Karp, S.; Jeon, J.H.; Schrunk, E.T. Parametric, cyclic aging and characterization studies for CO2 capture from flue gas and catalytic conversion to synthetic natural gas using a dual functional material (DFM). J. CO2 Util. 2018, 27, 390–397. [Google Scholar] [CrossRef]






| Entry | Catalysts | Additives | Time (h) | Conv. (%) a | Select. (%) a |
|---|---|---|---|---|---|
| 1 b | POP-TPP | n-Bu4NBr | 24 | 52.1 | >99.0 |
| 2 c | Co/POP-TPP | n-Bu4NBr | 24 | 95.6 | 99 |
| 3 c | Co/TPP | n-Bu4NBr | 24 | 97.5 | 99 |
| 4 | Co/POP-TPP | None | 24 | 9.7 | 99 |
| 5 | Co/TPP | None | 24 | 18.5 | 99 |
| 6 | None | n-Bu4NBr | 24 | 34 | 99 |
| 7 d | Co/POP-TPP | n-Bu4NBr | 96 | 96.1 | 99 |
| 8 e | Co/POP-TPP | n-Bu4NBr | 24 | 88.9 | 99 |
| 9 f | Zn/POP-TPP | n-Bu4NBr | 24 | 93.2 | >99.0 |
| 10 f | Zn/TPP | n-Bu4NBr | 24 | 93.5 | >99.0 |
| 11 g | Mg/POP-TPP | n-Bu4NBr | 24 | 80.5 | >99.0 |
| 12 g | Mg/TPP | n-Bu4NBr | 24 | 99.3 | >99.0 |
| 13 h | Co/POP-TPP | n-Bu4NBr | 24 | 93.6 | 99 |
| DFM | Condition (°C) | Ref. | |
|---|---|---|---|
| Absorption | Reaction | ||
| Ni-CaO/Al2O3 | 280–520 10% CO2/Ar | 280–520 10% H2/Ar | [55] |
| Ni-Na2CO3/Al2O3 | 280–520 10% CO2/Ar | 280–520 10% H2/Ar | |
| Ni-Na2O/Al2O3 | 320 7.5% CO2/N2 and 7.5% CO2, 4.5% O2, 15% H2O/N2 | 320 15% H2/N2 | [52] |
| Ru-Na2O/γ-Al2O3 | 320 15% CO2/N2 | 320 20% H2/N2 | [57] |
| Rh-CaO/γ-Al2O3 | 320 10% CO2/N2 | 320 2% H2/N2 | |
| Ru-CaO/γ-Al2O3 | 280–400 1.4% CO2/Ar and 11% CO2/Ar | 280–400 10% H2/Ar | [54] |
| Ru-Na2CO3/γ-Al2O3 | 280–400 1.4% CO2/Ar and 11% CO2/A | 280–400 10% H2/Ar | |
| Ru-CaO/γ-Al2O3 | 320 10% CO2/air and 8% CO2/21% H2O/air | 320 5% H2/N2 | [51] |
| Ru-CaO/γ-Al2O3 | 320 10% CO2/N2 | 320 4% H2/N2 | [57] |
| Ru-CaO/γ-Al2O3 | 320 7.5% CO2, 4.5% O2, 15% H2O/N2 | 320 5% H2/N2 | [58] |
| Ru-Na2CO3/γ-Al2O3 | 320 7.5% CO2/N2 and 7.5% CO2, 4.5% O2, 15% H2O/N2 | 320 5% H2/N2 | [59] |
| Ru-Na2O/γ-Al2O3 | 250–350 7.5% CO2, 4.5% O2, 15% H2O/N2 | 250–350 15% H2/N2 | [60] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ning, H.; Li, Y.; Zhang, C. Recent Progress in the Integration of CO2 Capture and Utilization. Molecules 2023, 28, 4500. https://doi.org/10.3390/molecules28114500
Ning H, Li Y, Zhang C. Recent Progress in the Integration of CO2 Capture and Utilization. Molecules. 2023; 28(11):4500. https://doi.org/10.3390/molecules28114500
Chicago/Turabian StyleNing, Huanghao, Yongdan Li, and Cuijuan Zhang. 2023. "Recent Progress in the Integration of CO2 Capture and Utilization" Molecules 28, no. 11: 4500. https://doi.org/10.3390/molecules28114500
APA StyleNing, H., Li, Y., & Zhang, C. (2023). Recent Progress in the Integration of CO2 Capture and Utilization. Molecules, 28(11), 4500. https://doi.org/10.3390/molecules28114500