
Store-Operated Calcium Entry Inhibition and Plasma Membrane Calcium Pump Upregulation Contribute to the Maintenance of Resting Cytosolic Calcium Concentration in A1-like Astrocytes

 ,
, 
 and
and
Abstract
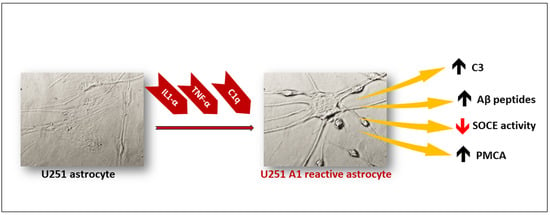
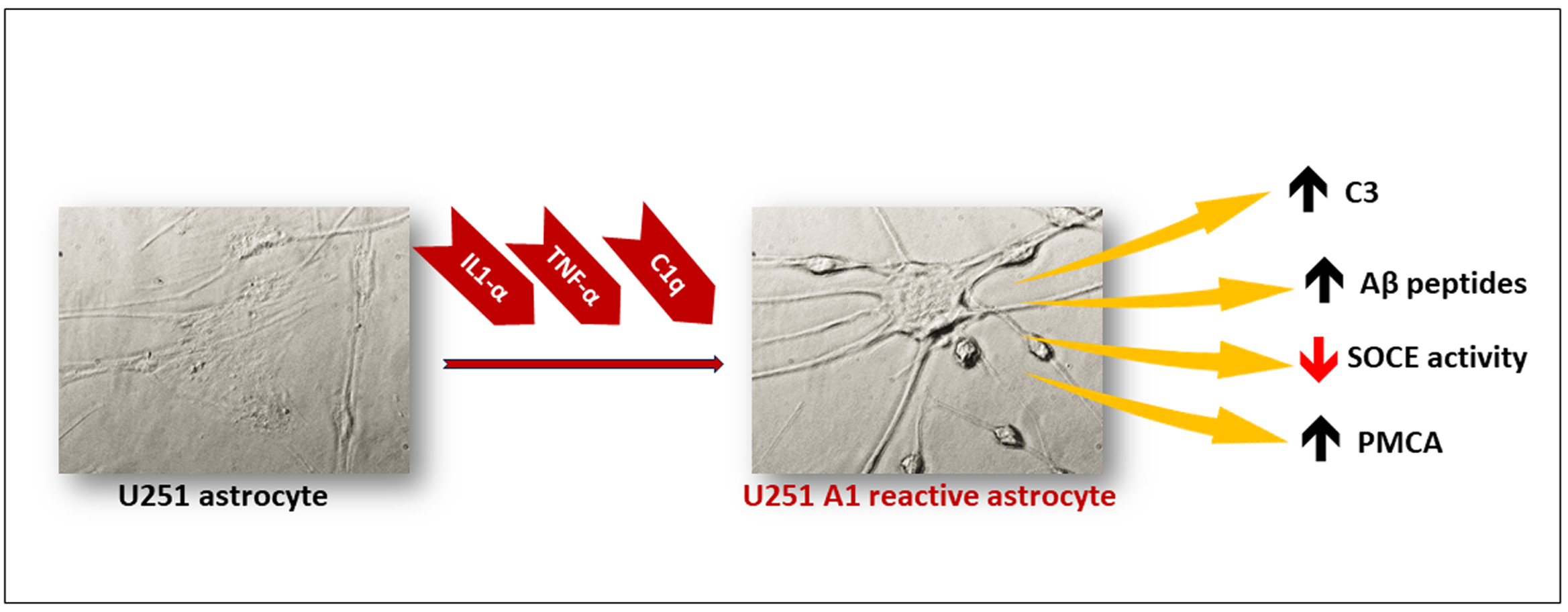
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
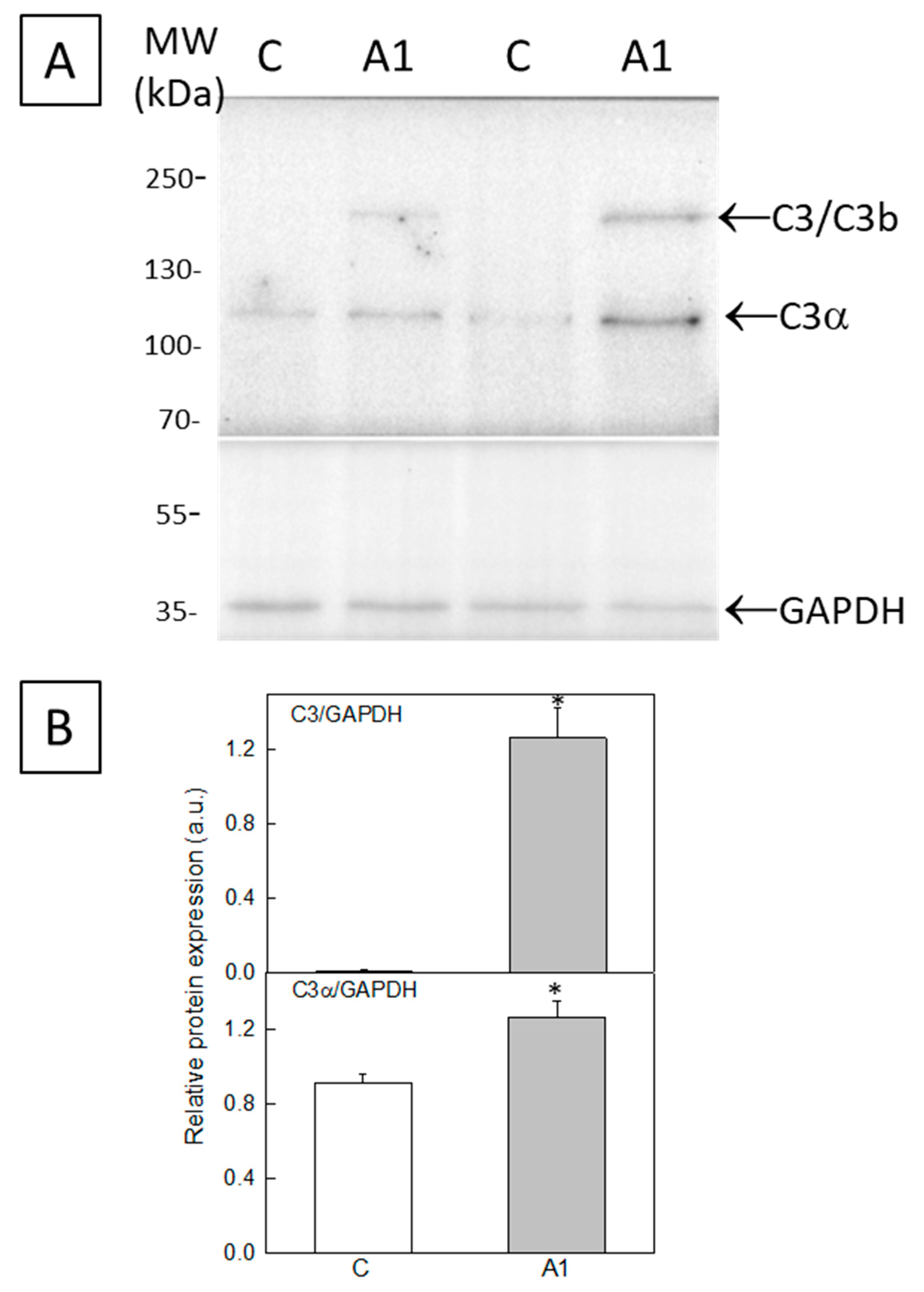
2.1. Selection of A1-like Astrocytes Using C3 Expression as Biomarker
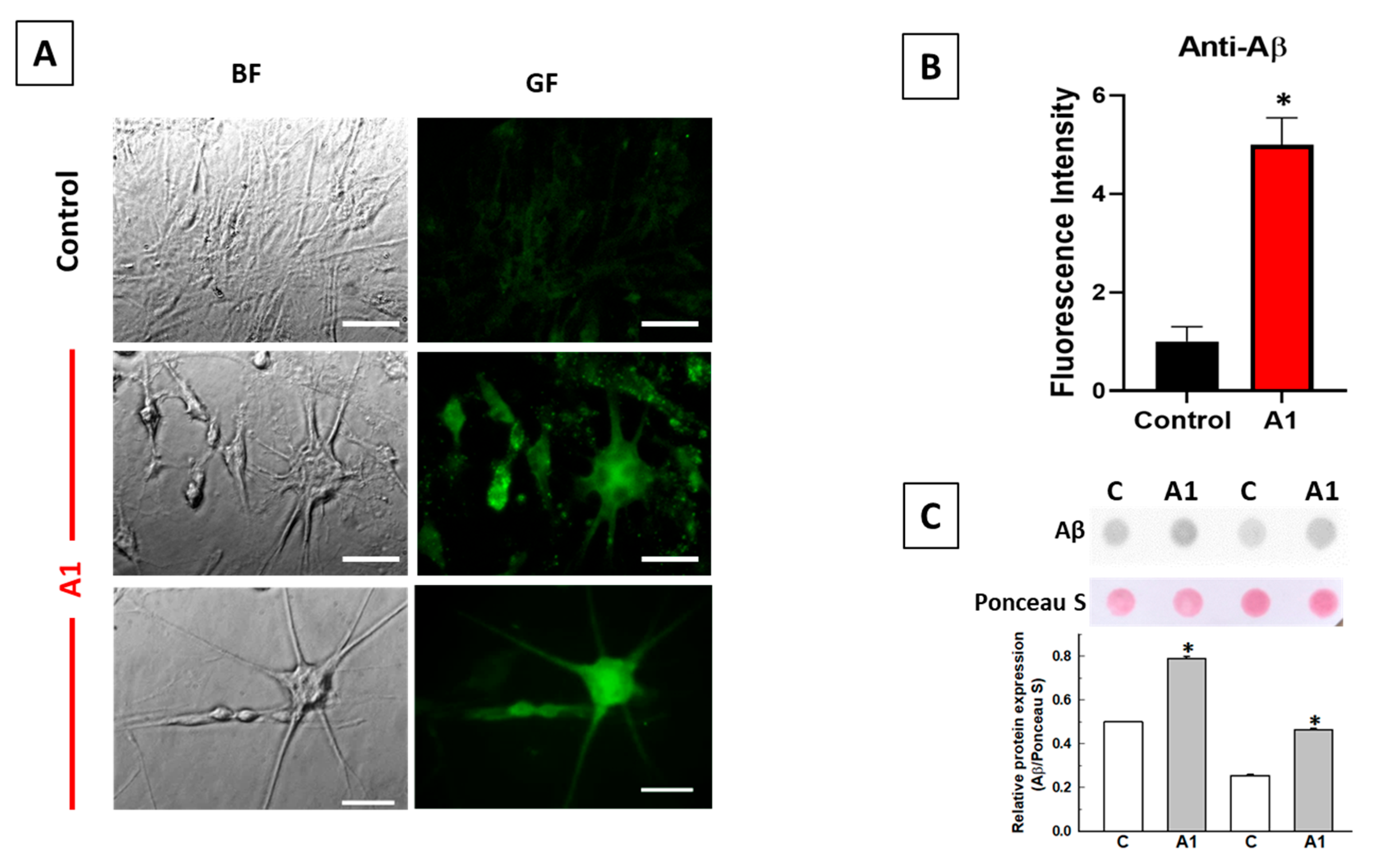
2.2. A1-like Astrocytes Express Higher Levels of Aβ Peptides Than Control U251 Astrocytes
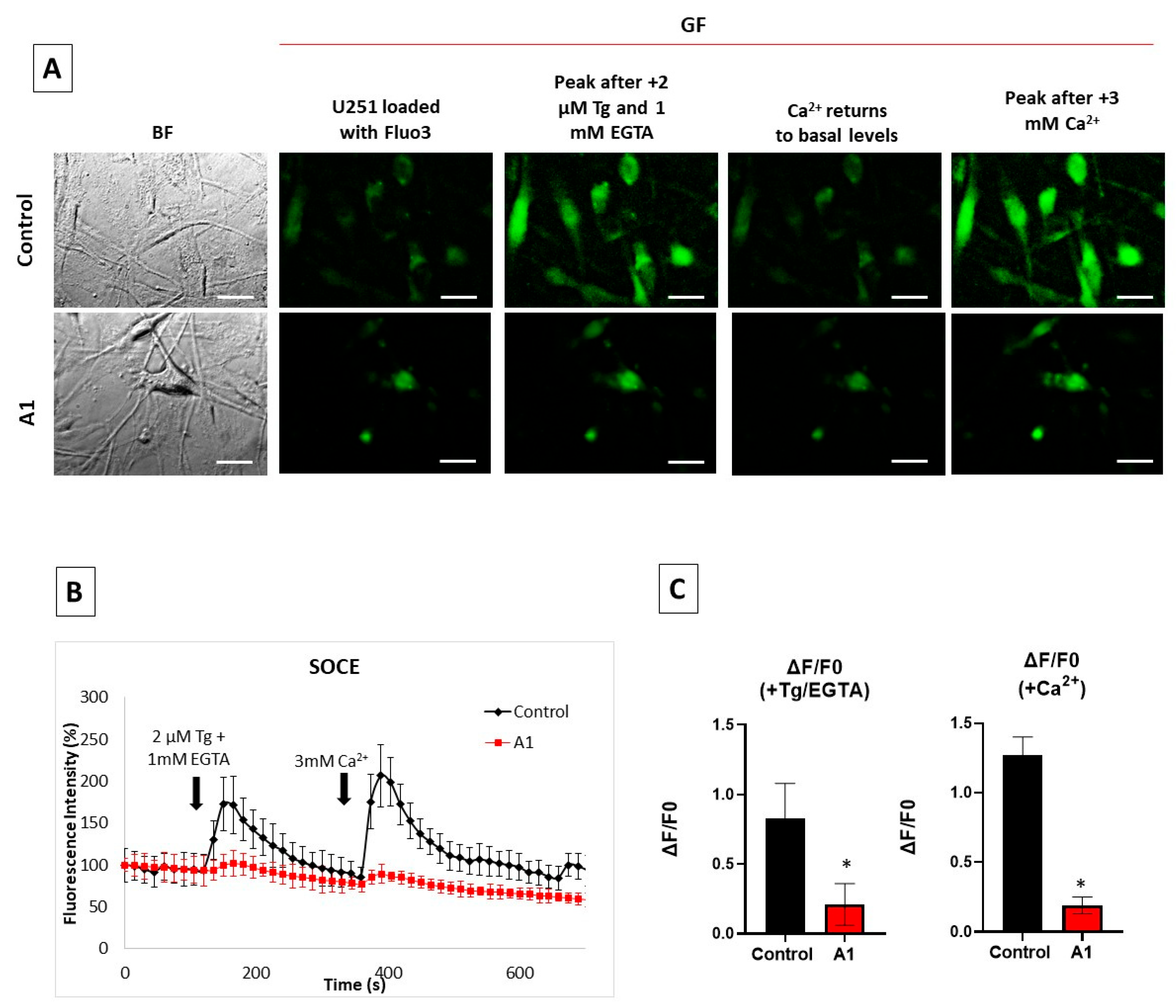
2.3. Store-Operated Calcium Entry (SOCE) Is Largely Decreased during the Transformation of U251 Astrocytes in A1-like Astrocytes
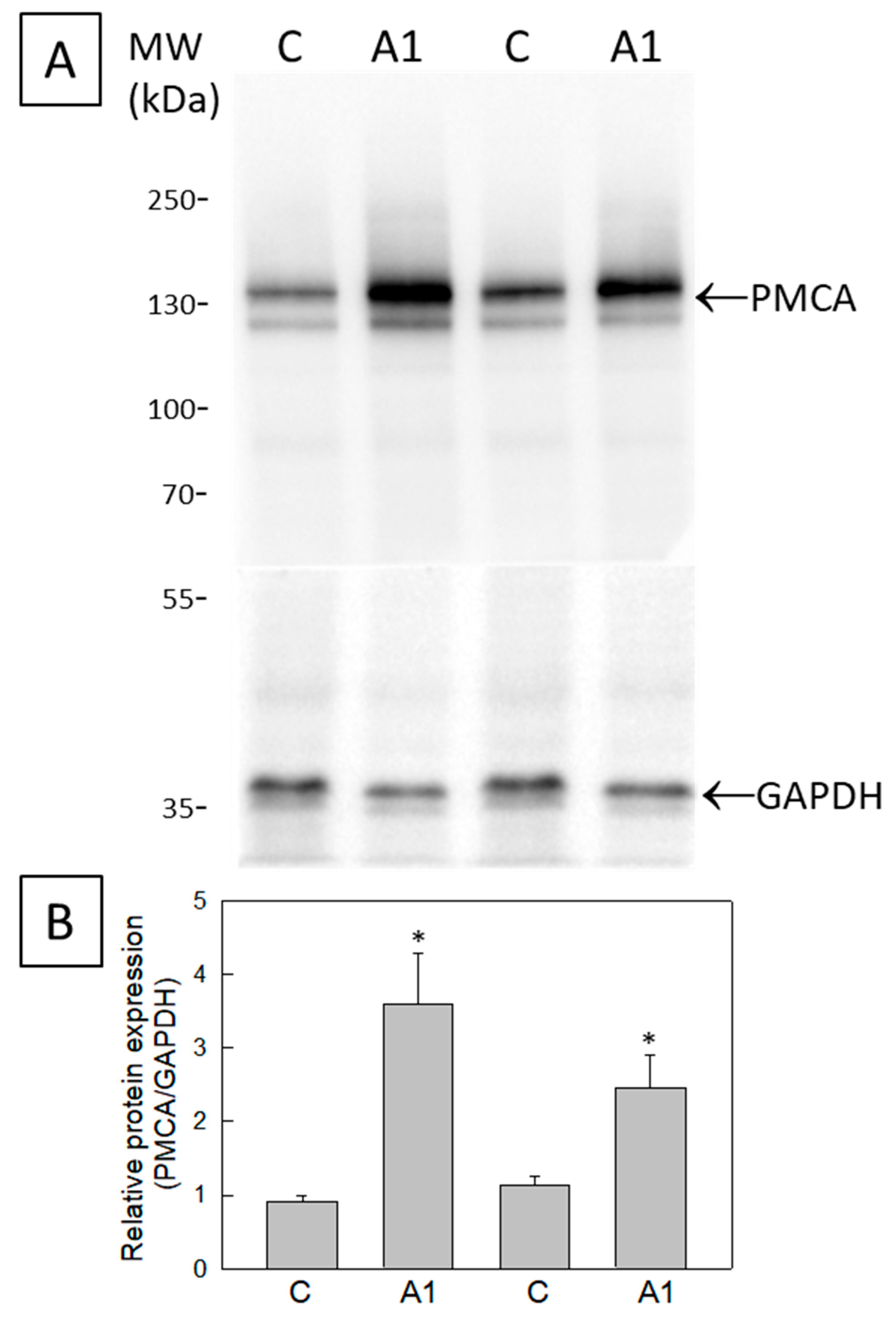
2.4. The Cytosolic Ca2+ Concentration Does Not Increase during the Transformation of U251 Astrocytes in A1-like Astrocytes, Which Express Higher Levels of PMCA Than the Control U251 Astrocytes
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Preparation of Reactive Astrocytes
4.3. U251 Cell Staining with C3 and Aβ Antibodies
4.4. Cell Homogenate Preparation, and Western Blot and Dot Blot Analyses
4.5. Intracellular Cytosolic Ca2+ Measurements
4.6. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| Aβ | amyloid β peptide |
| AD | Alzheimer’s disease |
| APP | amyloid β protein precursor |
| BACE1 | β-site APP-cleaving enzyme 1 |
| C1q | complement component 1q |
| DMEM | Dulbecco’s modified Eagle’s medium |
| EGTA | ethyleneglycoltetraacetic acid |
| ER | endoplasmic reticulum |
| Fluo3 AM | Fluo-3-pentaacetoxymethyl ester |
| Fura2 AM | Fura2 acetoxymethyl ester |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GF | green fluorescence |
| HEPES | N-[2-hydroxyethyl] piperazine-N′-[2-ethanesulfonic acid] |
| IL-1α | interleukin-1α |
| NPA | 3-nitropropionic acid |
| PBS | phosphate-buffered saline |
| PBST | PBS supplemented with 0.2% 4-(1,1,3,3-tetramethyl butyl) phenyl-polyethylene glycol (Triton X-100®) |
| PMCA | plasma membrane calcium pump |
| PSEN | presenilin |
| PVDF | polyvinylidene difluoride |
| RF | red fluorescence |
