Since Cu-BTC samples in the current study had a high percentage of internal surface, as discussed above, pore diffusion was believed to the main factor in affecting the transport of Pb(II) ions inside the structures. The binding of adsorbates to the active adsorption sites is a complex process and is influenced by a variety of factors. The current study focused on two important factors: (a) the availability of active adsorption sites on the surface of Cu-BTC MOFs, and (b) the nature of these active adsorption sites in terms of their homogeneity/heterogeneity.
2.2.1. Adsorption Kinetic Study
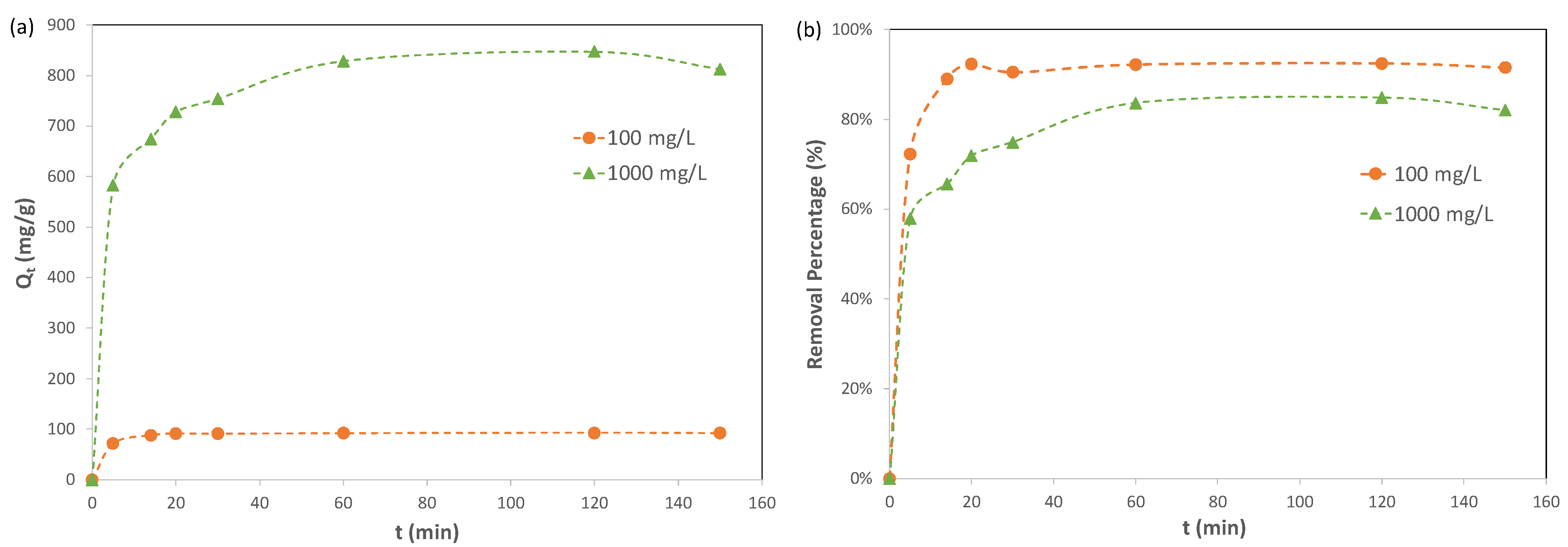
Adsorption kinetics describes the rate of adsorbate uptake on an adsorbent. In the current study, the adsorption kinetics of Pb(II) ions on Cu-BTC MOFs were investigated for two lead solutions with initial concentrations of Pb(II) ions of 100 mg/L and 1000 mg/L, respectively. The results are included in
Figure 4. For the 100 mg/L solution, the adsorption reached equilibrium at 20 min, and the adsorption capacity of Pb(II) ions on Cu-BTC MOFs at equilibrium,
qe, had an averaged value of 91.98 mg/g, equivalent to 92% removal. For the 1000 mg/L solution, the results indicated that the adsorption took a longer time to reach equilibrium, namely, 60 min. However, Cu-BTC MOFs displayed a much higher adsorption capacity of Pb(II) ions at 829.6 mg/g in this solution, while the removal percent was lower, with a value of 83%.
Exploring the adsorption mechanism using kinetic data is common practice in the literature [
10,
11,
12,
14,
15,
17,
19], and certain mathematic models are employed to fit the kinetic data. Equations (1) and (2) represents two of the commonly used models, namely, the “pseudo first order” model and the “pseudo second order” model.
where
(mg/g) is the adsorption capacity at equilibrium, and
(mg/g) is the adsorption capacity at a given adsorption time
t (min).
(min
−1) and
(g/mg/min) are the rate constants of the pseudo first order adsorption and the pseudo second order adsorption, respectively.
Figure 5 shows the plots of the kinetic data for the adsorption of Pb(II) on Cu-BTC using the pseudo first order and the pseudo second order models, and the corresponding results are summarized in
Table 2. It is noteworthy that only pre-equilibrium data points were used for the plots, which included those from 0 to 20 min for the 100 mg/L solution and those from 0 to 60 min for the 1000 mg/L solution. Such steps were taken because a close inspection of the kinetic data fitting practice [
29,
30] indicated that the inclusion of post-equilibrium data would introduce a methodological bias. As a result, the pseudo second order kinetics model has been widely and unfairly favored as the dominant model.
For both solutions being studied, the results shown in
Figure 5 and in
Table 2 indicated that the pseudo second order model was still favored over the pseudo first order model to describe the adsorption kinetics of Pb(II) ions on Cu-BTC MOFs. This was based on the comparison made for the coefficient of determination, or R
2, between these two models. For the 1000 mg/L solution, the R
2 value was clearly favored for the pseudo second order model, though the R
2 value for the 100 mg/L solution was only slightly favored for the pseudo second order model. In addition to the R
2 values, the pseudo second order model also predicted the adsorption capacities (
qe) of 93.46 mg/g and 833.3 mg/g for the two solutions, respectively, which were closer to the experimental values (91.98 mg/g and 829.6 mg/g) than those derived from the pseudo first order model (78.58 mg/g and 814.4 mg/g). Therefore, the pseudo second order model is more suitable to describe the adsorption kinetics of Pb(II) ions on Cu-BTC MOFs.
As described above, the second step in an adsorption process can be characterized in the context of binding between adsorbates and adsorbents. Based on the nature of such interactions, adsorption is generally classified as either physisorption (characteristic of weak van der Waals forces) or chemisorption (characteristic of covalent bonding). In many published works [
11,
12,
14,
17], pseudo first order and pseudo second order kinetic models were used as main tools to identify these two different types of adsorption. Claims were made based on the assumption that if the pseudo first order model displays a better fit for the experimental results, then adsorption is a physical process, while if the pseudo second order model fits better, then it is a chemical adsorption. In the current study, caution was taken for interpreting the kinetic results in this manner. This is because (a) the two models as expressed by Equations (1) and (2) were obtained from empirical differential equations, and there was lack of clear evidence that they were derived based on theories of adsorbate–adsorbent interaction; and (b) even though some known chemisorption cases in the literature [
8,
13] are described well by the pseudo second order model, the opposite is not always true. With such caution being taken, we used these two mathematic models to examine the adsorption based on the fundamentals of kinetic theories, that is, the limiting steps in the adsorption could be identified from the kinetic data.
The adsorption and desorption of a solute A in a solution can be represented by an interface reaction as follows:
where
and
are the adsorption and desorption rate constants, respectively, the subscripts (
l) and (
s) denote the liquid and solid phases, respectively, and
S and
AS represent the active sites capable of adsorbing adsorbate
A and those sites occupied by
A on the surface of the adsorbent, respectively.
Since the adsorption kinetics of Pb(II) ions on Cu-BTC MOFs agreed better with the pseudo second order model, one of the reasonable interpretations for such an observation is that the rate of adsorption in this case was being controlled by two limiting species: (a) the number of adsorbates near the adsorbent’s surface (not necessarily equal to the overall number of adsorbates in the bulk solution), and (b) the number of active adsorption sites on the adsorbent’s surface. The former was affected by the first adsorption step, namely, the transport of the adsorbates to the exterior of the adsorbents, while the latter was affected by the second adsorption step, namely, the binding of adsorbates to the adsorption sites. Therefore, both the transport and the binding steps were rate-limiting in the adsorption kinetics of Pb(II) ions on Cu-BTC MOFs. Since micropore surface areas in the Cu-BTC MOFs of this study accounted for the majority of the total surface areas, as shown in
Table 2, pore diffusion rather than film diffusion is believed to have controlled the transport of Pb(II) ions in this case. Therefore, most transport must occur within the pores of Cu-BTC MOFs rather than their external surface. With regard to the fact that the number of active adsorption sites on the Cu-BTC’s surface also controls the rate of adsorption, it could be indicative of heterogeneous rather than homogenous adsorption occurring on the Cu-BTC MOFs. In other words, adsorption occurs only on the Cu-BTC’s surface with preferred sites and those sites become scarce when adsorption progresses. As a result, Pb(II) ions must travel further into the pores to reach those unoccupied preferred sites.