| ROI | region of interest |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| SEM | standard error of the mean |
| SOCE | store-operated calcium entry |
| STIM1 | stromal interaction molecule 1 |
| Tg | thapsigargin |
| TNFα | tumor necrosis factor α |
References
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.; MacLean, A.G. New advances on glial activation in health and disease. World J. Virol. 2015, 4, 42–55. [Google Scholar] [CrossRef]
- Lopez-Sanchez, C.; Garcia-Martinez, V.; Poejo, J.; Garcia-Lopez, V.; Salazar, J.; Gutierrez-Merino, C. Early reactive A1 astrocytes induction by the neurotoxin 3-nitropropionic acid in rat brain. Int. J. Mol. Sci. 2020, 21, 3609. [Google Scholar] [CrossRef]
- Lopez-Sanchez, C.; Poejo, J.; Garcia-Lopez, V.; Salazar, J.; Garcia-Martinez, V.; Gutierrez-Merino, C. Kaempferol prevents the activation of complement C3 protein and the generation of reactive A1 astrocytes that mediate rat brain degeneration induced by 3-nitropropionic acid. Food Chem. Toxicol. 2022, 164, 113017. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sun, Q.; Yang, J.; Wang, G.; Zhao, F.; Chen, Y.; Jin, Y. Reactive astrocytes induced by 2-chloroethanol modulate microglia polarization through IL-1β, TNF-α, and iNOS upregulation. Food Chem. Toxicol. 2021, 157, 112550. [Google Scholar] [CrossRef] [PubMed]
- Machein, U.; Lieb, K.; Hüll, M.; Fiebich, B.L. IL-1β and TNFα, but not IL-6, induce a1-antichymotrypsin expression in the human astrocytoma cell line U373 MG. Neuroreport 1995, 6, 2283–2286. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Leiter, L.M.; McPhee, J.; Cahill, C.M.; Zhan, S.S.; Potter, H.; Nilsson, L.N. Translation of the Alzheimer amyloid precursor protein mRNA is upregulated by interleukin-1 through 50-untranslated region sequences. J. Biol. Chem. 1999, 274, 6421–6431. [Google Scholar] [CrossRef] [Green Version]
- Blasko, I.; Veerhuis, R.; Stampfer-Kountchev, M.; Saurwein-Teissl, M.; Eikelenboom, P.; Grubeck-Loebenstein, B. Costimulatory effects of interferon-γ and interleukin-1β or tumor necrosis factor a on the synthesis of Aβ1–40 and Aβ1–42 by human astrocytes. Neurobiol. Dis. 2000, 7, 682–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Aβ production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflammation 2011, 8, 150. [Google Scholar] [CrossRef] [Green Version]