2.2.2. Adsorption Equilibrium Study
From an equilibrium point of view, adsorption from a solution results in the continuous uptake of adsorbates until the concentration of adsorbates remaining in the solution is in equilibrium with that at the surface of adsorbents. In the literature, such an equilibrium is described in terms of an adsorption isotherm, in which the uptake of adsorbates per until weight of adsorbent, defined above as adsorption capacity (
qe), is plotted against the concentration of adsorbates remaining in solution (
Ce) when equilibrium is reached. In the current study, measurements were made on a series of lead solutions with the initial concentration (
C0) ranging from 10 mg/L to 1500 mg/L. After equilibrium was reached,
Ce was determined by AAS, from which
qe was calculated, as described in
Section 3.4. In many studies [
11,
12,
14,
19,
21], however, most of the data collected for adsorption isotherms fell in the range of unchanged
qe, which means that in those solution, adsorbents were saturated with adsorbates. This practice may cause some unintended bias toward the data points prior to saturation being reached if caution is not taken. The current study included the solutions in the range of 10–50 mg/L with an increment of 10 mg/L for adsorption measurements in addition to those with concentrations between 100 and 1500 mg/L. This assured that enough data in the low concentration range were included in the equilibrium study.
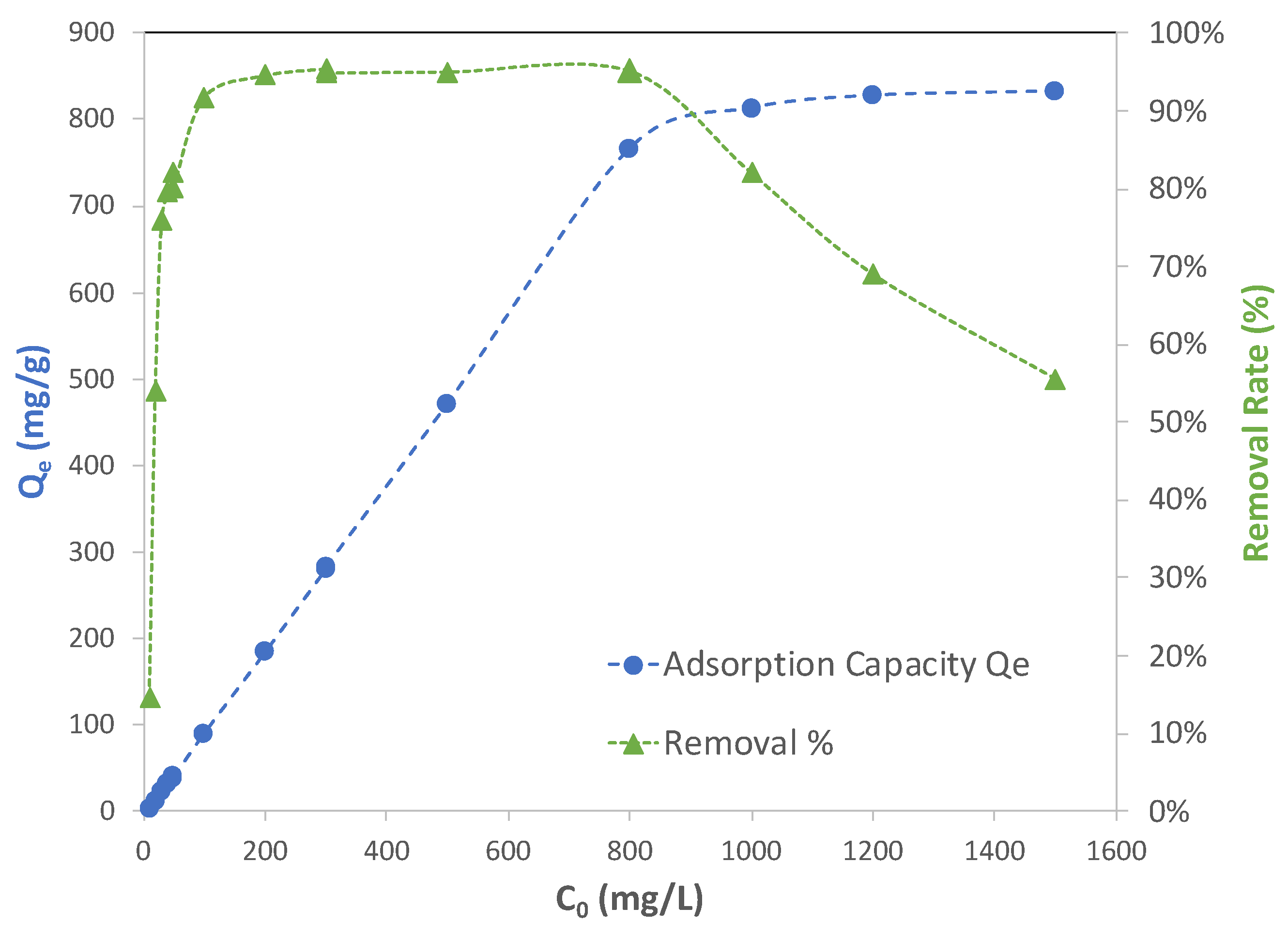
Figure 6 illustrates how adsorption capacity (
qe) and removal percent changed with the initial concentration of lead solution (
C0). The blue dot curve in the figure shows that adsorption capacity started at a low value, meaning that Cu-BTC MOFs were not being fully utilized but increased until they reached a plateau after the initial concentration reached 800 mg/L. This indicates that at this concentration, Cu-BTC MOFs were close to maximizing their capacity of adsorbing Pb(II) ions. At concentrations higher than 800 mg/L, Cu-BTC MOFs left most of the excess Pb(II) ions unadsorbed. The adsorption performance of Cu-BTC MOFs could also be seen from the removal percent results, which are represented by the green triangle curve in
Figure 6. The MOFs displayed the highest removal of near 95% for the solutions between 100 mg/L and 800 mg/L, and beyond 800 mg/L, the removal percent declined. Since Cu-BTC MOFs only reached their maximum capacity of adsorption at the concentration of 800 mg/L, this led to an expectation that the MOFs in the solutions with a lower concentration than 800 mg/L would carry a comparable level of capacity to that in the 800 mg/L solution. However, it is seen from
Figure 6 that the removal percent started at only about 15% for the solutions of 10 mg/L and then increased until it starts to plateau at the concentration of 100 mg/L.
The equilibrium results shown in
Figure 6 corroborated the interpretation made earlier from the kinetic data in terms of the two steps involved in adsorption, including the transport of adsorbates (mainly by pore diffusion) and the binding between adsorbate and adsorbent. The transport of adsorbates in solution is driven by a departure from an equilibrium condition, under which the concentration of adsorbates remaining in the bulk solution is equal to that at the surface of adsorbents. This is the “driving force” of the transport. On the other hand, when adsorbates travel in the fluid, passing through the pores inside the adsorbents until they find vacant adsorption sites, they also experience resistance. For given adsorbates, such a resistance force could be affected by the chemical and physical properties of adsorbents, such as the nature of their surface charges, their surface chemistry, as well as the geometry and the size of pores of the adsorbents. When adsorbents are porous, especially with micropores in the majority, pore diffusion rather than film diffusion is in control of the transport, as it was in the samples of the current study. In such cases, the resistance will become more prominent, and thus it must be taken into consideration when adsorption is being investigated. In simplifying analyses of such transport activity involving both driving and resistance forces, it is reasonably assumed that the driving force has a strong correlation with the concentration of adsorbates. Since the resistance is mainly affected by the property of adsorbents, it is justified to assume that it has negligible a correlation with the concentration of adsorbates.