- Khakh, B.S.; McCarthy, K.D. Astrocyte calcium signaling: From observations to functions and the challenges therein. Cold Spring Harb. Perspect. Biol. 2015, 7, a020404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.; Kosuri, P.; Arancio, O. Picomolar Amyloid-β Peptides Enhance Spontaneous Astrocyte Calcium Transients. J. Alzheimers Dis. 2014, 38, 49–62. [Google Scholar] [CrossRef]
- Angulo, M.C.; Kozlov, A.S.; Charpak, S.; Audinat, E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J. Neurosci. 2004, 24, 6920–6927. [Google Scholar] [CrossRef] [Green Version]
- Fellin, T.; Pascual, O.; Gobbo, S.; Pozzan, T.; Haydon, P.G.; Carmignoto, G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 2004, 43, 729–743. [Google Scholar] [CrossRef] [Green Version]
- Kuchibhotla, K.V.; Lattarulo, C.R.; Hyman, B.T.; Bacskai, B.J. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 2009, 323, 1211–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delekate, A.; Fuchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef] [Green Version]
- Poejo, J.; Salazar, J.; Mata, A.M.; Gutierrez-Merino, C. The Relevance of Amyloid β-Calmodulin Complexation in Neurons and Brain Degeneration in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4976. [Google Scholar] [CrossRef]
- D’Andrea, M.R.; Nagele, R.G.; Wang, H.Y.; Peterson, P.A.; Lee, D.H. Evidence that neurons accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer’s disease. Histopathology 2001, 38, 120–134. [Google Scholar] [CrossRef]
- Leissring, M.A.; Akbari, Y.; Fanger, C.M.; Cahalan, M.D.; Mattson, M.P.; LaFerla, F.M. Capacitative Calcium Entry Deficits and Elevated Luminal Calcium Content in Mutant Presenilin-1 Knockin Mice. J. Cell Biol. 2000, 149, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Yoo, A.S.; Cheng, I.; Chung, S.; Grenfell, T.Z.; Lee, H.; Pack-Chung, E.; Handler, M.; Shen, J.; Xia, W.; Tesco, G.; et al. Presenilin-Mediated Modulation of Capacitative Calcium Entry. Neuron 2000, 27, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease—A therapeutic opportunity? Biochem. Biophys. Res. Commun. 2017, 483, 998–1004. [Google Scholar] [CrossRef] [Green Version]
- Leissring, M.A.; Parker, I.; LaFerla, F.M. Presenilin-2 Mutations Modulate Amplitude and Kinetics of Inositol 1,4,5-Trisphosphate-mediated Calcium Signals. J. Biol. Chem. 1999, 274, 32535–32538. [Google Scholar] [CrossRef] [Green Version]
- Chavez, S.E.; O’Day, D.H. Calmodulin binds to and regulates the activity of beta-secretase (BACE1). Curr. Res. Alzheimers Dis. 2007, 1, 37–47. [Google Scholar]
- Giliberto, L.; Borghi, R.; Piccini, A.; Mangerini, R.; Sorbi, S.; Cirmena, G.; Garuti, A.; Ghetti, B.; Tagliavini, F.; Mughal, M.R.; et al. Mutant Presenilin 1 Increases the Expression and Activity of BACE1. J. Biol. Chem. 2009, 284, 9027–9038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeiger, W.; Vetrivel, K.S.; Buggia-Prévot, V.; Nguyen, P.D.; Wagner, S.L.; Villereal, M.L.; Thinakaran, G. Ca2+ Influx through Store-operated Ca2+ Channels Reduces Alzheimer Disease β-Amyloid Peptide Secretion. J. Biol. Chem. 2013, 288, 26955–26966. [Google Scholar] [CrossRef] [PubMed]
- Poejo, J.; Orantos-Aguilera, Y.; Martin-Romero, F.J.; Mata, A.M.; Gutierrez-Merino, C. Internalized Amyloid-β (1-42) Peptide Inhibits the Store-Operated Calcium Entry in HT-22 Cells. Int. J. Mol. Sci. 2022, 23, 12678. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Taylor, A.M.W.; Nagai, J.; Golshani, P.; Evans, C.J.; Coppola, G.; Khakh, B.S. Reducing astrocyte calcium signaling in vivo alters striatal microcircuits and causes repetitive behavior. Neuron 2018, 99, 1170–1187.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, C.; Hérault, K.; Oheim, M.; Maldera, S.; Vialou, V.; Cauli, B.; Li, D. Astrocytes respond to a neurotoxic Aβ fragment with state-dependent Ca2+ alteration and multiphasic transmitter release. Acta Neuropathol. Commun. 2021, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Nadler, Y.; Alexandrovich, A.; Grigoriadis, N.; Hartmann, T.; Rao, K.S.J.; Shohami, E.; Stein, R. Increased expression of the gamma-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia 2008, 56, 552–567. [Google Scholar] [CrossRef] [PubMed]