Overall, the net effect between the driving force and the resistance force will result in adsorbates moving through the internal space of porous adsorbents and reaching the active sites for adsorption. To better illustrate how such a net effect on adsorbates determines adsorption, a schematic diagram is shown in
Figure 7. The net effect on adsorbates is directly attributed to the amount of surface area (therefore the number of active adsorption sites) that adsorbates can reach, namely, reachable surface area (ReSA), as represented by a blue dashed line in the figure. As the initial concentration of adsorbates increases, so does the net effect. Therefore, the reachable surface area increases with concentration. At the low concentration range, however, the net effect is not strong enough to drive adsorbates to reach such an amount of surface areas to achieve full adsorption (as defined by the highest removal percent that the samples can achieve in this regard). The threshold for full adsorption is linked to the so-called full-adsorption surface area (FaSA), which is represented by a blue dotted line in
Figure 7. The full-adsorption surface area is directly proportional to the initial concentration of adsorbates. Within the range of low concentrations, the reachable surface area (ReSA) is smaller than the full-adsorption surface area (FaSA), as illustrated in
Figure 7. As a result, the removal percent within this range, as represented by the green line, is not able to reach its highest level. Such a range corresponded to 10–100 mg/L for Cu-BTC MOFs, as previously shown in
Figure 6. For these solutions, pore diffusion clearly played a dominate role in the “under-adsorption” performance of the MOFs. After the concentration became high enough, which was 100 mg/L for Pb(II) adsorption on Cu-BTC, the net effect was then strong enough so that the reachable surface area surpassed the full-adsorption surface area. As expected, adsorption arrived at the highest rate. As the concentration continued to increase, adsorbates were capable of reaching more surface area in such a way as to maintain at the highest rate of adsorption. Therefore, “full adsorption” was used to characterize the performance within this range. For Pb(II) adsorption on Cu-BTC, this corresponded to the concentration range between 100 and 800 mg/L, as shown in
Figure 6, where
qe continued to rise, and the removal percent remained at the highest level. As the concentration moved even higher, however, adsorbates already used the total accessible surface area (TASA) existing in the adsorbents as represented by the blue solid line in
Figure 7. Therefore, after that, the adsorption capacity plateaued, and the removal percent started to decline. This was observed for the adsorption of Pb(II) on Cu-BTC in the solution beyond 800 mg/L. Since the surface areas were directly related to the number of active adsorption sites, the “under-adsorption” performance within this concentration range was clearly due to the fact the availability of active sites limited adsorption in these solutions, which was different than the “under-adsorption” observed in the low concentration range, as mentioned earlier.