- Frost, G.R.; Li, Y.M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef] [Green Version]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.Y.; Hyman, B.T.; Bacskai, B.J. Aβ Plaques Lead to Aberrant Regulation of Calcium Homeostasis in Vivo Resulting in Structural and Functional Disruption of Neuronal Networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J. Calcium Signalling and Alzheimer’s Disease. Neurochem. Res. 2011, 36, 1149–1156. [Google Scholar] [CrossRef]
- Fresu, L.; Dehpour, A.; Genazzani, A.A.; Carafoli, E.; Guerini, D. Plasma membrane calcium ATPase isoforms in astrocytes. Glia 1999, 28, 150–155. [Google Scholar] [CrossRef]
- Prà, I.D.; Whitfileld, J.F.; Pacchiana, R.; Bonafini, C.; Talacchi, A.; Chakravarthy, B.; Armato, U.; Chiarini, A. The amyloid-β42 proxy, amyloid-β25-35, induces normal human cerebral astrocytes to produce amyloid-β42. J. Alzheimers Dis. 2011, 24, 335–347. [Google Scholar] [CrossRef]
- Kim, K.S.; Miller, D.L.; Sapienza, V.J.; Chen, C.M.J.; Bai, C.; Grundke-Iqbal, I.; Currie, C.J.; Wisniewski, H.M. Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyloid peptide. Neurosci. Res. Commun. 1988, 2, 121–130. [Google Scholar]
- Hatami, A.; Albay, R., 3rd; Monjazeb, S.; Milton, S.; Glabe, C. Monoclonal antibodies against Abeta42 fibrils distinguish multiple aggregation state polymorphisms in vitro and in Alzheimer disease brain. J. Biol. Chem. 2014, 289, 32131–32143. [Google Scholar] [CrossRef] [Green Version]
- Baghallab, I.; Reyes-Ruiz, J.M.; Abulnaja, K.; Huwait, E.; Glabe, C. Epitomic Characterization of the Specificity of the Anti-Amyloid Aβ Monoclonal Antibodies 6E10 and 4G8. J. Alzheimers Dis. 2018, 66, 1235–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Laird, F.M.; Cai, H.; Savonenko, A.V.; Farah, M.H.; He, K.; Melnikova, T.; Wen, H.; Chiang, H.C.; Xu, G.; Koliatsos, V.E.; et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J. Neurosci. 