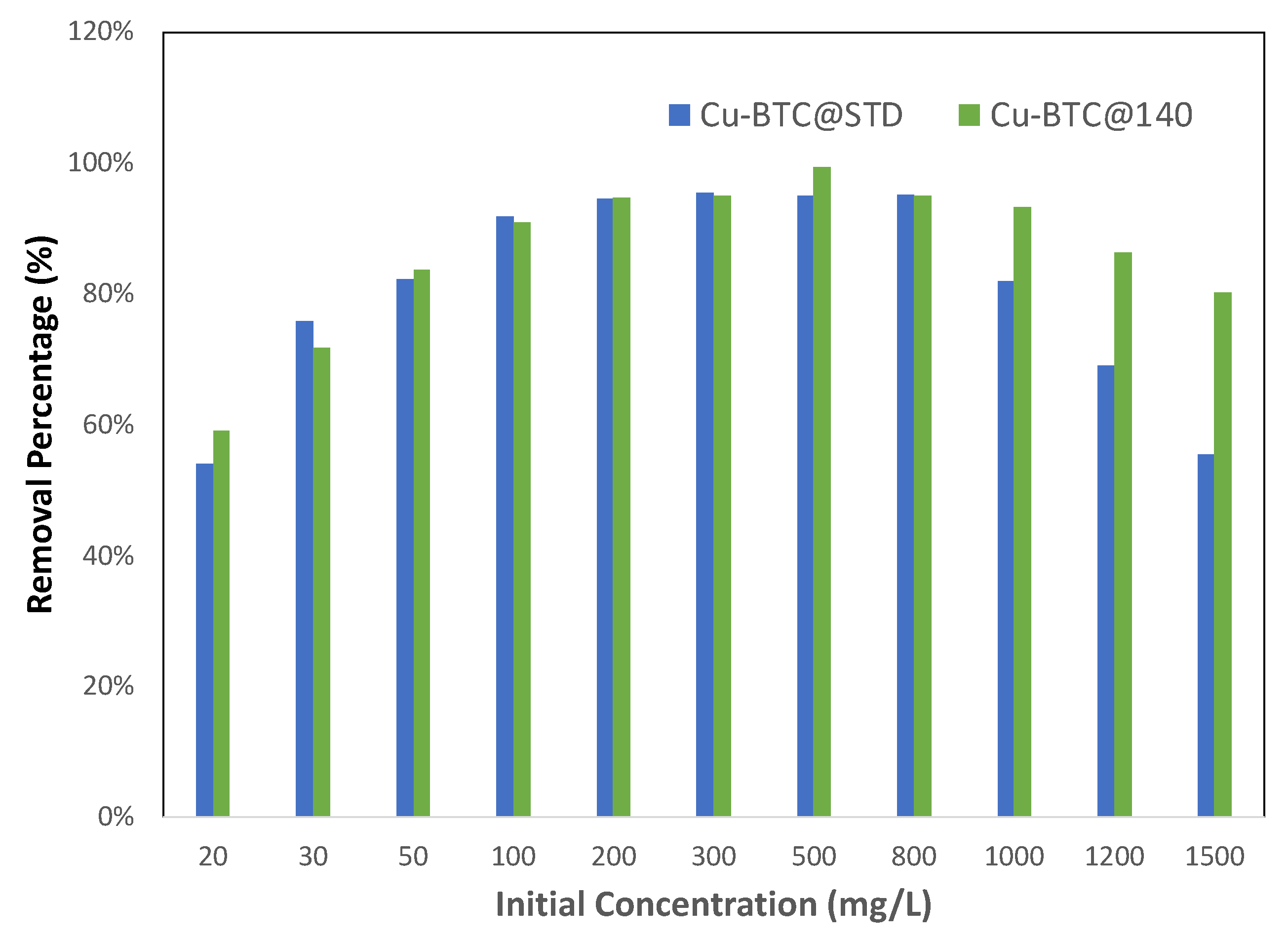
To further examine the role that the availability of active sites plays in adsorption, equilibrium measurements were also made to compare between two sets of Cu-BTC MOFs with different surface areas. The results are shown in
Figure 8. As stated in
Section 3, Cu-BTC@STD is a commercial product, while Cu-BTC@140 is our lab-made sample synthesized at 140 °C by using microwave method. As shown in
Table 1, both samples contained a majority of micropores in their structure with similar pore sizes. However, their surface areas were quite different, with Cu-BTC@STD at 714 m
2/g vs. Cu-BTC@140 at 1930 m
2/g. With the Pb(II) concentration of 100 mg/L and below, as illustrated in
Figure 8, Cu-BTC@140 showed a very similar adsorption performance to Cu-BTC@STD in terms of the removal percent, both of which climbed along with an increase in the concentration until it peaked at 100 mg/L. The reachable surface areas in both MOFs were comparable due to their similarity in pore dimensions, but they were lower than the full-adsorption surface areas for those concentrations. Such similar behavior between these two MOFs is consistent with the analysis made earlier, further indicative that the resistance experienced by adsorbates in the pore diffusion, rather than the number of active adsorption sites, played a determining role in the adsorption performance within this range of concentration. For the concentration between 100 and 800 mg/L, the reachable surface areas in both MOFs became larger than the full-adsorption surface areas, which means that adsorbates in both MOFs were able to reach enough surface area to be adsorbed so that their removal percent was maintained at a peak level. The difference in adsorption performance between these two MOFs appeared in the region beyond 800 mg/L. Cu-BTC@140 contained a larger total available surface area and thus more active adsorption sites than Cu-BTC@STD. As a result, the former was able to adsorb more Pb(II) ions, keeping its removal percent at the peak level until 1000 mg/L, while the latter already started to decline in its removal percent. After 1000 mg/L, a decrease in the removal percent in the Cu-BTC@140 sample also appeared, but such a decline occurred at a much slower pace than did the Cu-BTC@STD sample. Such an observation provided further corroborating evidence that the availability of active sites limits adsorption in the step where adsorbates attempt to bind to adsorbents.
Equilibrium data for the adsorption of Pb(II) ions on Cu-BTC MOFs were also further studied by fitting them with numerical models. At the first, the adsorption isotherm of Pb(II) ions on Cu-BTC MOFs was plotted between the adsorption capacity (
qe) against the equilibrium concentration of adsorbates (
Ce), and the results are shown in
Figure 9a. As the equilibrium concentration increased, there was a sharp increase in the adsorption capacity at the initial stage (referred to as the “acceleration stage”), followed by a much slower second stage (“slowdown stage”). Two of the most-commonly used models for fitting isotherms are the Langmuir isotherm model [
31] and the Freundlich isotherm model [
32].