2005, 25, 11693–11709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandel, E.R.; Schwartz, J.H.; Jessell, T.M. Principles of Neural Science, 4th ed.; McGraw-Hill Medical Press: New York, NY, USA, 2000. [Google Scholar]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Raven, F.; Ward, J.F.; Zhen, S.; Zhang, S.; Sun, H.; Miller, S.J.; Choi, S.H.; Tanzi, R.E.; Zhang, C. Upregulation of Alzheimer’s Disease Amyloid-β Protein Precursor in Astrocytes Both in vitro and in vivo. J. Alzheimers Dis. 2020, 76, 1071–1082. [Google Scholar] [CrossRef]
- Schieb, H.; Kratzin, H.; Jahn, O.; Mobius, W.; Rabe, S.; Staufenbiel, M.; Wiltfang, J.; Klafki, H.W. β-Amyloid peptide variants in brains and cerebrospinal fluid from amyloid precursor protein (APP) transgenic mice: Comparison with human Alzheimer amyloid. J. Biol. Chem. 2011, 286, 33747–33758. [Google Scholar] [CrossRef] [Green Version]
- Moore, B.D.; Chakrabarty, P.; Levites, Y.; Kukar, T.L.; Baine, A.M.; Moroni, T.; Ladd, T.B.; Das, P.; Dickson, D.W.; Golde, T.E. Overlapping profiles of Aβ peptides in the Alzheimer’s disease and pathological aging brains. Alzheimers Res. Ther. 2012, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Bayer, T.A.; Wirths, O. Focusing the amyloid cascade hypothesis on N-truncated Aβ peptides as drug targets against Alzheimer’s disease. Acta Neuropathol. 2014, 127, 787–801. [Google Scholar] [CrossRef] [Green Version]
- Oberstein, T.J.; Spitzer, P.; Klafki, H.W.; Linning, P.; Neff, F.; Knölker, H.J.; Lewczuk, P.; Wiltfang, J.; Kornhuber, J.; Maler, J.M. Astrocytes and microglia but not neurons preferentially generate N-terminally truncated Aβ peptides. Neurobiol. Dis. 2015, 73, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Akama, K.T.; Krafft, G.A.; Chromy, B.A.; Van Eldik, L.J. Amyloid-beta peptide activates cultured astrocytes: Morphological alterations, cytokine induction and nitric oxide release. Brain Res. 1998, 785, 195–206. [Google Scholar] [CrossRef]
- Craft, J.M.; Watterson, D.M.; Frautschy, S.A.; Eldik, L.J.V. Aminopyridazines inhibit β-amyloid-induced glial activation and neuronal damage in vivo. Neurobiol. Aging 2004, 25, 1283–1292. [Google Scholar] [CrossRef] [Green Version]
- White, J.A.; Manelli, A.M.; Holmberg, K.H.; Van Eldik, L.J.; LaDu, M.J. Differential effects of oligomeric and fibrillar amyloid-β1–42 on astrocyte-mediated inflammation. Neurobiol. Dis. 2005, 18, 459–465. [Google Scholar] [CrossRef]
- Jin, S.M.; Cho, H.J.; Kim, Y.W.; Hwang, J.Y.; Mook-Jung, I. Aβ-induced Ca2+ influx regulates astrocytic BACE1 expression via calcineurin/NFAT4 signals. Biochem. Biophys. Res. Commun. 2012, 425, 649–655. [Google Scholar] [CrossRef]
- Herculano-Houzel, S. The glia/neuron ratio: How it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia 2014, 62, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Perez-Alvarez, A.; Navarrete, M.; Covelo, A.; Martin, E.D.; Araque, A. Structural and functional plasticity of astrocyte processes and dendritic spine interactions. J. Neurosci. 2014, 34, 12738–12744. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.; Moro, D.H.; Mouffle, C.; Didienne, S.; Hepp, R.; Pfrieger, F.W.; Mangin, J.M.; Legendre, P.; Martin, C.; Luquet, S.; et al. Mapping astrocyte activity domains by light sheet imaging and spatio-temporal correlation screening. NeuroImage 2020, 220, 117069. [Google Scholar] [CrossRef] [PubMed]
- Stafford, N.; Wilson, C.; Oceandy, D.; Neyses, L.; Cartwright, E.J. The Plasma Membrane Calcium ATPases and Their Role as Major New Players in Human Disease. Physiol. Rev. 2017, 97, 1089–1125. [Google Scholar] [CrossRef] [Green Version]
- Strehler, E.E.; Thayer, S.A. Evidence for a role of plasma membrane calcium pumps in neurodegenerative disease: Recent developments. Neurosci Lett. 2018, 663, 39–47. [Google Scholar] [CrossRef]
- Fortalezas, S.; Marques-da-Silva, D.; Gutierrez-Merino, C. Creatine Protects Against Cytosolic Calcium Dysregulation, Mitochondrial Depolarization and Increase of Reactive Oxygen Species Production in Rotenone-Induced Cell Death of Cerebellar Granule Neurons. Neurotox. Res. 2018, 34, 717–732. [Google Scholar] [CrossRef]
- Fortalezas, S.; Poejo, J.; Samhan-Arias, A.K.; Gutierrez-Merino, C. Cholesterol-Rich Plasma Membrane Submicrodomains Can Be a Major Extramitochondrial Source of Reactive Oxygen Species in Partially Depolarized Mature Cerebellar Granule Neurons in Culture. J. Neurphysiol. Neurol. Dis. 2019, 5, 1–22. [Google Scholar] [CrossRef]