The Langmuir model assumes that the surface of adsorbents is homogeneous, that only a monolayer of adsorbates is formed on the surface of adsorbents, and that adsorbed molecules do not interact with one another. The linearized form of the Langmuir isotherm is given by Equation (4):
where
qe and
Ce are the same quantities as defined previously,
KL (L/mg) is the Langmuir constant that reflects the affinity of adsorbate and adsorbent, and
qm (mg/g) is the maximum adsorption capacity.
Unlike the Langmuir model, the Freundlich model was established based on the assumption that multilayer adsorption occurs on a heterogeneous adsorbent surface. It also recognizes the interaction of adsorbed molecules. The linearized form of the Freundlich isotherm is given by Equation (5):
where
KF (unit depends on
n) is the Freundlich constant, which is related to the adsorption capacity, and
n is an empirical number related to the adsorption intensity.
A third numerical model, the Temkin model [
33], was also used for the isotherm analysis in this study. Similar to the Freundlich model, this model recognizes multilayer adsorption and the interaction in the layer of adsorbed molecules. However, the Temkin model assumes that the intensity of such an interaction, expressed as heat of adsorption, decays linearly with increasing surface coverage instead of logarithmically, as implied in the Freundlich equation. The Temkin model is described by Equation (6):
where
AT (L/g) is the Temkin constant, related to the equilibrium binding constant corresponding to the maximum binding energy, and
bT (J/mol) is the heat of adsorption.
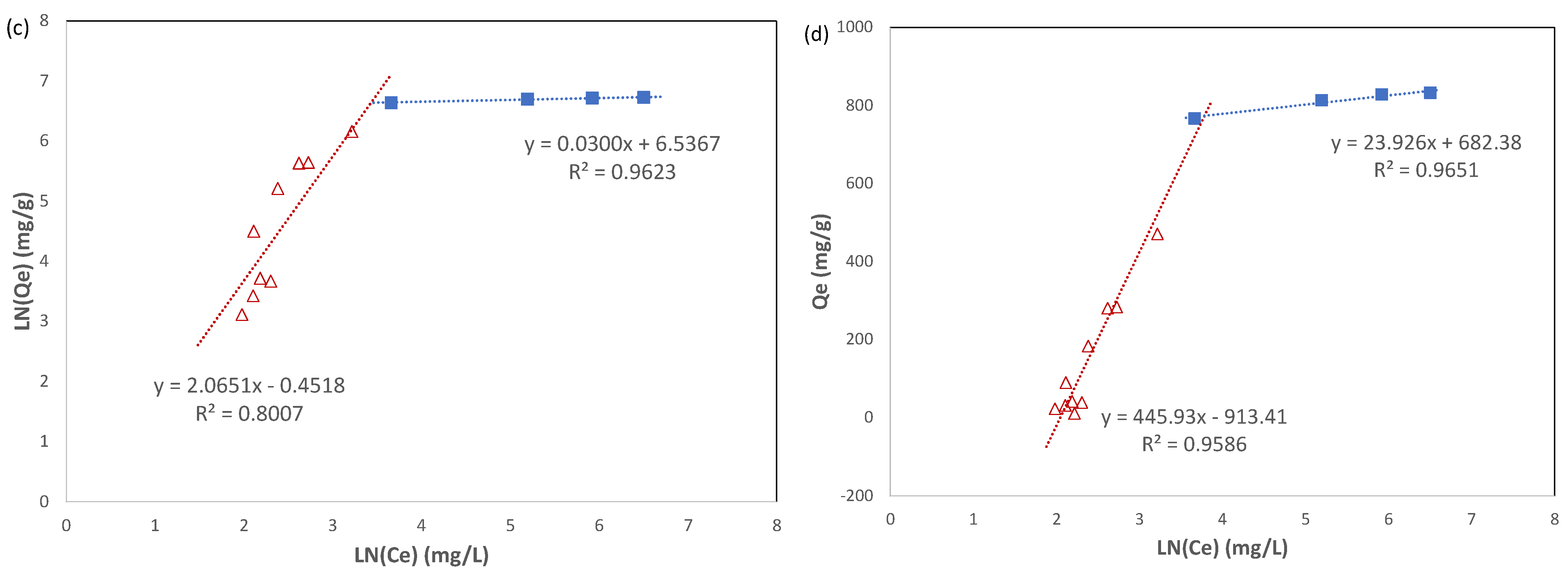
The experimental adsorption isotherm data included in
Figure 9a were fitted with the three numerical models, as described above, and the results are shown in
Figure 9b–d. The data fitting was performed for each individual stage of the adsorption isotherm, the acceleration stage and the slowdown stage, instead of over the whole range. The calculated constants for each model and their coefficient of determination (R
2) are summarized in
Table 3.