- Poejo, J.; Salazar, J.; Mata, A.M.; Gutierrez-Merino, C. Binding of Amyloid β(1–42)-calmodulin Complexes to Plasma Membrane Lipid Rafts in Cerebellar Granule Neurons Alters Resting Cytosolic Calcium Homeostasis. Int. J. Mol. Sci. 2021, 22, 1984. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.P.Y.; Li, G.; Auston, D.A. Chapter 5—Practical Aspects of Measuring Intracellular Calcium Signals with Fluorescent Indicators. In Methods in Cell Biology; Academic Press: Cambridge, MA, USA, 2010. [Google Scholar] [CrossRef]
- McCormack, J.G.; Cobbold, P.H. Cellular Calcium: A Practical Approach—The Practical Approach Series; Oxford University Press: Oxford, UK, 1991. [Google Scholar]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poejo, J.; Berrocal, M.; Saez, L.; Gutierrez-Merino, C.; Mata, A.M. Store-Operated Calcium Entry Inhibition and Plasma Membrane Calcium Pump Upregulation Contribute to the Maintenance of Resting Cytosolic Calcium Concentration in A1-like Astrocytes. Molecules 2023, 28, 5363. https://doi.org/10.3390/molecules28145363
Poejo J, Berrocal M, Saez L, Gutierrez-Merino C, Mata AM. Store-Operated Calcium Entry Inhibition and Plasma Membrane Calcium Pump Upregulation Contribute to the Maintenance of Resting Cytosolic Calcium Concentration in A1-like Astrocytes. Molecules. 2023; 28(14):5363. https://doi.org/10.3390/molecules28145363
Chicago/Turabian StylePoejo, Joana, María Berrocal, Lucía Saez, Carlos Gutierrez-Merino, and Ana M. Mata. 2023. "Store-Operated Calcium Entry Inhibition and Plasma Membrane Calcium Pump Upregulation Contribute to the Maintenance of Resting Cytosolic Calcium Concentration in A1-like Astrocytes" Molecules 28, no. 14: 5363. https://doi.org/10.3390/molecules28145363
APA StylePoejo, J., Berrocal, M., Saez, L., Gutierrez-Merino, C., & Mata, A. M. (2023). Store-Operated Calcium Entry Inhibition and Plasma Membrane Calcium Pump Upregulation Contribute to the Maintenance of Resting Cytosolic Calcium Concentration in A1-like Astrocytes. Molecules, 28(14), 5363. https://doi.org/10.3390/molecules28145363