In the acceleration stage, the coefficients of determination (R
2) for the Freundlich model and the Temkin model were 0.8007 and 0.9586, respectively, which were superior to that for the Langmuir model, which was 0.1698. Therefore, the Freundlich model and the Temkin model were more satisfactory to describe this stage of adsorption. This supports the scenario that the uptake of Pb(II) ions on Cu-BTC MOFs is multilayer adsorption with preferential binding sites on the adsorbent surface. The fitting results also confirmed that Pb(II) ions in the adsorbed layer have interaction with one another as well as with Cu-BTC MOFs. Since the Temkin model was a better fit with the experiment data in the acceleration stage compared to the Freundlich model, it suggests to some degree that the heat of adsorption, indicative of the intensity of interaction among those adsorbed on Cu-BTC MOFs, decays linearly along with the increasing coverage of the adsorbents. As adsorbates are built up by layers, such an interaction continues to weaken until it does not result in a net gain between adsorbates being adsorbed and those leaving the adsorbents. The model-related constants are also included in
Table 3. Because of the roughness of these models, however, caution has been taken for interpretating the physical meanings of these constants merely based on their magnitude, unless such claims could be verified by other methods.
In the slowdown stage of adsorption, the comparison among all three models showed that the Langmuir isotherm had the best fit to the experimental data, with the coefficient of determination (R
2) at 1. This suggests that a monolayer of Pb(II) ions was formed during this stage of adsorption. However, it occurred to such a very limited extent that the adsorption capacity only increased lightly during this stage, as shown in
Figure 9a. The maximum uptake capacity (
qm) for Pb(II) ions derived from the Langmuir equation was 833.3 mg g
−1, which was quite close to the experimental result as well as the one predicted from the kinetic model. In the literature [
10,
11,
17,
21], the maximum uptake capacity for the adsorption of Pb(II) ions on Cu-BTC MOFs ranges from 220 to 950 mg/g. The results in the current study showed that the Cu-BTC synthesized by the microwave method carried a high capacity for the adsorption of Pb(II) ions.
Most adsorption occurring in solution, especially with heavy metal ions being involved, is dependent on the pH of the solution, since pH not only affects the surface charge of adsorbents but also the form of heavy metal ions. Therefore, examining the effect of pH on adsorption could provide insight into the adsorption mechanism.
Figure 10 shows how the adsorption of Pb(II) ions on Cu-BTC MOFs changed with various pH values of the solution. For pH < 3, there was little adsorption between Pb(II) ions and Cu-BTC MOFs. However, above this threshold value, the results showed that a significant amount of Pb(II) ions was adsorbed, and the removal % was comparable among all the solutions being studied. For transition divalent metals, such as Pb(II) in the current study, the most dominant species in their salt solutions are cations, such as Pb
2+ and Pb(OH)
+ [
13,
34], unless the solutions are adjusted to be basic. Positively charged hydronium ions (H
3O
+) also co-exist in the solution. Therefore, these metal cations may have to compete with hydronium ions for the preferential binding sites on the adsorbent. What is observed from
Figure 10 validated such a scenario for the adsorption of Pb(II) ions on Cu-BTC MOFs. At the pH of 2, there was a large amount of hydronium ions existing in the solution. Hydronium ions have advantage over Pb(II) ions so that they occupy most of the active adsorption sites on Cu-BTC MOFs. When this happens, the surface charge of Cu-BTC might be also changed from being negative to positive. As a result, Pb(II) ions could be repulsed from, instead of being attracted to, the surface of the MOFs. That is why few of the Pb(II) ions were adsorbed at pH = 2. When the pH value was larger, however, the concentration of hydronium ions became lower. As a result, more of the preferential binding sites may have remained available for Pb(II) ions to be adsorbed to, and at the same time, the surface charge of Cu-BTC MOFs may also have remained negative, so that additional layers of Pb(II) ions could be adsorbed, even after hydronium ions were adsorbed. Therefore, the pH results further supported that there are preferential binding sites on the surface of Cu-TBC. As for the chemical nature of those preferential binding sites, it was reported in the literature [
8,
13] that they would be the negative regions at the carboxylic acid group on Cu-BTC MOFs, but this is beyond the scope of the current study.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}